 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
In eukaryotes, DNA is wrapped around proteins called histones and is condensed into chromatin. Post-translational modification of histones can result in changes in gene expression. One of the most well-studied histone modifications is the methylation of lysine 4 on histone H3 (H3K4). This residue can be mono-, di- or tri-methylated and these varying methylation states have been associated with different levels of gene expression. Understanding exactly what the purpose of these methylation states is, in terms of gene expression, has been a topic of much research in recent years. Enzymes that can add (methyltransferases) and remove (demethylases) these modifications are of particular interest. The first demethylase discovered, LSD1, is the most well-classified and has been implicated in contributing to human cancers and to DNA damage response pathways. Currently, there are limited methods for accurately studying the activity of demethylases in vitro or in vivo. In this work, we present MassSQUIRM (mass spectrometric quantitation using isotopic reductive methylation), a quantitative method for studying the activity of demethylases capable of removing mono- and di-methyl marks from lysine residues. We focus specifically on LSD1 due to its potential as a prime therapeutic target for human disease. This quantitative approach will enable better characterization of the activity of LSD1 and other chromatin modifying enzymes in vitro, in vivo or in response to inhibitors.
Keywords: :
Introduction
As a result of the sequencing of the human genome, it has become apparent that the complexity of an organism does not necessarily correlate with the size of its genome. This has sparked an interest in discovering exactly how genes are regulated on levels other than primary genomic sequence. Epigenetics has arisen as a field that addresses this concern by focusing on post-transcriptional methods of gene expression control including DNA methylation, histone post-translational modifications (PTMs) and non-coding RNAs.Citation1 Modifications made to proteins post-translationally can affect their function, location or longevity. One set of highly modified proteins important to gene expression is histones. DNA is wrapped around histones, which form nucleosomes before being condensed into chromatin and ultimately chromosomes.
Histone PTMs have been shown to be very important to gene expression. Some modifications serve to signal the recruitment of chromatin modifying enzymes while some serve to alter the interaction between the histones and DNA allowing or prohibiting the access of transcription machinery.Citation2 Some more common histone modifications include methylation, acetylation and phosphorylation. Of these common modifications, methylation is by far the most complex. Both lysine and arginine residues can be modified by mono- or di-methylation and lysine residues can be tri-methylated as well. These varying states of methylation can be associated with both active and inactive genes. The complexity and importance of methylation, on histones in particular, has stirred much interest in the enzymes capable of adding (methyltransferases) and removing (demethylases) these modifications.
The first demethylase was discovered in 2004 and was termed Lysine Specific Demethylase 1 or LSD1.Citation3 LSD1 is a flavin-dependent amine oxidase that can remove mono- and di-methyl marks from H3K4 primarily, H3K9 under certain conditions and some non-histone substrates such as p53.Citation4–Citation6 It has been shown to be part of many protein complexes including CoREST, NuRD and AR/ER.Citation7 LSD1 is also associated with gene repression and has been suggested to be important in initiating myc-induced transcription in cancers.Citation3,Citation8–Citation11
Structural and biochemical studies have led to the development of numerous LSD1 inactivators that have the potential to be keen therapeutic tools, much like the successful deacetylase inhibitors currently in use.Citation12 In addition, the mechanism of LSD1 indicates that it is an excellent candidate for suicide inactivators. Many monoamine oxidase (MAO) inhibitors have been suggested as potential LSD1 suicide inactivators.Citation13 Several different assays are used to study the activity of LSD1 in the presence and absence of these various inhibitors in order to determine their efficiency.
Currently, histone demethylase activity can be accurately measured using a handful of assays. One assay involves synthetic methylation of histone substrates using a recombinant methyltransferase and isotopically heavy S-adenosyl methionine.Citation14 The removal of tritiated methyl groups from histone substrates is monitored by autoradiography. Also, due to the formation of formaldehyde in certain demethylase reactions, formaldehyde release can be followed to determine demethylase activity. While these methods are useful in determining if an enzyme is acting as a demethylase, they are not quantitative in nature. Another common method for following demethylase activity is western blotting using antibodies specific to certain modifications.Citation15 While western blotting can be used as a semi-quantitative technique, the antibodies used for detection of histone modifications are often non-specific making absolute quantification difficult.Citation16 Using antibody-based methods to quantify different methyl states of a single residue is especially difficult due to the similarity in structure of mono-, di- and tri-methylated lysines.Citation17
As an alternative, mass spectrometry is often used as a tool for studying the activity of demethylases on synthetic peptides.Citation18 This technique provides a rapid method for highly sensitive analysis of full products of demethylase reactions. One caveat to using mass spectrometry for such an analysis is that it is, at best, semi-quantitative because differentially modified peptides cannot be equally compared. For example, a mono-methylated peptide will not necessarily ionize the same as a tri-methylated species of the exact same sequence. This leads to a difficulty in using mass spectrometry-based assays to quantify the efficiency of demethylases.
To address the issues of differential ionization and sample variation, it is common to use isotopically heavy chemistry to modify peptides. For example, H4 lysine acetylation has been quantified by using isotopically heavy acetic anhydride to synthetically acetylate all lysine residues, and recent applications of this approach have incorporated MALDI mass spectrometers, allowing for more straightforward data analysis.Citation19,Citation20 We present an assay which employs reductive methylation, using deuterated formaldehyde, followed by MADLI mass spectrometry, to differentiate between various lysine methylation states. This method accounts for methyl states within a sample, allowing sample-to-sample relative comparison.
Reductive methylation is commonly used as a method to improve the stability of proteins for analysis by x-ray crystallography.Citation21 During this process, lysine residues are chemically di-methylated using the following reactions to alkylate and then reduce amines:
(1)
(1)
(2)
(2)
In this paper, we use a combination of reductive methylation, isotopic labeling and mass spectrometry to quantitatively measure the activity of LSD1 alone and in the presence of an MAO inactivator. We have termed our assay MassSQUIRM (Mass Spectrometric Quantitation Using Isotopic Reductive Methylation). MassSQUIRM will prove invaluable to the further study of the mechanism of not only LSD1 but also many methyltransferase and demethylase enzymes and the efficiency of their proposed inhibitors. Effective LSD1 inactivators have the potential to become targeted therapies with high specificity that could potentially treat LSD1-associated diseases, such as cancer, with minimal side effects.
Results
Reductive methylation is an efficient method for modifying lysine residues.
We chose to use reductive methylation to address the issue of differential ionization of methylated peptides. Ordinarily, methylation is seen in mass spectrometric data as an addition of 14 Da with mono-methylation (one open circle, ) and 28 Da with di-methylation (two open circles, ). It might seem logical to compare the peak areas of the three monoisotopic peaks seen in in order to quantify their abundance. This would lead us to believe that the unmethylated peptide (corresponding to the green monoisotopic peak) is in higher abundance than the mono-methylated (red) and di-methylated (blue) peptides. This, however, is not necessarily a correct assumption. It is possible that the green peptide is differentially ionized and is thus recorded by the mass spectrometer detector at a higher level. For this reason, we cannot compare these three peaks in a quantitative manner.
Reductive methylation is a reaction involving formaldehyde that results in di-methylated lysine residues (EquationEquations 1(1)
(1) and Equation2
(2)
(2) ). Using this technique, we can convert all three peptides in to the same chemical species and thus cause them to ionize identically (). In making all peptides the same chemical species, we can compare their peak heights to each other in a quantitative manner. Unfortunately, making all peptides the same chemical species causes them to be contained in a single monoisotopic peak, from which we cannot decipher the original methylation state of each individual peptide ().
To solve this dilemma, we used deuterated formaldehyde in our reactions. Reductive methylation performed with heavy formaldehyde results in the addition of up to two deuterated methyl groups to lysine residues (). Pre-existing methylated lysines will be isotopically light (open circles), while methyl groups added by reductive methylation will be isotopically heavy (closed circles) (). Once peptides are converted to the dimethylation state by reductive methylation, we can compare their peak areas in a quantitative manner. Peaks from differentially modified peptides will be separated by 2 or 4 Da based on their original modification state. A peptide that exists in the di-methyl state in the original sample will have two light methyl groups (14 Da methyl + 14 Da methyl = 28 Da) and will be the lowest m/z peak. Peptides that contain one methyl group in the initial sample will have an additional, deuterated methyl group added upon reductive methylation (14 Da methyl + 16 Da heavy methyl = 30 Da). Any peptides unmodified in the original samples will have an addition of two heavy methyl groups from reductive methylation (16 Da heavy methyl + 16 Da heavy methyl = 32 Da). Thus, the originally unmodified peptide will actually occur at the highest m/z in the cluster while the originally di-methylated species will be the lowest (). The isotopic overlap that results from these peaks being in such close proximity is addressed in later sections.
To determine the efficiency of the reductive methylation reaction using both heavy and light formaldehyde, we subjected a synthetic peptide, containing four unmodified lysine residues, to both forms of the reaction. We found that the reaction is ∼100% efficient when using light or heavy formaldehyde (). Reductive methylation also occurs on the N-terminus of the peptide as is shown in . We observed an addition of ten methyl groups to the peptide (two for each lysine and two on the N-terminal residue) totaling 140 Da with light formaldehyde and 160 Da with heavy formaldehyde. In an effort to show the utility of MassSQUIRM on full length proteins, we resolved recombinant full-length histone H3 by SDS-PAGE and performed reductive methylation on the gel band. Once again, we observed nearly 100% conversion of lysines to the di-methyl state using heavy formaldehyde (). In solution digestion of the same sample resulted in similar results.
When using mass spectrometry to quantify biological samples, it is prudent to take into consideration the dynamic range capabilities of both your experiment and the instrument that will be used for analysis because a linear response is needed for accurate quantification. To determine the dynamic range of our assay, we used three different forms of a synthetic peptide: un-, mono- and di-methylated. A mono-methylated peptide was mixed with un- and di-methylated peptides at the following ratios: 1:1, 1:2, 1:4, 1:8, 1:16 and 1:32. Using this method, we determined a dynamic range of 1:8 ( and squares). To check the dynamic range following reductive methylation chemistry, we incubated the same peptides in the same ratios but exposed them to MassSQUIRM analysis. We determined the peak ratios as mentioned above with added compensation for isotopic overlap as described in the next section. We determined the dynamic range following reductive methylation to be 1:8 as well ( and diamonds).
MassSQUIRM accurately corrects for differences in ionization.
In , we show a mass spectrum of equimolar amounts of un-, mono- and di-methylated histone H3 peptide, which demonstrates the variable efficiencies of ionization for these methylated peptides. This unequal efficiency of ionization is problematic for performing quantitative mass spectrometry. As a proof of principle, we compared conventional and MassSQUIRM-based mass spectrometric analysis on a synthetic histone H3 peptide dimethylated on Lys4 following an enzymatic demethylation reaction with LSD1. Conventional mass spectrometric analysis of the products of this reaction showed high levels of unmethylated peptide and lower levels of the original di-methylated peptide ( and D). However, MassSQUIRM analysis of this same demethylase reaction revealed the presence of significant levels of mono-methylated peptide as a reaction product ( and D). Thus, MassSQUIRM provided for the detection of a reaction intermediate that would not have been observed by conventional mass spectrometric analysis. The detection of the mono-methyl peptide using MassSQUIRM, but not conventional mass spectrometry, is in agreement with the observed reduced ionization efficiency of monomethylated peptides shown in .
Development of a method for compensating for isotopic overlap when using MassSQUIRM.
When using mass spectrometry for relative quantification, it is most common to use the lowest mass or monoisotopic, peak as a reference ( and 12C). In addition to the monoisotopic peak, a given peptide will show a series of peaks representing naturally occurring isotopes, which are termed the isotopic envelope ( and all other peaks). When two peptides are similar in mass and are being relatively compared, the isotopic envelopes can overlap and complicate quantification. As can be seen in , one caveat with the use of deuterated formaldehyde in reductive methylation is that some isotopic overlap occurs between the different peptides. For example, the second monoisotopic peak in (one open and one closed circle) contains some overlap from the isotopic envelope of the first peptide (noted by both red and blue). In order to compensate for this overlap, we first determined the peak area (A) ratio of 13C2 and 13C4 isotopes relative to the monoisotopic peak for the peptide with light dimethylation ().
This gave us the ratio of peptide existing in these isotopic states (r1 and r2) specific to our experiment and mass spectrometer. We then used this information to determine the following formulas for quantifying the amount of peptide existing in each modification state in a sample:
(5)
(5)
(6)
(6)
(7)
(7)
As an example, we have shown an LSD1 treated H3K4 peptide following heavy reductive methylation (0.25 µg LSD1 + 0.25 µg H3K4me2 from ) that exists in multiple states of methylation (). This method was used to generate all quantitative data presented in this paper.
LSD1 activity can be quantitatively measured using MassSQUIRM.
Once we had optimized the heavy reductive methylation reaction, we decided to use it to determine the activity of a demethylase. Even though LSD1 is has been extensively studied, a reliable method for quantitatively studying its demethylase activity has proven elusive. For this reason, we chose to use LSD1 to test the ability of MassSQUIRM as a method for determining demethylase activity in vitro. Histone demethylase assays were performed using a synthetic H3K4me2 peptide and varying concentrations of LSD1. Samples were then subjected to MassSQUIRM (). The resulting mass spectra showed an increase in LSD1 activity as LSD1 concentration was increased from zero to 0.5 µg. A shift of peptide methylation state from completely di-methylated to 97% un-methylated was observed when using 0.5 µg LSD1 (). These results indicate that MassSQUIRM is an appropriate method for measuring the activity of LSD1 by quantitatively following methylation levels in vitro.
To emphasize the importance of our assay, we repeated the above experiment using phenyethylhydrazine, an MAO inhibitor known to inhibit LSD1 activity. We chose to use 0.125 µg LSD1 for this assay because it yielded a mixed population of modifications in our initial experiments (). Triplicate samples of peptide alone, peptide with LSD1 and peptide with LSD1 with phenylethylhydrazine were subjected to histone demethylation assays. Following these assays, samples were analyzed by MassSQUIRM. Upon addition of LSD1, we observed a shift of spectra to greater mass, indicating a mixed population of methylation states ( and middle relative to bottom part). When samples were treated with LSD1 and its inhibitor, the shift disappeared ( and top part) indicating inhibition of LSD1 activity. Samples containing peptide alone ( and bottom part) were used to determine the experimental r1 and r2 values used to quantify the results. Under these conditions, phenylethylhydrazine inhibits LSD1 activity by 96% ().
Discussion
The recent discovery of lysine demethylases has led to an overwhelming amount of correspondence in a very short amount of time (reviewed by Agger et al).Citation22 Currently, demethylase activity can only be measured semi-quantitatively by methods such as autoradiography, formaldehyde release and western blotting. Although mass spectrometry is commonly used to identify post-translational modifications (PTMs), it can also provide an excellent method for attaining quantitative measurements. Because lysine methylation can occur in three different states, it is possible to have four different populations (un-, mon-, di- and tri-) in a single sample. Deciphering, quantifying and comparing these different methyl states can be challenging. An assay with the ability to follow each of these different methyl states quantitatively within a population does not currently exist. Most currently available mass spectrometric methods used to quantify changes in PTM status of a peptide are complex and expensive.Citation23 In this work, we present a straightforward and inexpensive mass spectrometry-based method for quantifying the activity of demethylases acting on mono- and di-methyl lysines.
Since the initial discovery of LSD1, more than twenty human demethylase enzymes have been identified.Citation7 Several efforts to classify these enzymes in detail are currently underway. Many demethylases have already been implicated as potential cancer diagnostic and prognostic indicators while some have been implicated as potential therapeutic agents.Citation24 Since, most histone demethylase mechanisms involve redox chemistry, they are prime candidates for suicide inactivators.Citation13 We have shown that phenyethylhydrazine, an MAO inhibitor, serves to inhibit LSD1 activity by 96% (). Further studies of this inactivator could lead to its use in inhibiting LSD1 in some cancers. Our assay can be used to further classify the effect of this drug on different diseases involving LSD1, possibly leading to rapid turnaround for therapeutic use.
We chose to test our assay on LSD1, specifically, due to the existence of a wealth of literature suggesting that it is important in many diseases and the availability of a number of LSD1 inhibitors.Citation3,Citation12,Citation25–Citation29 Overexpression of LSD1 in prostate cancer, poorly differentiated neuroblastoma and estrogen receptor (ER)-negative breast cancer has been associated with aggressive forms of these diseases.Citation5,Citation24,Citation30,Citation31 There have also been some promising results indicating that LSD1 inhibitors, in combination with other drugs, lead to slower growth of colon cancer in mouse xenograft models.Citation32 All signs point to LSD1 being an excellent target for drug development.
LSD1 exists as a component of several complexes, many of which contain histone deacetylase enzymes.Citation33–Citation36 Deacetylase inhibitors have been used successfully in clinical trials and could be used in combination with demethylase inhibitors to provide potent treatment for some diseases.Citation37 Interestingly, when LSD1 interacts with androgen receptor, its specificity changes from H3K4 to H3K9 although this interaction has not been verified in vitro.Citation5 Current assays exist that allow the study of interactions between LSD1 and its associating proteins, yet these assays merely represent a qualitative measure of the effect of these interactions on LSD1 activity. When studying protein complexes, it is common to combine interacting proteins in vitro and perform enzymatic assays to try and reconstruct the optimal complex. These results are used to design inhibitors that may affect a component of a complex that is not necessarily the ultimate target but that would have the same biological effect.Citation38 For this reason, our assay will be useful in designing new drugs to target diseases that involve LSD1 and its various complexes.
Although LSD1 is the most commonly studied demethylase enzyme to date, there are several other demethylases that have not been as well characterized. Some have already been shown to be important factors in human disease.Citation24 Members of the JMJD1 family of lysine demethylases remove mono- and di-methyl marks from H3K9 resulting in loss of a repressive mark.Citation39 Thus, these enzymes are most likely associated with activating genes but limited analysis of them has been performed. More extensive studies of these enzymes will be undertaken before more definite conclusions can be made about their role in human disease.
We present MassSQUIRM as an inexpensive and quantitative method for comprehensive study of the activity of demethylases involved in mono- and di-methylation. MassSQUIRM offers quantitation not only of the product of the reactions of these enzymes but also their intermediates. This assay will be a powerful tool in studying the mechanism of LSD1 and possibly its interacting partners. It will also serve as a useful tool in classifying many newly discovered lysine demethylase enzymes such as PHF8 and could be used for certain methyltransferase enzymes.Citation40–Citation47 We are currently developing the chemistry to extend this approach for the quantitative measurement of lysine tri-methylation. MassSQUIRM is the first assay of its kind to offer a quantitative method for studying LSD1 activity, thus its impact on the field has the potential to be quite extensive.
Materials and Methods
MassSQUIRM.
Lysine residues were chemically di-methylated using a reductive methylation technique adapted from Rayment, et al. Briefly, 5 µg of synthetic histone H3 peptide (1ART KQT ARK STG GKA PRK QLC) was resuspended in 50 mM sodium phosphate pH 7.4 then 0.12 mg of borane dimethylamine (Sigma) and 3.2 mM isotopically light formaldehyde (Sigma) or isotopically heavy d2-formaldehyde (Cambridge Isotope Laboratories) were sequentially added. This reaction was incubated for 2 h at 4°C. Fresh aliquots of borane dimethylamine and formaldehyde were added and the reaction was again incubated at 4°C for 2 h. A final aliquot of borane dimethylamine was added and the reaction was incubated at 4°C for ∼16 h. The reaction was quenched with 80 mM Tris-Cl, pH 7.5. Peptides were incubated with POROS R2 20 micron beads (Applied Biosystems), collected with a C18 ZipTip (Millipore) and spotted for MALDI analysis in 2,5-dihydroxybenzoic acid. Mass spectra of peptides were collected with a MALDI-prOTOF mass spectrometer (PerkinElmerSciex).Citation48,Citation49 Spectra were viewed and peak areas extracted using MoverZ software (Genomic Solutions). Reaction products were verified by MS2 with a Thermo LTQ XL mass spectrometer coupled to a NanoLC-2D liquid chromatography system (Eksigent). Un-, mono- and di-methylated K27 versions of synthetic histone H3 peptide (22SKA ARK SAP STG G) were used for dynamic range experiments.
For the isotopically heavy reductive methylation analysis of histone H3.2, the following adaptations were used. Recombinant human histone H3.2 was purified essentially as described.Citation50 To test the efficiency of the isotopically heavy reductive methylation on this full-length histone, 1 µg of histone H3.2 was resolved by SDS-PAGE in two lanes with a 4–20% Tris-glycine gel (Invitrogen) and visualized by Coomassie staining. The two gel bands containing histone H3.2 were excised, and Coomassie was removed by serial washes with 50% methanol/50 mM ammonium bicarbonate.Citation20,Citation49 For isotopically heavy reductive methylation, a solution of 50 mM sodium phosphate (pH 7.4), 0.12 mg borane dimethylamine and 3.2 mM isotopically heavy d2-formaldehyde (Cambridge Isotope Laboratories) was added to one gel band, while 50 mM sodium phosphate (pH 7.4) alone was added to the control gel band. This reaction was incubated for 2 h at 4°C. An equivalent aliquot of the borane dimethylamine/d2-formaldehyde solution was added and the reaction was again incubated at 4°C for 2 h. A final aliquot of the borane dimethylamine/d2-formaldehyde solution was added and the reaction was incubated at 4°C for ∼16 h. The reaction was quenched with 80 mM Tris-Cl, pH 7.5. The gel bands were washed three times (5 min per wash) with 0.1 M ammonium bicarbonate. Trypsin (75 ng) was added to the gel bands for 15 h at 37°C.Citation20,Citation49 Tryptic peptides were collected with POROS R2 20 micron beads (Applied Biosystems), isolated with C18 ZipTips (Millipore) and analyzed by MALDI mass spectrometry as described above.
Demethylase assay.
The demethylase reaction consisted of 0.25 µg H3K4me2-biotin peptide (1ARTKme2QTA RKS TGG KAP RKQ LYKbio) plus 15.6, 31.3, 62.5, 125, 250 or 500 ng recombinant LSD1 (BPS Biosciences) in the following reaction buffer: 50 mM Tris-Cl pH 8.5, 50 mM KCl, 5 mM MgCl2, 5% glycerol. The reaction proceeded for 2 h at 37°C. Reaction products were collected with POROS R2 20 micron beads for 15 min at room temperature, loaded into a C18 ZipTip, eluted in 40 µL 70% acetonitrile/0.1% TFA, lyophilized and subjected to MassSQUIRM as described above.
LSD1 inhibition assay.
Inhibition experiments were performed in triplicate under demethylase assay conditions as described above using 0.25 µg of H3K4me2-bio peptide alone, peptide with 125 ng GST-LSD1 (prepared in-house) and peptide with LSD1 and 16.7 mM phenylethylhydrazine inhibitor (Chem Services). As described above, peptides were isolated with POROS R2 20 micron beads and ZipTips prior to Mass SQUIRM analysis.
Abbreviations
| massSQUIRM | = | Mass spectrometric quantitation using isotopic reductive methylation |
| MAO | = | monoamine oxidase |
| PTM | = | post-translational modification |
Figures and Tables
Figure 1 Reductive methylation of histone H3. (A) The N-terminus of H3 is shown as being un- (green), mono- (red) or di- (blue) methylated at lysine 4. On a MALDI mass spectrometer, addition of a single methyl group is recorded as an addition of 14 Da while addition of two methyl groups is recorded as an addition of 28 Da. Variation in the chemical composition of each peptide leads to differential ionization making quantification complex. (B) Reductive methylation converts all lysine residues to the di-methyl state, which causes all peptides to be identical in mass and ionize similarly; however, it also makes the original methylation states indistinguishable. (C) The use of heavy formaldehyde in the reductive methylation reaction allows retention of the identity of the original, isotopically light, methylation (open circles). In addition, any lysines not already modified will undergo reductive methylation forcing them to an isotopically heavy di-methyl state (closed circles). This conversion allows each peptide to ionize the same while retaining the identity of the original peptide (light vs. heavy methylation).
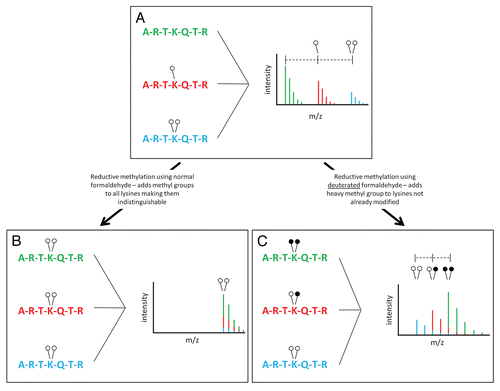
Figure 2 Efficiency and dynamic range of MassSQUIRM. (A) A synthetic peptide containing four unmodified lysine residues (lower part) was exposed to reductive methylation using either light formaldehyde (middle part) or heavy formaldehyde (top part). The resulting spectra show a ∼100% conversion of all lysine residues, as well as the N-terminus, to the di-methyl state. Heavy formaldehyde showed a peak 20 Da larger than that of the light formaldehyde as would be expected. (B) MassSQUIRM is efficient in-gel with full-length proteins. A recombinant version of full-length human H3.2 was subjected to in-gel tryptic digestion either with (top part) or without (bottom part) prior in-gel isotopically heavy reductive methylation. Resulting peptides were analyzed by MALDI-TOF mass spectrometry. A ∼100% conversion to heavy di-methylated peptide was observed. (C) Un-, mono- and di-methylated synthetic peptides were normalized to a 1:1:1 mixture and mono-methylated peptide concentration was varied at the theoretical ratios indicated (untreated sample). The same peptide mixtures were separately treated with MassSQUIRM (treated sample). The linear dynamic range was determined to be 1:8 for both treated (◆) and untreated (■) samples. Open circles indicate light methylation while closed circles indicate heavy methylation.
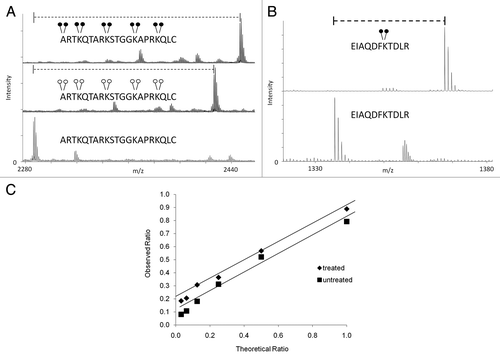
Figure 3 MassSQUIRM accurately corrects for differences in ionization. (A) Equimolar amounts of an H3K27 peptide with 0, 1 or 2 methyl groups were mixed and analyzed by MALDI mass spectrometry. The un-, mono- and di-methylated peptides ionize at different efficiencies. (B) A synthetic peptide representing H3K4me2 was incubated with the histone demethylase LSD1 and the resulting peptides were analyzed by conventional mass spectro-metry. (C) The same reaction was performed as in (B) but peptides were analyzed using MassSQUIRM. (D) MassSQUIRM allows quantification of mono-methyl peptide species that was omitted in conventional mass spectrometry analysis due to differential ionization seen in (A). Open circles indicate light methylation while closed circles indicate heavy methylation.
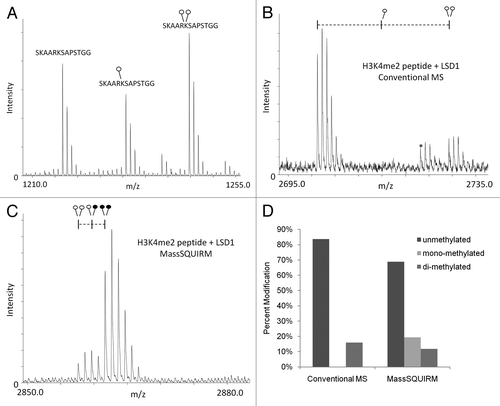
Figure 4 MassSQUIRM can be used to successfully quantify differentially modified peptides. (A) A di-methylated synthetic peptide was analyzed using mass spectrometry and the resulting isotopic envelope was used to determine r1 and r2 from Equationequations 3(3)
(3) and Equation4
(4)
(4) . This peptide corresponds to that noted in blue in (inset). (B) The same synthetic peptide was incubated with 125 ng GST-LSD1 in demethylase buffer for two hours at 37°C. Samples were then subjected to MassSQUIRM analysis. A mixed population of overlapping peaks represents three different methylation states as seen in (inset). Peptides initially mono-methylated occur at a mass 2 Da heavier than di-methylated peptides due to the addition of one heavy methyl group during reductive methylation. Similarly, peptides initially unmodified will occur 4 Da heavier than di-methylated peptides due to the addition of two heavy methyl groups. Areas under the monoisotopic peaks were noted as A1, A2 and A3. These values were used to determine Equationequations 5
(5)
(5) –Equation7
(7)
(7) , which take into account the isotopic overlap noted in (A). Open circles indicate light methylation while closed circles indicate heavy methylation.
 , which take into account the isotopic overlap noted in (A). Open circles indicate light methylation while closed circles indicate heavy methylation.](/cms/asset/ba4c1e64-100b-48fb-8706-c4ea784be143/kepi_a_10914531_f0004.gif)
Figure 5 LSD1 activity can be measured using MassSQUIRM. (A) 0.25 µg of a synthetic H3K4me2 peptide was subjected to varying concentrations of recombinant LSD1 in demethylase buffer for 2 h at 37°C and analyzed with MassSQUIRM. (B) Quantification of methylation levels was determined using r1 and r2 values from peptide alone samples (bottom panel) and Equationequations 5(5)
(5) –Equation7
(7)
(7) . Open circles indicate light methylation while closed circles indicate heavy methylation.
 . Open circles indicate light methylation while closed circles indicate heavy methylation.](/cms/asset/0530c4fd-abf3-461b-b32f-ce0ec59b8cee/kepi_a_10914531_f0005.gif)
Figure 6 LSD1 inhibitor efficiency can be measured quantitatively using MassSQUIRM. (A) Demethylation reactions were carried out using 125 ng of LSD1 and 0.25 µg H3K4me2 peptide in the presence (top part) or absence (middle part) of 16.7 mM of the LSD1 inhibitor phenylethylhydrazine. (B) Quantification of methylation levels was determined using r1 and r2 values from peptide alone samples (A, bottom part) and Equationequations 5(5)
(5) –Equation7
(7)
(7) . Open circles indicate light methylation while closed circles indicate heavy methylation.
 . Open circles indicate light methylation while closed circles indicate heavy methylation.](/cms/asset/89360544-d393-46a8-b40b-cdf330a324f5/kepi_a_10914531_f0006.gif)
Acknowledgements
We thank Samuel Mackintosh (UAMS Proteomics Core Facility) and Matthew Sekedat (The Rockefeller University, Laboratory of Mass Spectrometry and Gaseous Ion Chemistry) for mass spectrometric support. We also thank Dr. Frank Millett and Dr. Qin Yan for helpful discussion. Funding for this project was provided by NIH grants P20RR015569, P20RR016460 and R01DA025755.
References
- Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell 2007; 128:635 - 638
- Kouzarides T. Chromatin modifications and their function. Cell 2007; 128:693 - 705
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004; 119:941 - 953
- Forneris F, Binda C, Vanoni MA, Battaglioli E, Mattevi A. Human histone demethylase LSD1 reads the histone code. J Biol Chem 2005; 280:41360 - 41365
- Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005; 437:436 - 439
- Nicholson TB, Chen T. LSD1 demethylates histone and non-histone proteins. Epigenetics 2009; 4:129 - 132
- Mosammaparast N, Shi Y. Reversal of histone methylation: biochemical and molecular mechanisms of histone demethylases. Annu Rev Biochem 79:155 - 179
- Amente S, Bertoni A, Morano A, Lania L, Avvedimento EV, Majello B. LSD1-mediated demethylation of histone H3 lysine 4 triggers Myc-induced transcription. Oncogen 29:3691 - 3702
- Lakowski B, Roelens I, Jacob S. CoREST-like complexes regulate chromatin modification and neuronal gene expression. J Mol Neurosci 2006; 29:227 - 239
- Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell 2005; 19:857 - 864
- Lim S, Metzger E, Schule R, Kirfel J, Buettner R. Epigenetic regulation of cancer growth by histone demethylases. Int J Cancer 2010; 127:1991 - 1998
- Culhane JC, Cole PA. LSD1 and the chemistry of histone demethylation. Curr Opin Chem Biol 2007; 11:561 - 568
- Cole PA. Chemical probes for histone-modifying enzymes. Nat Chem Biol 2008; 4:590 - 597
- Chellappan SP. Chromatin protocols 2009; New York, NY Humana Press
- Yamane K, Tateishi K, Klose RJ, Fang J, Fabrizio LA, Erdjument-Bromage H, et al. PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol Cell 2007; 25:801 - 812
- Bordeaux J, Welsh A, Agarwal S, Killiam E, Baquero M, Hanna J, et al. Antibody validation. Biotechniques 48:197 - 209
- Connor C, Cheung I, Simon A, Jakovcevski M, Weng Z, Akbarian S. A simple method for improving the specificity of anti-methyl histone antibodies. Epigenetics 2010; 5:392 - 395
- Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, et al. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 2006; 442:307 - 311
- Smith CM, Gafken PR, Zhang Z, Gottschling DE, Smith JB, Smith DL. Mass spectrometric quantification of acetylation at specific lysines within the amino-terminal tail of histone H4. Anal Biochem 2003; 316:23 - 33
- Tackett AJ, Dilworth DJ, Davey MJ, O'Donnell M, Aitchison JD, Rout MP, et al. Proteomic and genomic characterization of chromatin complexes at a boundary. J Cell Biol 2005; 169:35 - 47
- Rayment I, Rypniewski WR, Schmidt-Base K, Smith R, Tomchick DR, Benning MM, et al. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science 1993; 261:50 - 58
- Agger K, Christensen J, Cloos PA, Helin K. The emerging functions of histone demethylases. Curr Opin Genet Dev 2008; 18:159 - 168
- Schulze WX, Usadel B. Quantitation in mass-spectrometry-based proteomics. Annu Rev Plant Biol 61:491 - 516
- Lim S, Janzer A, Becker A, Zimmer A, Schule R, Buettner R, et al. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 31:512 - 520
- Culhane JC, Wang D, Yen PM, Cole PA. Comparative analysis of small molecules and histone substrate analogues as LSD1 lysine demethylase inhibitors. J Am Chem Soc 132:3164 - 3176
- Yang M, Culhane JC, Szewczuk LM, Jalili P, Ball HL, Machius M, et al. Structural basis for the inhibition of the LSD1 histone demethylase by the antidepressant trans-2-phenylcyclopropylamine. Biochemistry 2007; 46:8058 - 8065
- Yang M, Culhane JC, Szewczuk LM, Gocke CB, Brautigam CA, Tomchick DR, et al. Structural basis of histone demethylation by LSD1 revealed by suicide inactivation. Nat Struct Mol Biol 2007; 14:535 - 539
- Szewczuk LM, Culhane JC, Yang M, Majumdar A, Yu H, Cole PA. Mechanistic analysis of a suicide inactivator of histone demethylase LSD1. Biochemistry 2007; 46:6892 - 6902
- Culhane JC, Szewczuk LM, Liu X, Da G, Marmorstein R, Cole PA. A mechanism-based inactivator for histone demethylase LSD1. J Am Chem Soc 2006; 128:4536 - 4537
- Kahl P, Gullotti L, Heukamp LC, Wolf S, Friedrichs N, Vorreuther R, et al. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res 2006; 66:11341 - 11347
- Schulte JH, Lim S, Schramm A, Friedrichs N, Koster J, Versteeg R, et al. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res 2009; 69:2065 - 2071
- Huang Y, Stewart TM, Wu Y, Baylin SB, Marton LJ, Perkins B, et al. Novel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genes. Clin Cancer Res 2009; 15:7217 - 7228
- Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W, et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 2009; 138:660 - 672
- Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 2007; 446:882 - 887
- Lee MG, Norman J, Shilatifard A, Shiekhattar R. Physical and functional association of a trimethyl H3K4 demethylase and Ring6a/MBLR, a polycomb-like protein. Cell 2007; 128:877 - 887
- Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 2005; 437:432 - 435
- Beumer JH, Tawbi H. Role of Histone Deacetylases and Their Inhibitors in Cancer Biology and Treatment. Curr Clin Pharmacol 2010; 5:196 - 208
- Mavers M, Ruderman EM, Perlman H. Intracellular signal pathways: potential for therapies. Curr Rheumatol Rep 2009; 11:378 - 385
- Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, et al. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 2006; 125:483 - 495
- Fortschegger K, de Graaf P, Outchkourov NS, van Schaik FM, Timmers HT, Shiekhattar R. PHF8 targets histone methylation and RNA polymerase II to activate transcription. Mol Cell Biol 30:3286 - 3298
- Liu W, Tanasa B, Tyurina OV, Zhou TY, Gassmann R, Liu WT, et al. PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature 2010; 466:508 - 512
- Loenarz C, Ge W, Coleman ML, Rose NR, Cooper CD, Klose RJ, et al. PHF8, a gene associated with cleft lip/palate and mental retardation, encodes for an Nepsilon-dimethyl lysine demethylase. Hum Mol Genet 19:217 - 222
- Qi HH, Sarkissian M, Hu GQ, Wang Z, Bhattacharjee A, Gordon DB, et al. Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature 466:503 - 507
- Suganuma T, Workman JL. Features of the PHF8/KIAA1718 histone demethylase. Cell Res 2010; 20:861 - 862
- Yu L, Wang Y, Huang S, Wang J, Deng Z, Zhang Q, et al. Structural insights into a novel histone demethylase PHF8. Cell Res 20:166 - 173
- Yue WW, Hozjan V, Ge W, Loenarz C, Cooper CD, Schofield CJ, et al. Crystal structure of the PHF8 Jumonji domain, an Nepsilon-methyl lysine demethylase. FEBS Lett 584:825 - 830
- Zhu Z, Wang Y, Li X, Xu L, Wang X, Sun T, et al. PHF8 is a histone H3K9me2 demethylase regulating rRNA synthesis. Cell Res 20:794 - 801
- Collom SL, Jamakhandi AP, Tackett AJ, Radominska-Pandya A, Miller GP. CYP2E1 active site residues in substrate recognition sequence 5 identified by photoaffinity labeling and homology modeling. Arch Biochem Biophys 2007; 459:59 - 69
- Gradolatto A, Rogers RS, Lavender H, Taverna SD, Allis CD, Aitchison JD, et al. Saccharomyces cerevisiae Yta7 Regulates Histone Gene Expression. Genetics 2008; 179:291 - 304
- Kurumizaka H, Wolffe AP. Sin mutations of histone H3: influence on nucleosome core structure and function. Mol Cell Biol 1997; 17:6953 - 6969