Abstract
Epigenetic processes, such as DNA methylation, are known to regulate tissue specific gene expression. We explored this concept in the placenta to define whether DNA methylation is cell-type specific. Cytotrophoblasts and fibroblasts were isolated from normal midtrimester placentas. Using immunocytochemistry, we demonstrated 95% purity for cytotrophoblasts and 60-70% for fibroblasts. We compared DNA methylation profiles from cytotrophoblasts, fibroblasts and whole placental villi using bisulfite modified genomic DNA hybridized to the Illumina Methylation27 array. Euclidean cluster analysis of the DNA methylation profiles showed 2 main clusters, one containing cytotrophoblasts and placenta, the other fibroblasts. Differential methylation analysis identified 442 autosomal CpG sites that differed between cytotrophoblasts and fibroblasts, 315 between placenta and fibroblasts and 61 between placenta and cytotrophoblasts. Three candidate methylation differences were validated by targeted pyrosequencing assays. Pyrosequencing assays were developed for CpG sites less methylated in cytotrophoblasts than fibroblasts mapping to the promoter region of the beta subunit of human chorionic gonadotropin 5 (CGB5), as well as 2 CpG sites mapping to each of 2 tumor suppressor genes. Our data suggest that epigenetic regulation of gene expression is likely to be a key factor in the functional specificity of cytotrophoblasts. These data are proof of principle for cell-type specific epigenetic regulation in placenta and demonstrate that the methylation profile of placenta is mainly driven by cytotrophoblasts.
Introduction
Epigenetic mechanisms are stable, mitotically heritable chemical and conformational modifications of DNA or its associated histone proteins. These modifications regulate the access of transcriptional elements to the DNA sequence without altering the primary nucleotide sequence.Citation1 Since epigenetic modifications are important regulators of cellular differentiation,Citation2 differential patterns of epigenetic modifications can be demonstrated between tissue typesCitation3,Citation4 and stages of development.Citation5,Citation6
DNA methylation, one of the best studied post-transcriptional epigenetic modifications, is characterized by the addition of a methyl group to the nucleotide cytosine by DNA methyl transferases. In eukaryotes this addition generally occurs on cytosines followed by guanine (CpG).Citation7 When this chemical modification occurs in promoter regions of genes, transcription is usually altered.Citation5,Citation7 DNA methylation, like other epigenetic processes, is known to differ among cell types within certain tissues.Citation3,Citation8,Citation9 This has been demonstrated in several breast tissue cell-types,Citation8 among different white blood cell typesCitation3 and between embryonic and adult stem cells.Citation10 Thus, the degree of methylation measured in a tissue is an average of the methylation in all the existing cell types. Variations in the proportions of specific cell types among samples of the same tissue can make interpretation of methylation differences between such samples difficult.
The identification of differences in cell-specific methylation within the placenta could provide the basis for understanding the epigenetic regulation of placenta cell-type specific functions. Furthermore, such data could enhance the analysis and interpretation of placental studies of epigenetic variation in association with disease.
Our objective was to determine whether DNA methylation in the human placenta is cell type-specific. Epigenetic modifications have been found, in cancer cells, to confer the capacity for uncontrolled proliferation and invasion.Citation1,Citation11 In a similar but more organized fashion, the invasive extra-villous trophoblast (EVT) cells of the placenta invade the endometrium.Citation12 We hypothesized that this similarity in biological behavior between neoplasms and placenta would be translated into epigenetic marks that could be detected by DNA methylation profiling.
Therefore we undertook an analysis of genome-wide methylation profiles in second trimester placenta and compared it to cell type specific fractions of cytotrophoblasts and fibroblasts. Although the separation of cell types was incomplete, we were able to define cell type specific differences in DNA methylation.
Our DNA methylation profiling study of placenta and its cell types, cytotrophoblasts and fibroblasts, demonstrates cell-type specific differences. We have validated the methylation differences for three of the genomic regions identified through our screening approach thereby confirming epigenetic signatures that are specific for cell types in the placenta. Importantly, our data PAPERdemonstrate that epigenetic studies of placental villi will reflect mainly the profile of cytotrophoblast cells.
Results
Placenta fractionation and cell population enrichment.
Sufficient amounts of cytotrophoblasts and fibroblasts were successfully separated by sequential enzymatic digestion and magnetic bead separation for the DNA methylation profiling experiments. However, the cell purity was much greater for cytotrophoblasts than for fibroblasts. Trypsin is a protease that cleaves trophoblastic cells from placental villi and results in collection of good quantities of excellent quality cytotrophoblasts, as previously reported.Citation13 In our experiment, we used a lower concentration of trypsin (0.25%) for an increased length of time to ensure gentle digestion and to limit cell membrane disruption and release of intra-cytoplasmic RNA and DNA. The proteolytic enzyme collagenase was then used to disrupt the extracellular matrix and enhance the release of mesenchymal type cells. Its use allowed us to collect significant amounts of fibroblasts, which were not obtained with trypsin digestion alone.
Placental cells can be identified by their size, the specific antigens that they express on the cell surface and the hormones that they produce. Cell fraction purity can be assessed using various modalities including immunocytochemistry or flow cytometry analysis; cytokeratin-7/vimentin-9 antibodies have been found to correlate well with trophoblast purity obtained by flow cytometry.Citation14 We demonstrated 95% purity of cytotrophoblasts and 60–70% in the fibroblast fraction, employing immunocytochemistry (). The fibroblast fraction was found to contain some debris; the contaminant cell population was cytotrophoblasts (immunostained by cytokeratin-7 antibody). For each placenta sample, three DNA aliquots were successfully extracted from cytotrophoblasts, fibroblasts and whole placentas.
DNA methylation analysis.
We hybridized sodium bisulfite modified DNA samples onto Illumina® Infinium Human Methylation27 BeadChip arrays. These arrays probe more than 27,000 CpG sites in the genome mostly mapping to promoter regions of ∼14,000 genes. Generally, each gene is represented by two probes, located in the promoter of the genes, one on each side of the putative transcription start site (TSS). Less frequently there are single or multiple probes. Single probes also map mainly to promoter regions whereas multiple probes generally map to promoter and intragenic sequences. The Illumina array was developed to include several cancer-related genes that are represented by two or more probes on the array.
A total of 21 DNA samples were hybridized to two Illumina array silica slides: three cell fractions for each one of the six placentas and a set of technical replicates (cytotrophoblast, fibroblast and placenta fractions of the same sample). Technical replicates were hybridized on different slides. One fibroblast sample was excluded due to suboptimal bisulfite conversion. We identified a strong correlation between methylation profiles of samples and their respective technical replicates (Placenta R2 = 0.989, fibroblast R2 = 0.982, cytotrophoblasts R2 = 0.986), indicating high reproducibility of these arrays.
Global methylation comparison among samples.
We tested for differences in global patterns of methylation between the two cell types. As expected, most probes in the array mapped to CpG islands in promoter regions and also, as expected, most had low methylation levels in all samples (interquartile range, ∼0.02–0.4; median, 0.08–0.16; mean, 0.24–0.27).Citation3,Citation15,Citation16 Correlations among all the arrays were similar even between the different types of samples [R2 = 0.90–0.95 (mean = 0.92)]. There was no significant variation with gestational age (R2 = 0.97) among our samples.
Non-hierarchical Euclidean cluster analysis was performed for 17 samples (). Samples were labeled by sample number, gestational age group (early versus late second trimester) and by cell type. There was no obvious clustering by sample number or by gestational age group. However, the methylation profiles for cytotrophoblasts tended to cluster together in a group with whole placenta. These profiles were distinct from the data obtained for the fibroblast fraction (). This observation suggests that methylation levels in non-fractionated placental samples are more likely to reflect the methylation levels of cytotrophoblasts than fibroblasts.
Cell type-specific differential methylation analysis.
Differential methylation analysis was carried out in order to identify representing CpGs in genomic regions with biologically meaningful differences in methylation between the two cell types. For this, we used the gene methylation selection criteria described in the Methods.
A total of 442 autosomal probes showed a methylation difference higher or equal to 20%, between fibroblasts and cytotrophoblasts (Wilcoxon rank-sum test p value < 0.05) (). In comparison, 315 probes were different, by the same criteria, between placentas and fibroblasts and 61 probes show the same type of difference between placentas and cytotrophoblasts. This is compatible with the Euclidean cluster analysis which demonstrated that placentas and cytotrophoblasts tended to cluster together but not with fibroblasts.
The 442 probes differentially methylated in fibroblasts versus cytotrophoblasts map to 375 autosomal genes; among those, 310 probes (265 genes) were more methylated in fibroblasts and 132 probes (111 genes) were more methylated in cytotrophoblasts. One of the genes, TP73, had probes in both groups; therefore, the sum of genes in both groups is more than the total of genes with differentially methylated probes. Supplemental Table 1 provides an annotated list of the genes selected as differentially methylated between cytotrophoblasts and fibroblasts by our criteria (> 20% difference in methylation).
Previous reports suggest that tissue-specific differentially methylated regions may be CpG content poor.Citation3,Citation9 Therefore, we looked at the relative proportion of cell type-specific differentially methylated probes on our list, mapping to CpG rich (CpG Island) versus CpG poor promoter regions. Only the probes that were more methylated in fibroblasts than in cytotrophoblasts showed an enrichment for CpG probes located in CpG poor regions relative to the same proportion in the arrays. In this group of probes, 170 map to CpG poor regions and 141 map to CpG islands (CpG rich) whereas in the array, the frequency of CpG poor regions is 0.27 (binomial p value < 0.001). In contrast, for probes more methylated in cytotrophoblasts there is no such enrichment, (32 map to CpG poor regions and 100 to CpG islands, p value = 0.5). Our cell type-specific methylation data for placenta suggest that tissue-specific differentially methylated regions may not always occur preferentially in CpG poor regions.
In the list of genes showing a difference in methylation between cytotrophoblasts and fibroblasts we sought to identify genes that were known to be either expressed or repressed in cytotrophoblasts. A lower level of methylation was found in cytotrophoblasts, as compared to fibroblasts, in probes mapping to the promoter regions of several genes coding for the β subunit of human Chorionic Gonadotrophin (β-hCG)—CGB. Placental human chorionic gonadotrophin (hCG) is known to be produced by cytotrophoblasts.Citation17 hCG is a glycoprotein with two subunits, α and β. The α subunit is encoded by the CGA gene on chromosome 6q14-q21 and it is common to all the members of a family of peptide hormones (LH, FSH, TSH and hCG). The β subunit is encoded by four genes, CGB7, 8, 5 and 3, all located in a 50 kb cluster on chromosome 19q13.3. Two other genes mapping to the same cluster, CGB1 and 2, encode two hypothetical, although not yet identified proteins.Citation18 The Illumina® array has probes targeting CpG sites mapping to the promoter region of CGB1 (two probes), CGB2 (two probes), CGB3 (one probe), CGB5 (two probes) and CGB8 (one probe). Our selection criteria detected differential hypomethylation in cytotrophoblasts for one of the two probes present in the promoter region of CGB1. Also differentially hypomethylated in cytotrophoblasts were the two probes mapping to CGB5 and the single probes mapping to the CGB8 and the CGB3. In contrast, the two probes mapping to CGB2 were not differentially methylated between the cell types, nor was the probe mapping to the α-subunit gene, CGA (). Since lower promoter methylation is often associated with higher gene expression, these data suggest that CGB trophoblast-specific expression is at least partially epigenetically regulated by promoter methylation.
We noted the presence of cancer-related genes in our gene list of differentially methylated genes between cytotrophoblasts and fibroblasts. In general, the CpGs mapping to the promoter regions of cancer genes encoding proteins with tumor suppressor function seem to be more methylated in the cytotrophoblast group.
To explore the possibility that tumor suppressor genes are more methylated in cytotrophoblasts than fibroblasts, we compiled a list of tumor suppressor genes and oncogenes for binomial enrichment analysis of two lists, i.e., higher or lower methylation of cytotrophoblasts versus fibroblasts. The only statistically significant enrichment was for probes mapping to tumor suppressor genes in the probes that are more methylated in cytotrophoblasts than fibroblasts. Of 26,493 autosomal probes in the array, 831 probes map to our list of tumor suppressor gene regions (corresponding to 276 genes). Of the 131 probes in the group with higher methylation in cytotrophoblasts than fibroblasts, 16 probes map to eight tumor suppressor genes (binomial test p value 4.52E-06). presents an annotated list of the probes and shows their methylation levels in cytotrophoblasts and fibroblasts for the genes represented by multiple probes. Two genes, APCCitation19,Citation20 and RASSF1,Citation21 have been previously reported to exhibit unique methylation patterns in placentas and in cancer. Others not previously reported (TP73, RASSF5, DAB2IP, PRKCDBP, MORF4L1) are also shown by our data to have probes in the promoter region that are more methylated in cytotrophoblasts than in fibroblasts. In contrast, of the probes with higher methylation in fibroblasts than cytotrophoblasts, only seven map to tumor suppressor genes. That is, there is no enrichment for these probes. Only one maps to a promoter region, the remainder map to introns. These findings highlight the overlapping biologic characteristics of placental tissue and tumorsCitation22,Citation23 and suggest that such characteristics are, at least in part, epigenetically regulated.
Targeted validation of APC, TP73 and CGB5.
We used pyrosequencing of bisulfite converted DNA to validate results of the array data for three probes selected as follows: The first, APC, maps to the promoter region of a tumor suppressor gene that previously was reported to be hypermethylated in placenta. The second, TP73, maps to the promoter of a putative tumor suppressor gene that has not previously been reported to be hypermethylated in placenta. For one of the CGB genes, CGB5, we chose to validate one of the two probes. The pyrosequencing assays measure the methylation of a variable number of CpG sites adjacent to the CpG site represented on the Illumina® arrays (See Sup. Table 2 for the details of each of the pyrosequencing assays).
We assessed the correlation, expressed as R2, between the methylation levels measured by the Illumina® arrays and by pyrosequencing, for the same CpG sites. This assessment gives a measure of the accuracy of the array methylation measurement in predicting the methylation value of each CpG as measured by a different method. The R2 for the methylation measure provided by the array and pyrosequencing for the three selected CpGs analyzed were 0.84, with a slope of 0.72 for the TP73 CpG site, 0.84, with a slope of 1.05 for the APC CpG site and 0.95, with a slope of 0.5 for the CGB5 CpG site.
We also assessed the accuracy of differences obtained from the array for the methylation of each specific CpG site in the cytotrophoblast fraction versus the same site in the fibroblast fraction of the same sample. The R2 for the methylation differences between the two cell fractions provided by the array and pyrosequencing for the three selected CpGs analyzed were 0.98, with a slope of 0.8 for the TP73 CpG site, 0.94, with a slope of 1.2 for the APC CpG site and 0.83, with a slope of 0.5 for the CGB5 CpG site. These data show a high correlation for the methylation values at three different CpG sites as determined by two independent methods.
Finally, we analyzed the accuracy of the array in determining the difference in methylation level of only one CpG as a predictor for parallel differences in the surrounding regional CpG sites. We assessed the absolute differences in methylation level between cytotrophoblasts and fibroblasts at each CpG site tested by the pyrosequencing assay versus the average of all CpG sites in the region. With the exception of one CpG site in each of the TP73 and APC target regions, all the CpG sites and the averages had a statistically significant difference between the values in the cytotrophoblasts and fibroblasts (p < 0.05 by Wilcoxon rank-sum test, n = 6 for each group) (). This shows that the differential methylation identified by the Illumina array analysis often extends beyond the CpG site represented in the array.
This validation of three sets of data obtained from the array analysis, by an independent molecular technology, supports the reliability of the microarray method we used to screen the genome for significant differences in DNA methylation between cytotrophoblasts and fibroblast cell types in placenta.
Discussion
Our experimental approach demonstrates for the first time, that although most genes present in the two main cell types of the placenta (cytotrophoblasts and fibroblasts) exhibit similar promoter methylation patterns, some specific genes show differential promoter methylation. Such differences may in part explain the underlying biologic basis of cell-specific differentiation within the developing human placenta.
We used serial enzymatic digestion followed by Ficoll gradient centrifugation and magnetic bead separation, to maximize the separation of the two main proliferating cell types within placental villi in the second trimester, namely villous cytotrophoblasts and mesenchymal fibroblasts. Most investigators use some, but not all, of these steps to obtain villous cytotrophoblast cells for trophoblast differentiation studies.Citation13 To the best of our knowledge, there are no published reports on the separation of large quantities of fibroblasts from placenta. These cells are more difficult to separate from the extracellular matrix, but they exhibit significant growth potential in culture. Therefore only small amounts are necessary to generate large colonies. We did not culture fibroblasts because cell culture conditions could potentially affect epigenetic marks and become a source of bias.Citation24
Our immunocytochemical (cytokeratin-7/vimentin antibodies) assessment of the remaining samples showed effective purification of villous cytotrophoblasts, but only enrichment of the fibroblast fraction, despite the addition of a double immunomagnetic bead separation step. Despite the protocol limitations, our exploratory data do demonstrate epigenetic differences between these two cell types. Although the data do not permit precise quantification of the methylation differences between cytotrophoblasts and fibroblasts, they do support the hypothesis of cell-type specific gene regulation by differential DNA methylation.
Genes that demonstrate cell-type specific differences in methylation in our study included hCG genes and tumor suppressor genes. Since hCG is produced mainly in cytotrophoblasts,Citation25 the lower promoter methylation levels of several genes coding for the hCG β subunit is consistent with epigenetically regulated cell specific expression.
Moreover, several aspects of early placental development, especially in extra-villous trophoblast cells, rely on rapid cell proliferation and invasion that is analogous to tumor behavior.Citation22–Citation24 Promoter regions of tumor suppressor genes have been found to be methylated in association with reduced expression in tumors.Citation26,Citation27 Some of these same promoter regions have also been reported to be methylated in placenta.Citation19–Citation21,Citation28 On the other hand, most normal tissues exhibit low methylation in these genomic regions, suggesting some molecular methylation signatures of tumor suppressor genes are shared by tumors and placenta. If this epigenetic signature is driven by the placental cell type with invasive behavior, it would be expected that methylation of tumor suppressor genes in cytotrophoblasts should be higher than in fibroblasts. In our list of selected probes with higher methylation in cytotrophoblasts, there was enrichment for CpG sites mapping to tumor suppressor gene promoter regions. Our data therefore suggest that repression, by promoter methylation, of tumor suppressor genes in cytotrophoblasts, facilitates extensive proliferation of this cell type within immature intermediate villi. Subsequent demethylation of these genes is one candidate pathway by which the third trimester placenta could reduce the proliferative potential of villous cytotrophoblasts near term.
There are several positive outcomes from our research. We have improved on previous efforts to separate populations of uncultured fibroblast cells from placenta. Further, we have generated a list of genomic regions for which there are clear cell type-specific DNA methylation differences. This list is likely to represent only a subset of such differences. Other differentially methylated probes may have been masked by the suboptimal fibroblast purification. New microarrays with better probe coverage of the genome will certainly extend this list.
In summary, we analyzed the DNA methylation patterns of cytotrophoblast and fibroblast enriched placental cell fractions for global and specific differences. Although the specific cell types do not demonstrate genome wide differentiation in DNA methylation, we identified several genomic regions for which the methylation pattern is significantly different, likely representing true biological differences between cytotrophoblasts and fibroblasts. The identification of such differences underscores the importance of epigenetic mechanisms in normal human placental development and provides a useful resource for the interpretation of placental DNA methylation data. Importantly, these data support the use of placental villi for epigenetic studies targeting cytotrophoblast cells. Further, they provide a framework from which to propose hypotheses regarding critical epigenetic modifications that drive normal placental development as well as important epigenetic alterations that could cause placental related diseases such as preeclampsia and intrauterine growth restriction. In severe placental insufficiency syndromes, complicated by severe early-onset preeclampsia or intrauterine growth restriction, histologic evidence of defective differentiation of the extravillous trophoblast or of the chorionic villi (comprising stroma, endothelium and a covering of villous trophoblast) has been found.Citation29 Defective epigenetic regulation affecting the differentiation of these different cell types may be a unifying mechanism that could illuminate currently unresolved questions about placental development.Citation30
The normal function of these tissues likely depends not only on the correct epigenetic profiles but also the normal proportion of the different cell types carrying each profile. Variations in the cell-specific profiles or the proportions of specific cell types among samples of the same tissue could result in abnormal development and/or function of this tissue. We undertook a study of cell specific methylation profiles in the placenta with the expectation that these data could provide a framework for understanding epigenetic regulation and cell-type specificity in the normal placentas. Such data would be expected to generate hypothesis-driven research into the epigenetic basis of normal placental development across gestation as well as the role of epigenetic alteration in diseases of the placenta and the fetus.
Materials and Methods
Sample selection and cell separation.
Samples of human placenta used in this study were obtained by the staff of our Research Centre for Women's and Infants' Health BioBank program at Mount Sinai Hospital, Toronto, Canada with written informed consent and Research Ethics Board approval. Villous cytotrophoblasts and mesenchymal core fibroblasts were isolated from singleton, healthy second trimester placentas at 14–15 weeks (n = 3) and at 18–19 weeks (n = 3) of gestation immediately following voluntary surgical termination of pregnancy. The tissues were placed in sterile phosphate buffered saline (PBS) and processed within 2 h of collection. The placental villi were dissected and sequentially digested prior to magnetic bead separation to obtain specific cell fractions.
Sequential enzymatic separation.
A preliminary wash [Mg2+ and Ca2+ free Hank's buffered salt solution (HBSS)] and dissection eliminated debris and clots. A 40 mg representative sample of placental villous tissue was subjected to serial trypsin digestion [0.25% trypsin (Invitrogen 27250-018) in HBSS]. Three to four digestions were necessary, each one consisting of 60 min of incubation in 50 mL of trypsin solution at 37°C, on a gentle shaker. The supernatant was collected, neutralized with 10% fetal bovine serum (FBS) and centrifuged (5 min, 362 g). The resulting cell pellet was further washed with a stabilizing PBS solution [PBS (Ca2+ and Mg2+) + 2% FBS].
The remaining placental tissue was rinsed in HBSS (Mg2+ and Ca2+ free) and subjected to two sequential collagenase (2 mg/mL) (SIGMA, C6885-1G) digestions of 25–30 min duration, in 25 ml of collagenase, at 37°C, on the slow shaker. The cells were then centrifuged and stabilized as described above.
The cells obtained from the trypsin only and trypsin collagenase digestions were analyzed separately as outlined below. The pellets obtained from the trypsin digestions were used for the isolation of cytotrophoblasts while the pellets from the collagenase digestions were enriched for fibroblasts.
Magnetic bead separation.
The stabilized cells were centrifuged (5 min, 362 g) and each individual pellet was suspended in 2 ml PBS and passed through a sterile metal mesh (Sigma, 200 µm) to remove undigested groups of cells. The samples were placed on a 4 cc Ficoll 1.077 column (GE Healthcare, 17-1440-02) and centrifuged for 10 min (805 g) to facilitate the density gradient separation. A layer of cells was obtained, collected, washed with MACS Buffer [PBS + 0.25% BSA + 2 mM EDTA (ethylenediaminetetraacetic acid)] and incubated for 30 min with magnetic-labeled, cell specific antibodies (20 µL per 107 cells) (MACS MicroBeads, Miltenyi Biotec). The antibodies used were CD-45 (MACS, 130-045-801) and anti-fibroblast (MACS, 130-050-601). Magnetic separation was performed according to the supplier's positive selection/depletion protocol: the cytotrophoblasts were obtained by depletion and the fibroblasts by positive selection.
The resulting cell fractions—cytotrophoblasts and fibroblasts—were stored in RLT Buffer + 10% β-mercaptoethanol (β-ME) at 4°C for the upcoming steps. A small fraction of cells (40 µL containing 2 × 105 cells) was plated with growth media (Dulbecco's modified Eagle medium nutrient mixture F12 [DMEM/F12 (Gibco 11039-021)] and Neomycin 0.5 ml/500 mL and 10% FBS) on a six-well plate. Subsequently, the plates were incubated in atmospheric conditions (O2/5% CO2 at 37°C) overnight in preparation for immunocytochemistry.Citation31
Immunocytochemistry protocol.
The plated cell fractions were fixed with 70% methanol for 30 min. The cells were then washed twice with PBS. The cells were blocked with Ready to Block reagent (DAKO Canada Inc.) for 30 min following the manufacturer's protocol. Specific antibodies (1:150 dilution) targeting cytotrophoblasts [cytokeratin-7 (DAKO, IR619)] and fibroblasts [vimentin-9 (DAKO, IR630)] were added. A secondary (anti-mouse) antibody was added, followed by DAB staining (KPL 54-10-00). The plates were allowed to incubate at room temperature and the positively stained cells were then counted under the microscope and expressed as a percentage of the total number of cells.
DNA extraction.
Genomic DNA was extracted from the whole placenta samples and the purified cell fractions of cytotrophoblasts and fibroblasts. The initial step of disruption was performed mechanically for the placenta samples (TissueRuptor, Qiagen) and chemically (β-ME) for the cytotrophoblasts and fibroblasts. The samples were homogenized in the QIAshredder spin column (Qiagen). The AllPrep DNA/RNA MiniKit (Qiagen, 80204) protocol was used to extract DNA from each sample.
Methylation arrays
Genomic DNA (1 µg) was sodium bisulfite modified using Imprint® DNA Modification Kit (Sigma) and hybridized to the Illumina® Infinium Human Methylation27 array. Each probe interrogates >27,000 highly-informative CpG sites covering >14,000 gene promoter regions, with an average of two probes per gene. Each glass slide includes arrays for 12 samples, which reduces inter array variation. As previously described in reference Citation24, labeling, hybridization, washing and scanning were carried out at the TCAG microarray facility following the manufacturer protocols. Beadstudio Methylation module v3.2.0 (Illumina) was used for background correction. For each probe a “detection p value” was generated as a measure of the reliability of each fluorescence measure. The CpG methylation percentage for each locus was determined by dividing the intensity of the methylated signal by the total signal intensity (methylated and un-methylated).
Global comparisons between samples.
Global comparisons were performed between all the samples by non-hierarchical Euclidean cluster analysis, using Partek Genomics Suite version 6.5.
Differential methylation analysis and enrichment analysis.
Probe specific differential methylation analysis was performed to identify differences between the cytotrophoblast and fibroblast fractions. Sex chromosome data was excluded. Given the non-normal distribution of the data, the unequal number of cytotrophoblast and fibroblast samples and the small sample size, we used non-parametric statistical analysis (Wilcoxon rank-sum test). Probes with detection p values >0.05 were excluded. We used a 20% methylation difference cut-off level in addition to a p value less than 0.05, to identify probes that are most likely to have a biological significant difference between the 2 cell types and partially to overcome the challenge presented by incomplete cell separation (see results).
In order to define the methylation patterns of cytotrophoblasts and fibroblasts as they relate to tumor suppressor genes, a search was performed in the Genecards database, V3.02.128, March 21 2010 (www.genecards.org) using the key words “tumor suppressor” in the fields “Summary” or “Function.” The resulting gene list was used for binomial enrichment analysis of our two lists of differentially methylated probes. For comparison, a similar strategy analysis was performed for oncogenes.
Pyrosequencing validation of selected regions.
Three differentially methylated CpG sites were selected for validation using universal biotinylated primer-based pyrosequencing methylation analysis of bisulfite converted DNA, as previously described in reference Citation24 and Citation32. Assays were designed using Pyromark Assay Design software version 2.0.1.15 (Qiagen) targeting a region flanking the Illumina CpG site by up to 100 basepairs. Details of the primers used are given in Supplemental Table 2. The same bisulfite converted DNA sample that was used for the Illumina arrays hybridization was likewise used for pyrosequencing. PCR amplicons were analyzed with the PyroMarq Q24 also from Qiagen, as specified by the manufacturer protocol. The results, provided in percent methylation, were calculated using PyroMarq Q24 software version 1.0.10. Bisulfite efficiency was controlled by the absence of non-converted C at C sites not followed by a G, (i.e., expected to be non-methylated) thus all converted to T after PCR.
Analysis of the pyrosequencing data included: (1) Pearson correlation between the methylation values, determined by the two methods, of the specific array targeted CpG sites (three assays x 11 samples); (2) Pearson correlation between the differences in methylation for each CpG site for each pair of related samples (three assays x 5 cytotropho-fibroblast pairs); (3) Differential analysis between methylation levels of cytotrophoblasts and fibroblasts, as determined by the pyrosequencing method, for each CpG site targeted by the pyrosequencing assay and for the site average, using the Wilcoxon rank-sum test.
Financial Support
Canadian Institutes of Health Research Grants (grant number MOP86758 to R.W.; MOP220231 to J.K.) and Rose Torno Chair to J.K. from Mount Sinai Hospital. José Carlos Ferreira is the recipient of a scholarship from FCT (Science and Technology Foundation), Lisbon, Portugal—SFRH/BD/28642/2006—funded by POPH (Operational Program for Human Potential) co-participated by FSE (European Social Fund) and by national funds from MCTES (Ministry of Science, Technology and Higher Education).
Conflict of Interest
There have been no involvements that might raise the question of bias in the work reported or in the conclusions, implications or opinions stated.
Ethics Approval
The Mount Sinai Hospital (Toronto, Ontario Canada) Research Ethics Board has reviewed and approved the collection of samples (MSH REB #04-0018-U).
Abbreviations
| β-ME | = | β-mercaptoethanol |
| DNMT | = | DNA methyl transferase |
| DMEM/F12 | = | Dulbecco's modified Eagle's medium nutrient mixture F12 |
| EDTA | = | ethylenediaminetetraacetic acid |
| EVT | = | extra-villous trophoblast |
| FBS | = | fetal bovine serum |
| FSH | = | follicle stimulating hormone |
| HBSS | = | Hank's buffered salt solution |
| hCG | = | human chorionic gonadotropin |
| LH | = | luteinizing hormone, PBS, phosphate buffered saline |
| TSH | = | thyroid stimulating hormone |
| TSS | = | transcription start site |
| TP73 | = | tumor protein p73 |
| CGB | = | chorionic gonadotropin, beta polypeptide |
| CGA | = | chorionic gonadotropin, α polypeptide |
| APC | = | adenomatous polyposis coli |
| RASSF1 | = | ras association (RalGDS/AF-6) domain family member 1 |
| RASSF5 | = | ras association (RalGDS/AF-6) domain family member 5 |
| DAB2IP | = | DAB2 interacting protein (DAB2-disabled homolog 2, mitogen-responsive phosphoprotein) |
| PRKCDBP | = | protein kinase C, delta binding protein |
| MORF4L1 | = | mortality factor 4 like 1 |
Figures and Tables
Figure 1 Immunocytochemistry of cells isolated from placenta. (A) Cytotrophoblasts stained positively for cytokeratin-7 and (B) did not stain for vimentin-9. The fibroblast fraction stained positively for both (C) cytokeratin-7 and (D) vimentin-9.
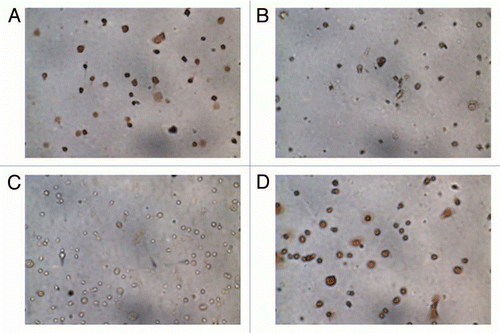
Figure 2 Cluster analysis for placenta, isolated cytotrophoblasts and fibroblasts. Non-hierarchical Euclidean cluster analysis was performed based on gestational age [Column A: 14–16 weeks (early); 17–18 weeks (late)], type of sample [Column B: fibroblast (Fibro), cytotrophoblast (Cyto) and whole placenta (Plac)] and sample number (Column C). The degree of similarity between two samples is given by the sum of the length of the horizontal lines between the samples. We chose the fourth branching to define the clusters (marked by the thick vertical line). There is no clustering by gestational age or sample. The sample type analysis reveals two main clusters: one composed of only cytotrophoblasts and placenta and the other contains mainly fibroblasts. The other three clusters contains only one sample each and may be due to the reduced number of significantly detected probes (24–25,000 vs. > 27,000) for these samples.
![Figure 2 Cluster analysis for placenta, isolated cytotrophoblasts and fibroblasts. Non-hierarchical Euclidean cluster analysis was performed based on gestational age [Column A: 14–16 weeks (early); 17–18 weeks (late)], type of sample [Column B: fibroblast (Fibro), cytotrophoblast (Cyto) and whole placenta (Plac)] and sample number (Column C). The degree of similarity between two samples is given by the sum of the length of the horizontal lines between the samples. We chose the fourth branching to define the clusters (marked by the thick vertical line). There is no clustering by gestational age or sample. The sample type analysis reveals two main clusters: one composed of only cytotrophoblasts and placenta and the other contains mainly fibroblasts. The other three clusters contains only one sample each and may be due to the reduced number of significantly detected probes (24–25,000 vs. > 27,000) for these samples.](/cms/asset/83eadad8-fc18-4fb9-8f1e-be4155629b68/kepi_a_10914196_f0002.gif)
Figure 3 Selection criteria of Differentially Methylated CpG Dinucleotides. (A) The distribution of DNA methylation differences is narrow between cytotrophoblasts and fibroblasts demonstrating that large methylation differences between the two cell types are rare. (B) The scatter plot shows the distribution of differences in methylation as a differential score where a value of >13 or <-13 is equal to a p value of <0.05. The probes with a significant difference in methylation higher than 20% are shown in black. Positive differences (higher cytotrophoblast methylation) in the left upper quadrant and negative differences (higher fibroblast methylation) in the right lower quadrant.
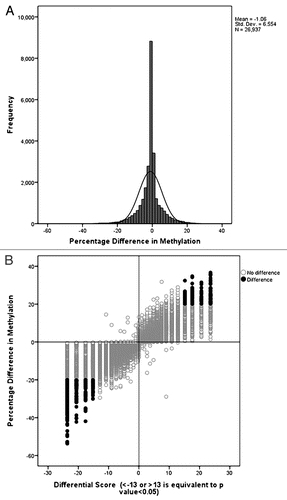
Figure 4 DNA methylation values of the CGB genes in cytotrophoblasts and fibroblasts. (A) CGB genes map to chromosome 19q (middle). At each CpG site there is lower methylation in cytotrophoblasts than in fibroblasts (top). The chromosome band containing the CGB genes is magnified (UCSC genome browser build 36.1 annotation) to illustrate the CpG islands and CpG probes as they relate to the genes (bottom). Six of eight probes show a reduction in methylation, five of these reach our stringent cut-off criteria: CGB3 (probe 1), CGB1, CGB5 and CGB8 (probes 5–8). (B) Box-plot of the methylation values of CpG sites mapping to the CGB genes. Each of the eight CpG probes mapping to CGB genes are represented. The methylation level is depicted on the y-axis. The x-axis shows the CGB gene probes in the order in which they appear in the genome (proximal to distal) but is not to scale. Some genes have two probes and are separated by the interrupted vertical lines. The solid horizontal arrows represent the TSS and point in the direction of transcription. Two CpG probes show no difference in methylation (white box) between the cytotrophoblasts (left) and fibroblasts (right). Probes are less methylated in cytotrophoblasts (left) then in fibroblasts (right). Here each of the array probes mapping to the region are represented by a boxplot. The statistically significant different probes (Wilcoxon rank-sum test, p < 0.05) reaching or passing the 20% cut-off are represented by a dark gray bar while the clear gray bar corresponds to the probe that has <20% difference in methylation but remains statistically significant.
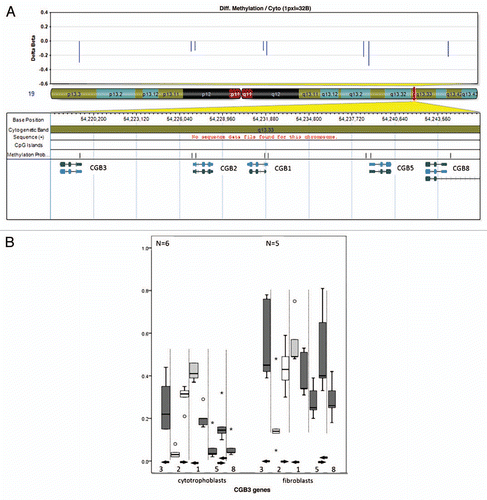
Figure 5 DNA methylation values of CpG sites mapping to tumor suppressor genes in cytotrophoblasts and fibroblasts. For each of the box plots the y-axis corresponds to methylation levels and the x-axis are the probes; the solid horizontal arrows indicate the direction of transcription. All the CpG sites are between 2,000 bp upstream and 500 bp downstream of the TSS (start point of the arrows). For each gene all the probes corresponding to a CpG Island that has at least one differentially methylated probe between cytotrophoblasts and fibroblasts are shown. Only dark grey boxes correspond to statistical significantly different probes (Wilcoxon rank-sum test, p < 0.05). (A) The solid vertical lines on the APC gene graph separate probes of two consecutive but distinct CpG islands. Of the 6 APC probes, one shows a non-statistically significant difference higher than 20% (light grey box). (B) DAP2IP, (C) PRKCDBP and (D) RASSF5 have multiple probes that show no difference in methylation (white boxes). (E) TP73 shows a non-statistically significant difference higher than 20% (light grey box). (F) All 3 probes corresponding to RASSF1 are statistically different (dark grey box).
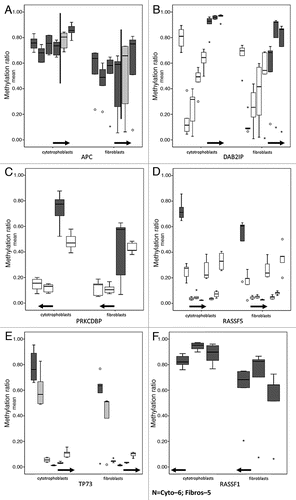
Figure 6 Pyrosequencing validation of DNA methylation measured by microarrays. Three CpG sites mapping to the promoter region of (A) CGB5 and the tumor suppressor genes (B) TP73 and (C) APC were analyzed. For each gene three box plots are shown. From left to right: the first corresponds to the values for each CpG site as measured in the pyrosequencing assay, the second to the average of the values of all the CpG sites for each pyrosequencing assay and the third plot corresponds to the methylation value measured by the array. The white boxes correspond to non-statistically significantly different CpG sites and the grey boxes to statistically significantly different CpG sites between cytotrophoblast and fibroblast samples. The light grey boxes show the CpG of the pyrosequencing target that corresponds to the one that was targeted by the array.
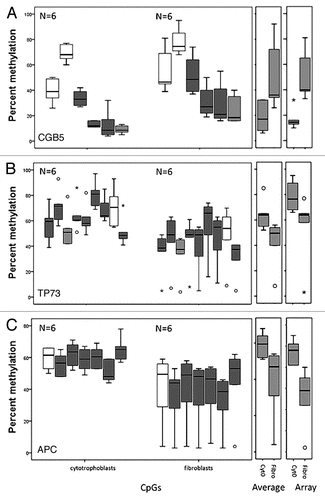
Table 1 Probes more methylated in cytotrophoblasts than fibroblasts mapping to tumor suppressor genes
Additional material
Download Zip (126 KB)Acknowledgements
The authors thank Youliang Lou and Chunhua Zhao for their technical assistance and the study participants, and the Research Centre for Women's and Infants' Health BioBank program of the CIHR Group in Development and Fetal Health (CIHR #MGC-13299), the Samuel Lunenfeld Research Institute and the Mount Sinai Hospital/University Health Network Department of Obstetrics & Gynaecology for the human specimens used in this study.
References
- Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature 2007; 447:433 - 440
- Persson J, Ekwall K. Chd1 remodelers maintain open chromatin and regulate the epigenetics of differentiation. Exp Cell Res 2010; 316:1316 - 1323
- Rakyan VK, Down TA, Thorne NP, Flicek P, Kulesha E, Graf S, et al. An integrated resource for genome-wide identification and analysis of human tissue-specific differentially methylated regions (tDMRs). Genome Res 2008; 18:1518 - 1529
- Ghosh S, Yates AJ, Fruhwald MC, Miecznikowski JC, Plass C, Smiraglia DJ. Tissue specific DNA methylation of CpG islands in normal human adult somatic tissues distinguishes neural from non-neural tissues. Epigenetics 2010; 5:527 - 538
- Bogdanovic O, Veenstra GJ. DNA methylation and methyl-CpG binding proteins: developmental requirements and function. Chromosoma 2009; 118:549 - 565
- Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 2010; 6:479 - 491
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16:6 - 21
- Bloushtain-Qimron N, Yao J, Snyder EL, Shipitsin M, Campbell LL, Mani SA, et al. Cell type-specific DNA methylation patterns in the human breast. Proc Natl Acad Sci USA 2008; 105:14076 - 14081
- Sakamoto H, Suzuki M, Abe T, Hosoyama T, Himeno E, Tanaka S, et al. Cell type-specific methylation profiles occurring disproportionately in CpG-less regions that delineate developmental similarity. Genes Cells 2007; 12:1123 - 1132
- Bloushtain-Qimron N, Yao J, Shipitsin M, Maruyama R, Polyak K. Epigenetic patterns of embryonic and adult stem cells. Cell Cycle 2009; 8:809 - 817
- Lima SC, Hernandez-Vargas H, Herceg Z. Epigenetic signatures in cancer: Implications for the control of cancer in the clinic. Curr Opin Mol Ther 2010; 12:316 - 324
- Knofler M. Critical growth factors and signalling pathways controlling human trophoblast invasion. Int J Dev Biol 2010; 54:269 - 280
- Bloxam DL, Bax CM, Bax BE. Culture of syncytiotrophoblast for the study of human placental transfer. Part I: Isolation and purification of cytotrophoblast. Placenta 1997; 18:93 - 98
- Maldonado-Estrada J, Menu E, Roques P, Barre-Sinoussi F, Chaouat G. Evaluation of Cytokeratin 7 as an accurate intracellular marker with which to assess the purity of human placental villous trophoblast cells by flow cytometry. J Immunol Methods 2004; 286:21 - 34
- Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 2008; 9:465 - 476
- Costello JF, Plass C. Methylation matters. J Med Genet 2001; 38:285 - 303
- Pierce JG, Parsons TF. Glycoprotein hormones: structure and function. Annu Rev Biochem 1981; 50:465 - 495
- Henke A, Gromoll J. New insights into the evolution of chorionic gonadotrophin. Mol Cell Endocrinol 2008; 291:11 - 19
- Wong NC, Novakovic B, Weinrich B, Dewi C, Andronikos R, Sibson M, et al. Methylation of the adenomatous polyposis coli (APC) gene in human placenta and hypermethylation in choriocarcinoma cells. Cancer Lett 2008; 268:56 - 62
- Novakovic B, Rakyan V, Ng HK, Manuelpillai U, Dewi C, Wong NC, et al. Specific tumour-associated methylation in normal human term placenta and first-trimester cytotrophoblasts. Mol Hum Reprod 2008; 14:547 - 554
- Chiu RW, Chim SS, Wong IH, Wong CS, Lee WS, To KF, et al. Hypermethylation of RASSF1A in human and rhesus placentas. Am J Pathol 2007; 170:941 - 950
- Ferretti C, Bruni L, Dangles-Marie V, Pecking AP, Bellet D. Molecular circuits shared by placental and cancer cells, and their implications in the proliferative, invasive and migratory capacities of trophoblasts. Hum Reprod Update 2007; 13:121 - 141
- Soundararajan R, Rao AJ. Trophoblast ‘pseudo-tumorigenesis’: significance and contributory factors. Reprod Biol Endocrinol 2004; 2:15
- Grafodatskaya D, Choufani S, Ferreira JC, Butcher DT, Lou Y, Zhao C, et al. EBV transformation and cell culturing destabilizes DNA methylation in human lymphoblastoid cell lines. Genomics 2010; 95:73 - 83
- Kovalevskaya G, Genbacev O, Fisher SJ, Caceres E, O'Connor JF. Trophoblast origin of hCG isoforms: cytotrophoblasts are the primary source of choriocarcinoma-like hCG. Mol Cell Endocrinol 2002; 194:147 - 155
- Dokras A. Epigenetic regulation of maspin expression in the human placenta. Molecular Human Reproduction 2006; 12:611 - 617
- Richter AM, Pfeifer GP, Dammann RH. The RASSF proteins in cancer; from epigenetic silencing to functional characterization. Biochim Biophys Acta 2009; 1796:114 - 128
- Lee JH, Byun DS, Lee MG, Ryu BK, Kang MJ, Chae KS, et al. Frequent epigenetic inactivation of hSRBC in gastric cancer and its implication in attenuated p53 response to stresses. Int J Cancer 2008; 122:1573 - 1584
- Kingdom J, Huppertz B, Seaward G, Kaufmann P. Development of the placental villous tree and its consequences for fetal growth. Eur J Obstet Gynecol Reprod Biol 2000; 92:35 - 43
- Aplin JD. Developmental cell biology of human villous trophoblast: current research problems. Int J Dev Biol 2010; 54:323 - 329
- Baczyk D, Drewlo S, Proctor L, Dunk C, Lye S, Kingdom J. Glial cell missing-1 transcription factor is required for the differentiation of the human trophoblast. Cell Death Differ 2009; 16:719 - 727
- Tost J, Gut IG. DNA methylation analysis by pyrosequencing. Nat Protoc 2007; 2:2265 - 2275