Abstract
Melanoma progression requires deregulation of gene expression by currently uncharacterized epigenetic mechanisms. A mouse model based on changes in cell microenvironment was developed by our group to study melanocyte malignant transformation. Melanoma cell lines (4C11- and 4C11+) were obtained as result of 5 sequential anchorage blockades of non-tumorigenic melan-a melanocytes. Melan-a cells submitted to 4 de-adhesion cycles were also established (4C), are non-tumorigenic and represent an intermediary phase of tumor progression. The aim of this work was to identify factors contributing to epigenetic modifications in early and later phases of malignant transformation induced by anchorage impediment. Epigenetic alterations occur early in tumorigenesis; 4C cell line shows changes in global and gene-specific DNA methylation and histone marks. Many histone modifications differ between melan-a, 4C, 4C11- (non-metastatic melanoma cell line) and 4C11+ (metastatic melanoma cell line) which could be associated with changes in gene and microRNA expression. These epigenetic alterations seem to play a key role in malignant transformation since melanocytes treated with 5-Aza-2'-deoxycytidine before each anchorage blockade do not transform. Some epigenetic changes seem to be also responsible for the maintenance of malignant phenotype, since melanoma cell lines (4C11- and 4C11+) treated in vitro with 5-Aza-2'-deoxycytidine or Trichostatin A showed reduction of tumor growth in vivo. Changes in gene expression reflecting cell adaptation to new environment were also observed. We propose a model in which sustained microenvironmental stress in melanocytes results in epigenetic reprogramming. Thus, after adaptation, cells may acquire epigenetic marks that could contribute to the establishment of a malignant phenotype.
Keywords: :
Introduction
Epigenetic modifications comprise DNA methylation, histone modifications, chromatin remodeling and mRNA silencing by microRNAs. These mechanisms are essential to normal development and tissue specific expression and their disruption often lead to inappropriate gene expression, contributing to diseases such as cancer.Citation1 Tumor cells normally present aberrant DNA methylation patterns with specific DNA hypermethylation in CpG islands of tumor suppressor promoters, which mostly lead to their inactivation. Besides that, global DNA hypomethylation is also very common in cancer cells compared to normal cells. This event is largely a consequence of DNA methylation loss in repetitive elements which contribute to genomic instability, transposon activation, loss of imprinting (LOI) and oncogene activation.Citation2–Citation4
DNA methylation is catalyzed by DNA methyltransferases (DNMTs), specifically DNMT1, 3A and 3B. Some studies suggest that their overexpression could be responsible for hypermethylation of tumor suppressor promoters in tumor cells, but until now this hypothesis is not clear.Citation5–Citation7 Posttranslational histone modifications, such as acetylation and methylation of lysine residues, have also been defined as epigenetic modifiers and have a crucial role in the control of gene expression. Overall, lysine acetylation is linked to gene activation whereas lysine methylation can be a marker for both gene activation, as in the case of H3K4 trimethylation, or repression, as in the case of H3K27 and H3K9 trimethylation. Enzymes catalyzing addition or removal of histone marks have been found altered in cancer cells as well as proteins responsible for identifying these histone marks.Citation8
The interaction between DNA methylation and histone modification is crucial during development and it is also important to maintain cellular identity.Citation9 At early stages in development, genes with role in embryonic development and lineage specification present a bivalent histone modification state, which contains both repression (trimethylated H3K27) and activation (trimethylated H3K4) histone marks. It was also proposed that this bivalent state is responsible for silencing these genes in stem cells while preserving their potential to become activated during a cell differentiation program.Citation10,Citation11 When bivalent marks are resolved during cell lineage specification, genes involved in development become activated at the same time that genes linked to alternative developmental pathways and pluripotency genes become inactivated. At this point, DNA methylation is also added to these regions resulting in long-term transcriptional silencing.Citation12 Consequently, since DNA methylation and histone modifications are important players in cell fate determination, it has been thought that they may participate at the early stages of cell malignant transformation. Since epigenetic mechanisms are potentially reversible and can change according to environmental variation (i.e., nutrition, inflammation or any other environmental damage or insult), aberrant epigenetic modifications caused by environmental alterations early in development could lead to changes in cell identity and behavior, originating diseases such as cancer. The hypothesis that early epigenetic changes in tissue-specific stem cells might provide an unifying view of cancer etiology has been described as the “Epigenetic progenitor model of cancer”.Citation13 This hypothesis could explain the fact that epigenetic changes are identified very early in cell malignant transformation, and even in normal tissues before tumors arise.Citation14–Citation16
Malignant melanoma is a complex disease that is influenced by many factors, including genetic and epigenetic alterations.Citation17 Melanoma is responsible for 80% of all skin cancer related deaths with no effective cure for metastatic disease. Patients diagnosed with metastatic melanoma have an overall median survival rate of six months.Citation18 Our group developed a mouse malignant transformation model to address questions associated with both early and late epigenetic events in melanoma genesis and to assess the impact of sustained stress conditions in epigenetic patterns. Different cell lines (i.e., 4C, 4C11− and 4C11+) were obtained as a result of sequential cycles of anchorage blockade of melan-a melanocytes.Citation19 Melan-a cells for 96 h cells submitted to two, three and four de-adhesion cycles gave rise to three different cell lineages, named 2C, 3C and 4C, respectively. In this study we used 4C cell line that is incapable to form tumors in vivo, present structural chromosomal abnormalities and is more resistant to anoikis than melan-a cells.Citation20,Citation21 Because 4C cell line presents important differences compared to parental lineage but is still non-tumorigenic we named it as a pre-malignant melanocyte lineage. Melanoma cell lines (i.e., 4C11− and 4C11+) were obtained after five cycles of melan-a cell line de-adhesion, followed by limiting dilution. All clones, randomly chosen, were able to form tumors in vivo. However, these melanoma cell lines presented different degree of aggressiveness and consequently distinct capacity of promoting tumor growth. The melanoma cell line 4C11− is non-metastatic while 4C11+ is able to form metastatic foci. These results were achieved by determining the number of metastatic colonies in the lung after intravenous inoculation of 4C11− and 4C11+ cells into caudal vein of syngeneic mice. Besides that, 4C11− melanoma cell line takes more time to grow in vivo than 4C11+ and is less invasive in Matrigel® than 4C11+ cell line (data not show).
Despite all characteristics described about these cell lines, it is also important to emphasize that melan-a, 4C, 4C11− and 4C11+ cells show differences in morphology, proliferation rate, gene and miRNA expression. Additionally, we have also shown that anchorage-independent culture conditions provided a stressful microenvironment to melan-a cells, with high levels of reactive oxygen species (ROS) that seems to regulate the expression of Dnmts and DNA methylation.Citation22 In this work we have analyzed global epigenetic changes in melan-a and its derived cells (4C, 4C11− and 4C11+) with the purpose to determine their relationship with malignant transformation induced only by changes in the cell microenvironment. In our study, changes in global and gene specific DNA methylation and histone modifications were identified in different steps of melanoma genesis interfering with both gene and microRNA expression. Moreover, melan-a melanocytes treated with the epigenetic drug 5-Aza-2′-deoxycytidine (DNMT inhibitor) immediately before each de-adhesion cycle were impaired to transform. Our main hypothesis is that anchorage blockade might promote epigenetic reprogramming that change the cell phenotype facilitating the adaptation to the new environmental conditions. Then, these reprogramming steps could lead to other events, such as genomic instability and changes in the expression of key genes, which could contribute to the acquisition of a malignant phenotype.
Results
Global DNA methylation levels are altered during melanoma genesis.
In order to establish how epigenetic events are involved in melanocyte malignant transformation and melanoma progression, the global content of DNA methylation was measured in the respective cell lines: (a) melan-a, representing non-tumorigenic skin melanocyte; (b) 4C, characterized as a pre-malignant stage of melanoma; (c) 4C11−, corresponding to non-metastatic melanoma and (d) 4C11+, corresponding to metastatic melanoma. Initially, the total amount of methylated cytosines in DNA was estimated by flow cytometry using a specific antibody. Reduced 5-methylcytosine relative amounts were observed during our model of melanoma progression (), with a significant decrease in DNA methylation in pre-malignant 4C and metastatic melanoma 4C11+ cell lines compared to melan-a melanocytes (). Another methodology used to evaluate the global DNA methylation content was through digestion of genomic DNA with the enzymes HpaII and MspI. Both enzymes have the same restriction site but HpaII is DNA methylation-sensitive and just digests the DNA when the CpG contained in its restriction site is non-methylated. On the other hand, MspI can digest the DNA independently of the CpG methylation status. Therefore, relative levels of DNA methylation were evaluated in the cell lines representing different steps of melanoma genesis. Using this approach, we observed that the relative amount of DNA methylation is reduced in 4C, 4C11− and 4C11+ when compared to melan-a melanocytes (). Taken together, these results indicate an inverse correlation between the degree of DNA methylation and melanocyte malignant transformation.
In the process of cell malignant transformation, it is observed an abnormal methylation profile with global hypomethylation and hypermethylation of specific sequences, such as tumor suppressor genes.Citation23 One of the main reasons for the reduction of total methylated cytosine content in the genome of cancer cells is the demethylation of repetitive elements. In order to investigate the methylation status of specific CpG sites present in some repetitive sequences we have used Ms-SNuPE assay. In a physiological condition, many repetitive sequences are usually methylated and the disruption of this status favors homologous recombination and increased genomic instability.Citation24 The analysis of three specific CpGs in A-repeats () and two CpGs in the retrovirus-C repetitive sequence () showed that these CpGs are progressively demethylated during melanoma genesis.
Changes in the expression of DNA methyltransferases during melanocyte malignant transformation.
To determine if the altered DNA methylation level observed during melanoma genesis is associated to differential expression of Dnmt1, 3a and 3b, we have evaluated mRNA and protein expression of these DNA methyltransferases. DNMT1 was classically characterized as the enzyme responsible for maintenance of DNA methylation profile during mitosis while DNMT3a and 3b play important roles in the establishment of new methylation patterns during embryogenesis. Despite the classification of maintenance or de novo DNA methyltransferases, all these three enzymes have a cooperative function and the disruption of the balance of their interaction can directly affect gene expression.Citation25
First, we have verified a significant increase of Dnmt1 mRNA and protein expression in cell lines representing different steps of melan-a malignant transformation ( and D). On the other hand, Dnmt3a mRNA and protein expression is only altered in the 4C11+ metastatic melanoma lineage, with an increased expression ( and E) while Dnmt3b mRNA and protein content were not statistically changed in the cell lines ( and F).
Therefore, these results demonstrate that the expression of DNA methyltransferases is changed during the process of malignant transformation of melan-a melanocytes suggesting that these events may contribute to the process of melanoma genesis. Furthermore, the alterations of DNA methyltransferases expression in this model may provide a connection with the new pattern of DNA methylation observed in 4C pre-malignant melanocyte, 4C11− non-metastatic melanoma and 4C11+ metastatic melanoma cell lines.
MeCP2 protein expression is significantly decreased during the process of melanoma progression.
MeCP2 is a protein that recognizes methylated DNA. Classically, MeCP2 was described as a factor that contributes with gene suppression, due to its function in recruiting repressive protein complexes to target genes.Citation26 Since we have shown global DNA methylation changes and DNA methyltransferases expression differences during melanoma genesis, our next step was to evaluate MeCP2 protein expression, in different lines that represent the process of melan-a malignant transformation. Using western blot, we have verified that MeCP2 expression is considerably reduced in pre-malignant 4C, in non aggressive 4C11− and in metastatic 4C11+ lineages when compared to melan-a melanocytes (). These results suggest that this protein is important in cancer initiation as well as in the maintenance of a malignant phenotype.
Histone modification changes during melanocyte malignant transformation.
Protein complexes involved in gene regulation are constituted by DNA methyltransferases and by enzymes that modify the histone code. Moreover, the molecules responsible for epigenetic changes in DNA normally act in synergy. For example, many histone methyltransferases (i.e., G9a, Suv39 h) can recruit DNMTs to stably silence coding genes and repetitive sequences.Citation27,Citation28
The histone marks and DNA methylation determine a biochemical signature that coordinate gene expression. Repressive histone marks and methylated DNA are implicated with gene silencing and heterochromatin and also contribute with cell differentiation. On the other hand, stem cells are characterized by the presence of histone bivalent marks which confer plasticity to this cell type.Citation29 In this context, after determining the alterations in DNA methyltransferase expression (), we have analyzed histone marks that could be contributing to the melan-a malignant transformation.
Using western blot, we have evaluated histone marks that can activate genes—trimethylated lysine 4 of histone 3 and acetylated lysine 16 of histone H4 (H3K4me3 and H4K16ac, respectively); repressive histone marks—trimethylated lysine 27, and trimethylated lysine 9 of histone 3 (H3K27me3 and H3K9me3, respectively) and a histone mark that is involved with both gene activation and repression—dimethylated lysine 4 of histone H3 (H3K4me2) (). We have verified an increase of H3K4me2, H4K16ac and H3K27me3 marks during the progression of melan-a malignant transformation. We have also observed an increase in H3K4me3 mark in 4C pre-malignant melanocyte lineage and in non-aggressive 4C11− melanoma cell line when compared to melan-a melanocytes. This increase was followed by a reduction in the metastatic melanoma 4C11+ cell line. A similar related pattern was also observed when the H3K9me3 mark was investigated, a significant increase in 4C, 4C11− and 4C11+ cell lines compared to melan-a melanocytes (). In a global manner, we have observed an increase in both repressive and active histone marks on the intermediate cell lineages (4C and 4C11−) in the process of melanoma genesis. Based on these results, we could conclude that there are important epigenetic modifications in the histone code in the initial steps of malignant transformation of melan-a melanocytes that may influence melanocyte malignant transformation.
Expression of epigenetic machinery components is altered during melanocyte malignant transformation.
Chromatin organization is very important for gene expression control. Chromatin structure assembly is carefully determined by enzymatic complexes that dictate if chromatin configuration will be open, favoring transcription process or if it will be closed and therefore repress transcription.Citation31 Imbalance of chromatin remodelers can disrupt nucleosome arrangement, protein expression and contribute to cancer development. In this manner, we have evaluated the expression of specific genes that are involved in chromatin remodeling. The differential gene expression was evaluated by microarray analysis, in the cell lines that represents melanoma progression (). We have determined that an important number of genes have similar expression in 4C and 4C11− lineages, including histone deacetylases (HDAC7a, Sirt1), methyltransferases (HMTs Dot1l, Setd1b, Setd2, Mll1, Nsd1), demethylases (HDM Jarid1b), Polycomb protein (Bmi1) and proteins containing methyl binding domains (MBDs 2 and 3). On the other hand, a group of genes had a progressive alteration in their expression during melanoma progression, such as histone acetyltransferase (HAT Crebbp), deacethylases (HDACs 5 and 8, Sirt2, 4 and 5), methyltransferase (HMT Prdm2), demethylases (HDM Jarid1c) and Polycomb protein (Cbx4) ().
We have also evaluated by western blot the expression of the proteins Ezh2 and Sirt1. These proteins are implicated in modifications of histone tails and, consequently, in chromatin structure. Sirt1 is a histone deacetylase and Ezh2 is a histone methyltransferase. Ezh2 and Sirt1 are associated with aberrant silencing of tumor suppressor genes. EZH2 is also implicated with repression of proteins of the epigenetic machinery, such as DNA methyltransferases.Citation32 On the other hand, SIRT1 is able to deacetylate many non-histone proteins such as p53.Citation33 Thus, after showing important alterations in histone marks during the process of melan-a melanocyte malignant transformation (), we have determined the expression of Ezh2 () and Sirt1 () during the tumor progression of melan-a melanocytes. Our results have shown that the expression of these enzymes increased significantly in the initial steps of melanoma genesis, represented by the cell lineages 4C and 4C11−, when compared to melan-a melanocytes. The levels of Ezh2 and Sirt1 in the metastatic 4C11+ melanoma cell line were similar to those of melan-a melanocytes.
Other gene studied was Chd1, which code for a chromatin remodeler protein. The expression of Chd1 is described as an important factor for stem cell pluripotency factor and is related with the maintenance of open chromatin. In human cells, Chd1 activity is linked to trimethylated H3K4 because of its capacity of selectively bind to this histone mark.Citation34 Chd1 mRNA expression in the different lineages of melanoma model was analyzed by real time PCR. We have observed that the expression of Chd1 was increased in pre-malignant 4C and non-metastatic melanoma 4C11− lineages and was reduced in the metastatic 4C11+ lineage when compared to melan-a melanocytes ().
As observed for Ezh2, Sirt1 and Chd1, many alterations observed in histone marks were also noted in the first steps of malignant transformation (4C and 4C11− cell lineages) (). Thereby, these changes may play an important role in the beginning of melan-a malignant transformation. Additionally, they reinforce the importance of our model to study cancer initiation and progression.
Treatment of cell lines with 5-Aza-CdR and TSA promotes phenotypic modifications with mRNA and microRNA expression changes.
After determining the epigenetic modifications that are associated to melanocyte malignant transformation, we have analyzed the effect of treatment with 5-Aza-CdR and TSA in cell morphology, protein-coding gene and miRNA expression in cell lines (melan-a, 4C, 4C11− and 4C11+) of our melanoma progression model ().
Initially, the cell lines were cultivated for 48 h with 10 µM 5-Aza-CdR or for 16 h with 40 nM TSA or without any drug. The combined treatment with 5-Aza-CdR followed by TSA was also performed (). Six hours after the end of treatments, images of the cells were acquired and evident phenotypic alterations were observed comparing the three drug treatments. Therefore, the less evident morphology change was observed in the lineages treated with TSA alone (; TSA), followed by intermediate changes in cells treated with 5-Aza-CdR (; Aza). The major alterations were verified in the lineages treated with the combination of these two drugs (; Aza + TSA).
Modification in cell morphology after 5-Aza-CdR treatment was accompanied by gene expression changes. The profile of gene expression determined by microarray revealed that a great number of genes had their expression increased after the treatment in the tumorigenic lines (4C11− and 4C11+) when compared with non-tumorigenic lines (ma and 4C) (). MiRNA profile expression was also analyzed. Cell lines were treated, or not, with 5-Aza-CdR, followed by TSA and miRNA expression was established by miRNA TLDA arrays. Many miRNAs had their expression increased after this treatment (), mainly in tumorigenic cell lines (4C11− and 4C11+).
Alterations in melanocyte epigenetic programs can affect malignant transformation.
In order to investigate the effects of epigenetic alterations in cell proliferation, melan-a melanocytes and their derived cell lines (4C, 4C11− and 4C11+) were treated with drugs that abrogate, in a progressive way, DNA methylation (5-Aza-CdR) and inhibit histone deacetylases (TSA). We have shown that combined use of these two drugs efficiently reduces the capacity of in vitro cell proliferation (). To evaluate the effects of epigenetic drugs in vivo with 4C11− and 4C11+ melanoma cell lines, these cells were treated or not (NT) in vitro with 5-Aza-CdR or TSA and injected into syngenic mice (). Interestingly, there was reduction of 4C11− tumor growth in vivo when these cells were treated with TSA, but not with 5-Aza-CdR (; 4C11−), while the same inhibitory effect was observed in the metastatic 4C11+ cells only when they were treated with 5-Aza-CdR (; 4C11+).
Furthermore, the same procedure resulting in the malignant transformation of melan-a melanocytes was repeated with cells treated with 5-Aza-CdR immediately before each de-adhesion cycle. We verified that no clone derived from melan-a cells submitted to 5 cycles of anchorage blockade, chosen randomly, was capable to develop tumors in vivo after this treatment (). Therefore, these results indicate that the epigenetic changes observed during melanoma genesis are important for cell reprogramming and tumor initiation and progression.
Tumor suppressor genes were upregulated after 5-Aza-CdR treatment in metastatic 4C11+ melanoma cell line.
As showed, 5-Aza-CdR treatment was capable to diminish 4C11+ melanoma cell line proliferation in vitro and in vivo. A possible explanation for this event could be the derepression of tumor suppressor genes abnormally silenced by DNA methylation. Genes involved with apoptosis (Fas), cell cycle arrest (Cdkn1c), negative regulation of JAK/Stat cascade (Socs2), multifunctional transport and tissue homeostasis (Apod), cell adhesion (Itgae), immune response (Bst2) and IFN response (Ifit1) are among the tumor suppressor genes upregulated in metastatic 4C11+ melanoma cell line after treatment with 5-Aza-CdR (). All of them, except Itgae and Cdkn1c, have their expression suppressed in the aggressive 4C11+ melanoma cell line compared to non-tumorigenic melanocytes (data not shown).
Discussion
An essential property of epigenetic marks is that they are dynamic and can be reprogrammed. In the past decades, the study of aberrant epigenetic modifications in human diseases has grown, especially in cancer research. However, little is known regarding the causal role of such mechanisms in the cell malignant transformation process, with some exceptions.Citation13 Tumors generally take many years to develop and disruption of epigenetic mechanisms as one of the major players in the etiology of cancer could provide an explanation for age dependency of disease incidence, which is not well explained by accumulation of mutations.Citation35 Additionally, aberrant epigenetic patterns could explain the relationship between cancer and environment injury, since chronic injury is one of the causes for increased cancer risk.Citation36–Citation38
In the present work, we have shown that epigenetic alterations have a key role in melanocyte malignant transformation induced by changes in cell microenvironment, specially the impediment of cell attachment. After submitting melan-a melanocytes to a stressful condition (successive anchorage blockade cycles), different melanoma cell lines were established. However, the cell malignant transformation was blocked and melanoma cell lines were not obtained when these adhesion impediment cycles were done in the presence of 5-Aza-2′-deoxycytidine (5-Aza-CdR) (). The 5-Aza-CdR drug is a nucleoside analog that promotes loss of DNA methylation by inhibiting DNA methyltransferases and may affect histone modifying enzymes since there are interactions between these proteins and DNMTs.Citation39 Modifications in epigenetic patterns were observed in the process that led to melanoma development, including the early steps of this process (represented by pre-malignant cell line 4C). We reported a progressive DNA hypomethylation () and significant alterations in histone marks () during melan-a melanocyte malignant transformation. Differential expression of enzymes that are responsible for DNA methylation (Dnmt1 and Dnmt3a) () and histone tails changes (i.e., Mll1, Ezh2 and Sirt1) (, respectively) were also observed during this process, as well the expression of other epigenetic machinery components (), such the methyl binding protein MeCP2 () and the chromatin remodeler Chd1 (). In addition, it was shown that a number of protein-coding genes and microRNAs are upregulated after 5-Aza-CdR treatment alone or combined with Trichostatin A (in the case of microRNAs) () suggesting their epigenetic control and their possible relationship with melanoma progression. Moreover, the same drugs were able to alter the phenotype () and in vitro proliferation capacity () of the melan-a, 4C, 4C11− and 4C11+ cell lines, and decrease their ability of in vivo tumor growth mediated by melanoma cell lines 4C11− and 4C11+ (). Interestingly, TSA treatment was more efficient in diminishing 4C11− tumor growth in vivo than 5-Aza-CdR. On the other hand, treatment with 5-Aza-CdR, but not TSA, was able to decrease 4C11+ tumor growth in vivo. Despite DNA methylation importance at the beginning of melanocyte malignant transformation as indicated in , histone modifications seem to be important to characterize the less aggressive melanoma phenotype better than DNA methylation. This scenario changes in metastatic melanoma since DNA methylation seems to be an additional mark to repress tumor suppressor genes. Tumor suppressor genes activated by 5-Aza-CdR treatment in 4C11+ melanoma cell line present properties linked to tumor growth control (). Moreover, these genes are related to human melanoma as well. According to Melanoma Molecular Map Project (available online at www.mmmp.org/MMMP/) there is a correlation between tumor suppressor genes identified in our study with those found aberrantly hypermethylated in human melanoma cell lines, uncultured melanoma tumor samples and/or serum from patients in the literature searched.Citation40–Citation46 Taken together, these results underscore the significance of our model to discovery novel epigenetic therapy responsive genes and miRNAs for the investigation of multiple markers in clinical trials for each step of human melanomagenesis and progression. This potential translational application from the basic research with our mouse melanoma model to clinical research might be especially important in metastatic disease.
Overall, the differences in the epigenetic profile of these cells (melan-a, 4C, 4C11− and 4C11+) suggest an epigenetic reprogramming caused by the sustained stress condition. It is possible that during malignant transformation, melan-a melanocytes undergo alterations conferring an undifferentiated state in order to reprogram the epigenetic marks and adapt the cells to new injury conditions. In contrast to melan-a cell line, that present an epithelial morphology, 4C cells show a mesenchymal morphology, which could indicate a less differentiated state ( and NT). The conversion of epithelial to mesenchymal phenotype, which characterize a process named epithelial to mesenchymal transition (EMT), is fundamental for embryonic development and is needed for final differentiation of specialized cell types. Furthermore, it has been shown that EMT is also involved in tumor progression and metastasis.Citation47 After EMT, tumor cells may reacquire an epithelial phenotype, in a process named mesenchymal to epithelial transition (MET). Additionally, the increase of Chd1 expression () correlates with 4C cells less differentiated phenotype. This chromatin remodeling factor recognizes H3K4 trimethylation mark, which is present in high levels in 4C cells (), and is required to maintain open chromatin state in stem cells. An open and highly accessible chromatin is a hallmark of stem cells and it is essential for their ability to give rise to any cell type (plurypotency).Citation48,Citation49 Besides the increase in H3K4 trimethylation (mark of gene activation and open chromatin) (), an increase in H3K27 trimethylation (mark of gene inactivation) () was also seen in the early steps of melanocyte malignant transformation. This observation might indicate other “stem cell like” features, since developmental genes are identified with both marks and this is important for determining cell fate.Citation10,Citation50 Another molecular change that corroborates with the hypothesis that a less differentiated phenotype is acquired in the beginning of melanocyte malignant transformation is the increased levels of Sirt1, which is related with pluripotency state. Calvanese and colleaguesCitation51 demonstrated that Sirt1 downregulation is needed for human embryonic stem cell differentiation. Low levels of Sirt1 allow the expression of key developmental genes, which are epigenetically silenced in first phases of embryogenesis. During melanoma progression, we have observed that Sirt1 expression increases in pre-malignant 4C cells compared to parental melan-a melanocytes. There was also increased expression of Mll1, a H3K4 methyltransferase, and Ezh2, a H3K27 methyltransferase, in pre-malignant 4C cells. The expression of MeCp2 protein is drastically reduced in the transition of the melanocytes to pre-malignant cells (). These results suggest that a decrease in MeCP2 protein that recognizes methylated CpGs and is implicated in gene repression occurs before global DNA hypomethylation.
Later in melan-a malignant transformation, when cells already show the full malignant phenotype, we can identify melanoma cell lines that are less or more aggressive. One example of a nonmetastatic melanoma cell line is 4C11−, which also have a mesenchymal phenotype and many epigenetic alterations linked to “stem cell phenotype” also found in 4C cells ( and , NT). Moreover, in both 4C and 4C11− cell lines, this morphology resembled an epithelial phenotype with 5-Aza-CdR and TSA treatment ( and 5-Aza-CdR and 5-Aza-CdR + TSA), indicating the participation of epigenetic mechanisms in epithelial to mesenchymal transition. According to this hypothesis, previous data from our group (Morais AS, unpublished data) revealed an increase of the key EMT regulator Snail1 in both 4C and 4C11− cell lines compared to melan-a cells. In addition, a global analysis demonstrated a similar expression of a significant number of important epigenetic machinery factors in 4C and 4C11− lineages (). However, some specific marks were acquired in this important step, when cells became tumorigenic. Among global epigenetic components analyzed in this model, increased levels of H3K9 trimethylation and H3K27 trimethylation (), and elevated expression of Sirt1 ( and respectively) characterized the conversion of 4C pre-malignant melanocyte cell line to tumorigenic, non-metastatic, 4C11− melanoma cells. In contrast, metastatic 4C11+ cells present the highest levels of DNA hypomethylation () and Dnmt3a expression ( and E), a decrease in H3K9 and H3K4 trimethylation (), and reduced expression of the histone deacetylase Sirt1, histone methyltransferase Ezh2 and remodeling factor Chd1 (, respectively) when compared to 4C11− cells. In addition, the number of protein-coding genes and microRNAs upregulated after 5-Aza-CdR treatment alone or in addition to Trichostatin A (in the case of microRNAs) increased in tumorigenic cells (4C11− and 4C11+) compared to the number of protein-coding genes and microRNAs upregulated after the same treatment in non-tumorigenic lineages (melan-a and 4C) (). Based on these results, the epigenetic deregulation of this group of genes and miRNAs might have an important role in melanoma development.
Previous work from our group has shown that the cycles of anchorage impediment promoted a sustained stress condition with high amounts of oxygen reactive species (ROS).Citation22 It is well established that ROS can promote DNA damage.Citation52 To restore DNA integrity and avoid gene mutations, molecules involved with the DNA repair machinery are activated. However, the proteins implicated in this process must be able to access the DNA and modifications in the chromatin structure are essential. In addition to that, chromatin should return to its original configuration after DNA repair, creating a dynamic process between opened and closed chromatin states.Citation53 The sustained oxidative stress condition provided by melan-a de-adhesion cycles could result in DNA damage and consequently in an opened chromatin in which DNA damage repair factors could bind to DNA lesions. In agreement with this hypothesis, it has been shown that chromatin context has a key role in DNA repair processes. The access-repair-restore theory proposed by SmerdonCitation54 showed that oxidative stress and other environment injuries can cause recruitment of DNA repair complexes preferentially to open chromatin regions.Citation55–Citation57 Additionally, an opened chromatin configuration caused by DNA damage might explain the “stem cell like” characteristics identified in pre-malignant 4C cells. The repair factors could then recruit chromatin remodeling proteins, histone modifying enzymes as well as DNMTs to DNA lesion in order to repress transcription and stop possible sequence reading errors. Corroborating this idea, O'Hagan and co-workers demonstratedCitation58 that normal repair of DNA breaks can cause recruitment of the epigenetic machinery linked with transcriptional inhibition, such as Sirt1 and Ezh2 as well as Dnmts. We hypothesize that successive stress conditions (in this case repetitive anchorage blockades) might cause stable epigenetic errors as a consequence of the recruitment of epigenetic machinery by DNA repair factors providing, with time, an aberrant epigenetic and gene expression profile (). Changes in epigenetic profile during the chromatin open state could affect developmental pathways and cell fate. This could lead to an incorrect differentiated state characterized by a malignant phenotype. Later on, tumor cells presenting high global hypomethylation of the DNA and genomic instability, loss of tumor suppressors as well as oncogene activation (by changes in DNA methylation and/or mutations) show a phenotype that is more adapted to new injuries caused by the microenvironment.
In conclusion, our findings significantly contribute to a better understanding of the melanoma physiopathology. This study has important clinical applications in addition to its conceptual contribution to this field. Differently from mutations, epigenetic modifications are reversible and this fact encouragesthe development of new classes of drugs. The model presented in our study provides an opportunity to test drugs (epigenetic drugs among others) that could target molecules that are implicated in epigenetic reprogramming during melanoma progression. We also believe that these new discoveries can open new avenues for a better treatment of melanoma and could also be applied to study other cancer types.
Materials and Methods
Cell culture and drug treatments.
Non-tumorigenic murine melanocyte lineage, melan-a (ma),Citation59 was kindly provided by Dr. Michel Rabinovitch (Department of Parasitology, UNIFESP, São Paulo, Brazil). Cells were cultured in RPMI (pH 6.9, Gibco, Carlsbad, CA) supplemented with 5% fetal bovine serum (FBS, Gibco BRL, Grand Island, NY), antibiotics (Gibco) and 200 nM of phorbol miristate acetate (PMA, Sigma-Aldrich, Saint Louis, MO) at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Pre-malignant melanocyte lineage 4C, non-metastatic melanoma cell line 4C11−, and metastatic melanoma cell line 4C11+, established after submitting melan-a cells to sequential cycles of anchorage blockadeCitation19 were cultured as melan-a cells, but in the absence of PMA. For anchorage independent cycles, melan-a cell line was plated on 1% agarose coated plates and cultured in this condition for 96 h. Small spheroids were then collected by decantation and cultured on standard condition. The de-adhesion cycles were repeated five times. After the last time, spheroids were counted and plated by limiting dilution originating the melanoma cell lines (i.e., 4C11− and 4C11+). Melan-a cells submitted to four de-adhesion cycles were also established. They are non-tumorigenic and were named 4C. Cells were treated with 10 µM of 5-Aza-2′-deoxycytidine (5-Aza-CdR, Calbiochem, Merck, Darmstadt, Germany) for 48 h and/or 40 nM of Trichostatin A (TSA, Calbiochem-Merck, Darmstadt, Germany) for 16 h.
5-methylcytosine relative content analyses.
Global DNA methylation level was evaluated by staining cells with a specific monoclonal antibody against 5-methylcytosine (Oncogene, La Jolla, CA, catalog #NA81). Cells were washed with PBS supplemented with 0.1% Tween-20 and 1% bovine serum albumin (PBS-TB), fixed with 0.25% paraformaldehyde at 37°C for 10 min, followed by 88% methanol at −20°C for at least 30 min. After washing, cells were treated with 2 N HCl at 37°C for 30 min, neutralized with 0.1 M sodium borate (pH 9.0), and blocked with 10% mouse serum for 20 min at 37°C. Then, cells were incubated with anti-5-MeC antibody (1:500) for 1 h at 37°C, followed by incubation with goat anti-mouse IgG conjugated with fluorescein (Kirkegaard & Perry Laboratories Inc., KPL, Gaithersburg, MD) for 45 min at room temperature. Finally, cells were analyzed by flow cytometry in a FACScan (FACScalibur; Becton Dickinson, San Juan, CA).
Digestion of genomic DNA with HpaII and MspI.
HpaII (catalog #FD0514) and MspI (#FD0544) have the same restriction site (CCGG); however HpaII is sensitive to DNA methylation. Two µg of genomic DNA were digested or not with 2 µl of each enzyme (Fast Digest, Fermentas, Glen Burnie, MD) in separated reactions. DNA was digested at 37°C for 16 h and after this period 1 µl of each enzyme was added and the tubes maintained at 37°C for one more hour. Samples were then resolved onto 0.8% agarose gel. The intensity of the band corresponding to intact genomic DNA in different samples was determined using ImageJ software. Percentage of methylation was calculated using the formula: relative global DNA methylation content = (HpaII − MspI) × 100/genomic DNA.
Methylation-sensitive single-nucleotide primer extension assay (Ms-SNuPE).
Average methylation at specific CpG sites present in repetitive elements (A-repeats and retrovirus-C long terminal repeats, RC-LTR) was quantified using methylation-sensitive single nucleotide primer extension assay (Ms-SNuPE) as previously described in reference Citation60. Two µl of genomic DNA treated with sodium bisulfite were amplified by PCR using primers for RC-LTR: 5′-GTT GAT ATT TTG TGT TTT AAG TGG TAA A-3′ and 5′-CTA ATT CTA AAA TAA AAT ATC CCT CC-3′ or primers for A-repeats: 5′-TGA TTT ATT TAT TAG AGG TTT TAG G-3′ and 5′-ACA TAA AAA AAC AAA CTA CCC-3′. PCR conditions were: denaturation at 94°C for 3 min, 35 cycles of 94°C for 1 min, 50°C for 1 min and 72°C for 1 min with an additional extension at 72°C for 10 min. PCR products were separated by electrophoresis through 2% agarose gel, purified from the gel and eluted in 40 µl of water. Single nucleotide extensions were obtained adding 4 µl of template, 1− PCR buffer, 1 µM of each SNuPE primer (described below), 1 µCi (32P) dCTP or (32P) dTTP, and 1 U of 1:1 Taq/TaqStart antibody (BD Biosciences Clontech, Palo Alto, CA). Reaction was then incubated at 95°C for 30 seconds, at 50°C for 30 seconds for RC-LTR and 53°C for 30 seconds for A-repeats, followed by an incubation at 72°C for 1 min and then combined with 5 µl of stop solution (95% formamide, 20 mM EDTA (pH 8.0), 0.05% bromophenol blue and 0.05% xylene cyanol) before being denatured at 94°C for 5 min and loaded onto a 15% denaturing polyacrylamide gel (Tris-borate-EDTA buffer, 14.25% acrylamide, 0.75% bis-acrylamide and 7 M urea). SNuPE primers for RC-LTRs: S1: 5′-TTG TAG TTA ATT AAG GAG TGA TA-3′ and S2: 5′-TTT GTG GTG TTT TTT TTG GT-3′; SNuPE primers for A-repeats: S1: 5′-TTT ATT TAT TAG AGG TTT TAG GGT TT-3′, S2: 5′-GAT TTT GTT GGT AAG GTA GTT-3′ and S3: 5′-GGA TTT GTG TTT TAG ATT AGG TT-3′. Methylation levels were quantified on a PhosphoImager Analysis System (Molecular Dynamics, Sunnyvale, CA). Methylation percentages in the figures represent the average of methylation levels in three different CpG sites for A-repeats and two different CpG sites for RC-LTR.
Methyl thiazol tetrazolium assay (MTT).
Cell viability in the presence of inhibitors (5-Aza-CdR and/or TSA) was determined using a standard methyl thiazol tetrazolium (MTT) assay (Sigma, St. Louis, MO) following standard protocol. The absorbance at 620 nm was recorded in each well using an ELISA microplate reader. Cells (ma, 4C, 4C11− and 4C11+) were cultured (2.5 × 104 cells/mL) in complete medium for 24 h on 96-well plates. Cell culture had the medium replaced by fresh medium containing 10% FBS and then were treated with 5, 10 and 20 µM of 5-Aza-CdR for 48 h and/or 10, 20 and 40 nM of TSA for 16 h. MTT assay was carried out before (T = 0) and after each treatment. Additionally, MTT assay was performed to evaluate the effect of the drug treatment (5-Aza-CdR and/or TSA) effects 24 h after treatment. Cells (ma, 4C, 4C11− and 4C11+) were also (2.5 × 103 cells/mL) cultured in complete medium for 24 h on 96-well plates and then treated as described above. After treatments, cells were washed, the medium replaced by fresh complete medium containing 5% FBS and MTT assay was performed immediately after each treatment (T = 0) and 24 h later (T = 24 h).
Cell image acquisition.
Cell images were acquired by an Olympus CK40/Olympus Singapore microscope using Imagepro 6.2/Media Cybernetics software®.
Western blot.
Whole-cell lysates were prepared using RIPA buffer (0.1% SDS, 0.5% NP40, 0.5% sodium deoxycolate and protease and phosphatase inhibitors). For nuclear extract isolation (for Dnmt3a western blot), cell pellets were re-suspended in a buffer containing 10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 0.1 mM EDTA, 0.5% NP40 and protease and phosphatase inhibitors. After incubating on ice for 5 min, samples were centrifuged (3,000 rpm, 5 min) and the supernatant removed (cytoplasmic extracts). Cells pellets were washed two times with solution containing 10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 0.1 mM EDTA and protease and phosphatase inhibitors. Then, nuclei were lysed in buffer with 400 mM NaCl, 7.5 mM MgCl2, 0.2 mM EDTA, 1 mM DTT and protease and phosphatase inhibitors. Samples were incubated on ice for 15 min (with intermitent shaking). Nuclear debris was pelleted at 13,000 g for 5 min at 4°C and supernatant containing nuclear proteins was collected. Protein extracts were resolved by SDS-PAGE and transferred onto PVDF membranes (Bio-Rad, Hercules, CA). Antibodies and the dilutions used were: mouse Dnmt3a (1:500, Imgenex, San Diego, CA, catalog #IMG-268A), mouse Dnmt3b (1:500, Imgenex, #IMG-184A), mouse Dnmt1 (1:1,000, Imgenex, #IMG-261A), mouse Pcna (1:1,500, Santa Cruz Biotechnology, Santa Cruz, CA, #sc-13057), mouse total Erk (1:1,000, Santa Cruz Biotechnology, #sc-94), mouse β-Actin (1:1,000, Santa Cruz Biotechnology, #sc-1616), mouse C23 (nucleolin, 1:400, NCL, Santa Cruz Biotechnology, #sc-13057) mouse pan-methyl H3K9 (1:1,000, Cell Signaling Technology, #4069), mouse tri-methyl H3K9 (1:1,000, Abcam, Cambridge, MA, #ab8898), mouse tri-methyl H3K27 (1:2,000, Upstate, Temecula, CA, #07-449), mouse di-methyl H3K4 (1:1,000, Cell Signaling Technology, ##9725), mouse tri-methyl H3K4 (1:1,000, Millipore, Temecula, CA, #17-614), mouse acetylated H4K16 (1:1,000, Millipore, Temecula, CA, #07-329), mouse Ezh2 (1:1,000, Cell Signaling Technology, #4905), mouse total H3 (1:1,000, Cell Signaling, #9715), mouse Sirt1 (1:1,000, Millipore, Temecula, CA, #07-131) and mouse MeCP2 (1:1,000, Calbiochem-Merck, Darmstadt, Germany). Followed by secondary antibody (Bio-Rad) incubations, signals were visualized by chemoluminescence using the SuperSignal West Pico® chemiluminescent substrate (Pierce, Thermo Scientific, Rockford).
mRNA expression analysis.
RNA was isolated from cell monolayers (melan-a, 4C, 4C11− and 4C11+) using TRIzol® (Invitrogen, Carlsbad, CA). cDNA was prepared from 1µg of RNA using random hexamer primers and OligodT (Superscript III first-strand synthesis system for RT-PCR, Invitrogen). For Dnmts mRNA expression analysis quantitative RT-PCR was performed in the MJ Research Bio-Rad Real Time PCR Opticon Engine® (MJ Research, Waltham, MO) using TaqMan® system and Kapa probe fast qPCR kit® (KapaBiosystems, Boston, MO) and specific primers (Eurofins MWG Operon, Huntsville, AL) and probes (Biosearch Technologies, Novato, CA): Dnmt3a (anti-sense: 5′-CCA GCT CTC CAA TGC CAA AG-3′, sense: 5′-AAT GCT ACC AAA GCA GCC GA-3′, probe: 5′ FAM-ATG AGC CTG AGT ATG AGG ATG GCC GG-BHQ-1 3′), Dnmt3b (anti-sense: 5′-AGA CAG CCG GGA GCT TGA C-3′, sense: 5′-CAG TCT TGG AGG CAA TCT GCA-3′, probe: 5′ FAM-AGA GCC AGT CTG CAC ACC AGA GAC CA-BHQ-1 3′), Dnmt1 (anti-sense: 5′-TCT GCC ATT TCT GCT CTC CA-3′, sense: 5′-AAA AGC CAA CGG TTG TCC C-3′, probe: 5′ FAM-CCA ACG GGA GCC GGC CAA-BHQ-1 3′), Pcna (anti-sense: 5′-TAG AAT TTT GGA CAT GCT GGT GA-3′, sense: 5′-TCG ACA CAT ACC GCT GCG-3′, probe: 5′ FAM-CCG CAA CCT GCC ATG GGC G-BHQ-1 3′). For Chd1 mRNA expression analysis, quantitative real-time PCR was performed using a Corbett Rotor-Gene 6000 detection system® with a Fast Rotor-Gene SYBER Green PCR Master Mix® (Qiagen, Dusseldorf, Germany). Specific primers were used as follows: Chd1 (sense: 5′-GAT CAC TGG AAC CCC TCT A-3′, anti-sense, 5′-GGA GGC TTG CAT AAC CAT-3′), Gapdh (sense: 5′-AAA TGG TGA AGG TCG GTG TG-3′, anti-sense, 5′-TGA AGG GGT CGT TGA TGG-3′).
Microarray-based gene expression analyses.
Total RNA was isolated as mentioned before and purified using RNEasy MinElute Cleanup® spin columns (QIAGEN, Dusseldorf, Germany) following the manufacturer's instructions. Only RNA samples with an A260/A280 nm ratio >2.0 were used for microarray hybridization. Briefly, 2 µg of total RNA was amplified by an in vitro transcription reaction and biotin-labeled. Arrays (GeneChip® Mouse Genome 430 2.0, format 49, Affymetrix Inc., Santa Clara, CA) were hybridized 16 h at 45°C and 60 rpm, washed, stained with streptavidin-phycoerythrin, scanned using an Affymetrix GeneChip® Scanner 3000 7G, and images were quantified by GCOS 1.4 software (Affymetrix), as described in the Affymetrix Expression Analysis Technical Manual. Sixteen microarrays were performed, including two biological replicates from the same phenotype.
Microarray data analyses.
Raw array data (.CEL file) were quantile normalized with background correction calculated with RMAExpress 1.0.5 (rmaexpress.bmbolstad.com). The normalized expression values (log2 scale) were filtered due to strict quality criteria and analyzed using MultiExperiment Viewer (MeV) 4.6 (www.tm4.org). Genes showing at least a two-fold change and a median false discovery rate (FDR) and a q-value threshold at 5% after SAM statistical analysis were chosen, and submitted to an unsupervised hierarchical clustering analysis (HCL) using the Pearson correlation metric distance to arrange samples according to similarity in their gene expression pattern.
miRNA expression analyses.
Total RNA was extracted as described before. MicroRNA expression profile of each cell line treated and non-treated with 5-AzaCdR and TSA were determined using TaqMan® Rodent MicroRNA Arrays (Sanger miRBase v10, card A and card B/Applied Biosystems, Foster City, CA) according to the manufacturer's instructions (Applied Biosystems' 7900 HT Micro Fluidic Cards). TLDA array A comprises miRNAs that are well-known, while Array B contains recently discovered miRNAs with the miR* sequences. The TLDA microfluid cards containing a total of 585 rodent microRNAs, were processed in the ABI 7900 HT Fast Real Time PCR System (Applied Biosystems) and analyzed with Real-Time StatMiner (Integromics, Philadelphia, PA). The miRNA expression was normalized with the endogenous control MammU6 determined in quadruplicate. The relative expression of miRNAs in cell lines treated and non-treated was determined by the 2-ΔΔCT method (p < 0.05 was considered as significant). miRNAs with ΔCt>35 were considered non-expressed. The differential expression was considered when 2-ΔΔCT >1.
Anchorage blockade cycles in the presence of 5-Aza-CdR.
The melan-a melanocyte lineage was treated 48 h with 10 µM 5-Aza-CdR. After treatment, cells were then cultured (105 cells/mL) on 1% agarose-coated Petri dishes for 96 h (for anchorage blockade) at 37°C in humidified 95% air 5% CO2 in RPMI pH 6.9 supplemented with 5% FBS and 200 nM PMA. Small spheroids were collected and cultured on dishes in adhesion condition. The anchorage blockade was repeated five times and after the last one, spheroids were counted and plated by limiting dilution (0.5–1 spheroid/well). After this procedure, clones were obtained and five of them, randomly chosen, were assayed for in vivo tumor formation.
In vivo tumor growth.
4C11− and 4C11+ melanoma cells (2 × 105 cells in PBS per animal), treated or not with 5-Aza-CdR or TSA were injected subcutaneously (s.c.) in the flank of 8 week-old C57BI/6 female mice. After 10 days of inoculation, tumor diameters were measured daily and tumor volume calculated as: d2 × D/2, where d corresponds to the smallest tumor diameter and D to the largest one. Each experimental group had at least 5 animals. All animal procedures used in this study were reviewed and approved by the Institutional Animal Care and Use Committee (approval number 458/08).
Statistical analysis.
For statistical analysis, non-parametric One-way ANOVA test followed by post-hoc test Dunnett or Tukey was used using GraphPad Prism® version 4 for Windows®. The significance level was established at p < 0.05; *p < 0.05 **p < 0.01; ***p < 0.001. Bars indicate +SE of a minimum of triplicate samples. A minimum of 3 independent experiments were performed for each assay.
Abbreviations
| 5-Aza-CdR | = | 5-Aza-2′-deoxycytidine |
| 5-MeC | = | 5-methylcytosine |
| Dnmt1 | = | maintenance DNA methyltransferase 1 |
| Dnmt3a | = | de novo DNA methyltransferase 3a |
| Dnmt3b | = | de novo DNA methyltransferase 3b |
| Ezh2 | = | enhance of zeste homologue-2 |
| Sirt1 | = | sirtuin-1 |
Figures and Tables
Figure 1 Progressive global DNA hypomethylation during melanocyte malignant transformation. (A) 5-methylcytosine (5-MeC) relative amount in the genome of cell lines representing different steps of melanocyte malignant transformation was measured by flow cytometry using a specific antibody against 5-MeC (Calbiochem). (B) Relative content of global DNA methylation was also analyzed by the relation between the genomic DNA digested by the enzymes HpaII and MspI. The methylation status of three specific CpGs of A-repeats (C) and two CpGs in retrovirus-C repetitive sequence (D) was determined by Ms-SNuPE in melan-a, 4C, 4C11− and 4C11+ cell lineages. S1, S2 and S3 represent each CpG analyzed. The C lanes represent primer extension reactions incubated in the presence of only [32P]dCTP; T lanes represent primer extension reactions incubated in the presence of only [32P]TTP. The percentage of methylation was determined by average of different CpG sites analyzed. In all analyses, the average and +SE of three independent experiments is plotted. Each experiment was performed in triplicate. For statistical analysis, a non-parametric one-way ANOVA test followed by post-test Dunnett (A and B) or Tukey (C and D) was used. Statistical significance was established at p < 0.05. Ma: non-tumorigenic melan-a melanocyte lineage; 4C: pre-malignant melanocyte lineage; 4C11−: non-metastatic melanoma cell line and 4C11+: metastatic melanoma cell line; *p < 0.05, **p < 0.01, ***p < 0.001.
![Figure 1 Progressive global DNA hypomethylation during melanocyte malignant transformation. (A) 5-methylcytosine (5-MeC) relative amount in the genome of cell lines representing different steps of melanocyte malignant transformation was measured by flow cytometry using a specific antibody against 5-MeC (Calbiochem). (B) Relative content of global DNA methylation was also analyzed by the relation between the genomic DNA digested by the enzymes HpaII and MspI. The methylation status of three specific CpGs of A-repeats (C) and two CpGs in retrovirus-C repetitive sequence (D) was determined by Ms-SNuPE in melan-a, 4C, 4C11− and 4C11+ cell lineages. S1, S2 and S3 represent each CpG analyzed. The C lanes represent primer extension reactions incubated in the presence of only [32P]dCTP; T lanes represent primer extension reactions incubated in the presence of only [32P]TTP. The percentage of methylation was determined by average of different CpG sites analyzed. In all analyses, the average and +SE of three independent experiments is plotted. Each experiment was performed in triplicate. For statistical analysis, a non-parametric one-way ANOVA test followed by post-test Dunnett (A and B) or Tukey (C and D) was used. Statistical significance was established at p < 0.05. Ma: non-tumorigenic melan-a melanocyte lineage; 4C: pre-malignant melanocyte lineage; 4C11−: non-metastatic melanoma cell line and 4C11+: metastatic melanoma cell line; *p < 0.05, **p < 0.01, ***p < 0.001.](/cms/asset/d930c76d-a976-4460-b4ca-828abe34ccf1/kepi_a_10914917_f0001.gif)
Figure 2 DNA methyltransferase expression is altered during melanoma genesis. The mRNA expression of Dnmt1 (A) Dnmt3a (B) and Dnmt3b (C) was quantified by real time RT-PC R in cell lines representing different stages of malignant transformation of melan-a melanocytes. The charts represent the averages of three independent experiments. For statistical analysis a non-parametric One-way ANOVA test followed by post-hoc test Tukey was used. The significance was established at p < 0.05. The total protein level of these enzymes was evaluated by western blot, using specific antibodies (Imgenex) for Dnmt1 (D), Dnmt3a (E) and Dnmt3b (F). The expression of Erk, nucleolin (C23) and Pcna was used as an internal control of protein extracts. Gel images are representing one of three biological replicates. Ma: non-tumorigenic melan-a melanocyte lineage; 4C: pre-malignant melanocyte lineage; 4C11−: non-metastatic melanoma cell line and 4C11+: metastatic melanoma cell line. **p < 0.01; ***p < 0.001.
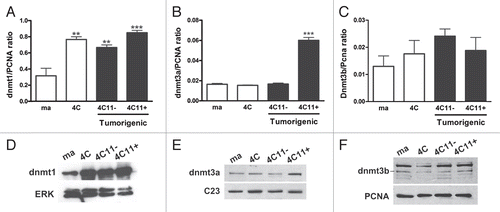
Figure 3 MeCP 2 expression is reduced during melanoma genesis. (A) MeCp2 protein expression was evaluated by western blot in the lineages that mimic different steps of melanocyte malignant transformation. Erk level was used as a control of protein extracts loading. (B) Median and +SE of the ratio between MeCP 2 and Erk expression are represented. The chart represents the averages of three independent experiments. For statistical analysis a non-parametric One-way ANOVA test followed by post-hoc test Tukey was used. The significance was established at p < 0.05. Ma: non-tumorigenic melan-a melanocyte lineage; 4C: pre-malignant melanocyte lineage; 4C11−: non-metastatic melanoma cell line and 4C11+: metastatic melanoma cell line; *p < 0.05, ***p < 0.001.
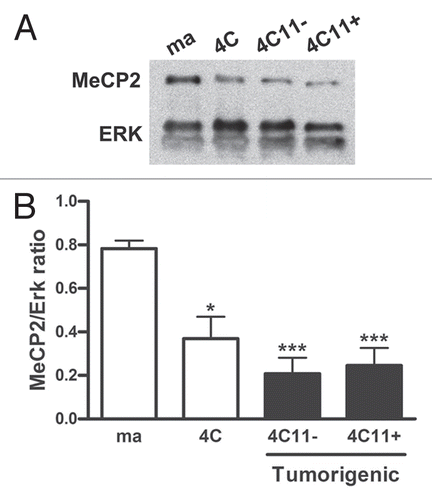
Figure 4 Histone mark analyses during melanocyte malignant transformation. Some histone marks were analyzed during the process of melanoma genesis. H3K4me2 mark, that could be mark for both, activation or repression, the activation marks H3K4me3 and H4K16ac and the repressive marks H3K9me3 and H3K27me3 were investigated by western blot. Total H3 histone expression was used as protein extracts control. The charts represent the ratio between histone marks and total H3 histone of one representative data of two independent experiments. Ma: non-tumorigenic melan-a melanocyte lineage; 4C: pre-malignant melanocyte lineage; 4C11−: non-metastatic melanoma cell line and 4C11+: metastatic melanoma cell line; *p < 0.05; **p < 0.01, ***p < 0.001.
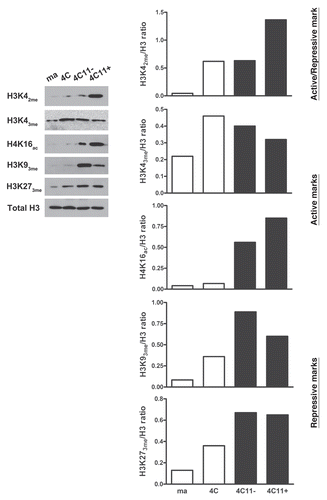
Figure 5 Evaluation of the expression of molecular marks that can model chromatin structure during melanoma progression. The expression of epigenetic machinery components was evaluated by microarray in cell lineages representing different steps of melanocyte malignant transformation. A heatmap for biological replicas of non-tumorigenic (ma and 4C) and tumorigenic (4C11− and 4C11+) cell lines was generated by HCL analysis showing some genes differentially expressed along melanoma genesis identified by SAM statistical analysis (FDR threshold at 5%). Green and red represent genes expressed at low and high level, respectively. The saturation of either color reflects the magnitude of the difference in expression level (A). Ezh2 (B) and Sirt1 (C) protein expression was evaluated by western blot and mRNA expression of Chd1 was measured by real time PCR (D), in the same lineages. Erk and Gapdh levels were used as protein extract control and reference genes, respectively. In the graphics are plotted the average between the histone modifying enzymes and Erk ratio or relative expression calculated by 2-ΔΔCT method +SE are plotted. All experiments were done at least three times, in an independent way. For statistical analysis a non-parametric One-way ANOVA test followed by post-hoc test Tukey was used. The significance was established at p < 0.05. Ma: non-tumorigenic melan-a melanocyte lineage; 4C: pre-malignant melanocyte lineage; 4C11−: non-metastatic melanoma cell line and 4C11+: metastatic melanoma cell line; *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 6 Alterations in cell phenotype, mRNA and miRNA expression after treatment with 5-Aza-CdR and TSA during melanoma genesis. The cell lines representing different steps of melanoma genesis were treated with 5-Aza-CdR and TSA in four different conditions, in RPMI medium with 10% FBS: (1) without any drug; (2) only with 10 µM 5-Aza-CdR for 48 h, (3) just with 40 nM TSA for 16 h and (4) with 10 µM 5-Aza-CdR for 48 h, followed by 40 nM TSA for 16 h. After 6 h, images were captured in an inverted microscope, with 20× magnification (A). Gene expression profile was determined by microarray in cell lines cultured as described in conditions 1 and 2 while miRNA expression patterns were established in lineages cultured in conditions 1 and 4. The number of genes upregulated after 5-Aza-CdR treatment and the number of miRNAs increased after 5-Aza-CdR and TSA treatment are indicated in the table (B). Ma: non-tumorigenic melan-a melanocyte lineage; 4C: pre-malignant melanocyte lineage; 4C11−: non-metastatic melanoma cell line and 4C11+: metastatic melanoma cell line; NT: not treated and Aza: 5-Aza-CdR. *These numbers indicate genes upregulated after 5azaCdR treatment in each cell line compared with non-treated cells. This analysis was performed in microarrays containing the whole mouse genome and considered FDR and q = 5%. # Values indicating number of miRNAs which expression was increased at least two times after 5azaCdR and TSA treatment. A total of 585 miRNAs were analyzed.
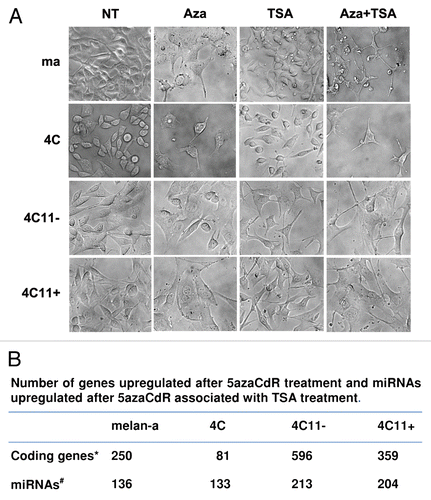
Figure 7 Aberrant epigenetic mechanisms alter the capacity of in vitro cell proliferation, in vivo tumor growth and the acquisition of a malignant phenotype. Treatment conditions of the lineages that mimic different stages of melanoma genesis in the experiments (A and B) were previously described in the legend of . The effect of the treatments in cell proliferation (A) and tumor volume (B) was evaluated, respectively, by MTT assay and subcutaneous inoculation of 2 × 105 4C11− and 4C11+ cells in the flank of 8-week-old C57BI/6 females (five animals per group). In vivo tumor growth of clones derived from melan-a melanocytes after 5 cycles of anchorage blockade in the presence (Cl4 Aza, Cl6 Aza, Cl13 Aza, Cl15 Aza and Cl22 Aza clones) or absence (4C11+ lineage) of 5-Aza-CdR (C). For statistical analysis a non-parametric Two-way ANOVA test followed by post-hoc test Bonferroni was used. The significance was established at p < 0.05. Ma: non-tumorigenic melan-a melanocyte lineage; 4C: pre-malignant melanocyte lineage; 4C11−: non-metastatic melanoma cell line and 4C11+: metastatic melanoma cell line; NT: not treated; Aza: 5-Aza-CdR and Cl: clone.
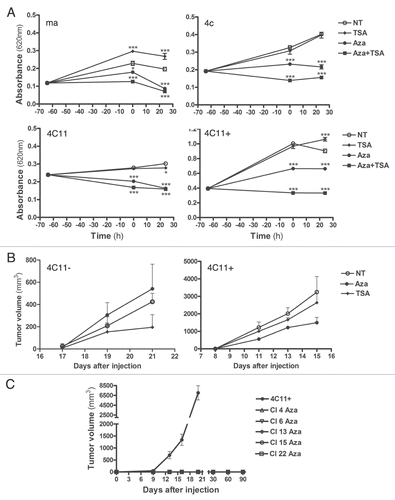
Figure 8 Epigenetic reprogramming associated with sustained stress conditions during melanocyte malignant transformation. We hypothesize that during malignant progression associated with sustained stress, an opened chromatin state occurs, allowing cells to undergo changes in gene expression. We can also observe features that are inherent of pluripotent cells such as the presence of H3K4me3 and H3K27me3 marks and increased expression of SIRT1 and Chd1. This indicates that, after sustained stress, normally differentiated cells acquire a less differentiated state in intermediate stages of progression (4C pre-malignant melanocytes and non-metastatic 4C11− melanoma cell line). Other epigenetic changes subsequently occur providing a more aggressive phenotype (4C11+ cell lineage). EMT: Epithelial to Mesenchymal Transition; MET: Mesenchymal to Epithelial Transition.
Table 1 Tumor suppressor genes upregulated by 5-Aza-CdR treatment in metastatic 4C11+ melanoma cell line
Acknowledgements
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (06/61293-1, to M.G.J.; 08/50366-3, to F.M.; 09/03335-8, to A.S.M., 09/51462-9, to C.F.S.), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (to F.M.M.) and The Maeve McNicholas Memorial Foundation (to F.F.C.).
References
- Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004; 429:457 - 463
- Ehrlich M. DNA hypomethylation, cancer, the immunodeficiency, centromeric region instability, facial anomalies syndrome and chromosomal rearrangements. J Nutr 2002; 132:2424 - 2429
- Narayan A, Ji W, Zhang XY, Marrogi A, Graff JR, Baylin SB, et al. Hypomethylation of pericentromeric DNA in breast adenocarcinomas. Int J Cancer 1998; 77:833 - 838
- Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, et al. Induction of tumors in mice by genomic hypomethylation. Science 2003; 300:489 - 492
- Wu J, Issa JP, Herman J, Bassett DE Jr, Nelkin BD, Baylin SB. Expression of an exogenous eukaryotic DNA methyltransferase gene induces transformation of NIH 3T3 cells. Proc Natl Acad Sci USA 1993; 90:8891 - 8895
- Robertson KD, Uzvolgyi E, LIang G, Talmadge C, Sumegi J, Gonzales FA, et al. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res 1999; 27:2291 - 2298
- Wang YA, Kamarova Y, Shen KC, Jiang Z, Hahn MJ, Wang Y, et al. DNA methyltransferase-3a interacts with p53 and represses p53-mediated gene expression. Cancer Biol Ther 2005; 4:1138 - 1143
- Chi P, Allis CD, Wang GG. Covalent histone modifications: miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer 2010; 10:457 - 469
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 2009; 10:295 - 304
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006; 125:315 - 326
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007; 448:553 - 560
- Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007; 447:425 - 432
- Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet 2006; 7:21 - 33
- Cui H, Cruz-Correa M, Giardiello FM, Hutcheon DF, Kafonek DR, Brandenburg S, et al. Loss of IGF2 imprinting: a potential marker of colorectal cancer risk. Science 2003; 299:1753 - 1755
- Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links aging and neoplasia in human colon. Nature Genet 1994; 7:536 - 540
- Baylin SB, Ohm JE. Epigenetic gene silencing in cancer—a mechanism for early oncogenic pathway addiction?. Nat Rev Cancer 2006; 6:107 - 116
- Dahl C, Guldberg P. The genome and epigenome of malignant melanoma. APMIS 2007; 115:1161 - 1176
- Houghton AN, Polsky D. Focus on melanoma. Cancer cell 2002; 2:275 - 278
- Oba-Shinjo SM, Correa M, Ricca TI, Molognoni F, PInhal MA, Neves IA, et al. Melanocyte transformation associated with substrate adhesion impediment. Neoplasia 2006; 8:231 - 241
- Ricca TI, Liang G, Suenaga AP, Han SW, Jones PA, Jasiulionis MG. Tissue inhibitor of metalloproteinase 1 expression associated with gene demethylation confers anoikis resistance in early phases of melanocyte malignant transformation. Transl Oncol 2009; 2:329 - 340
- Silva AG, Graves HA, Guffei A, Ricca TI, Mortara RA, Jasiulionis MG, Mai S. Telomere-centromere-driven genomic instability contributes to karyotype evolution in a mouse model of melanoma. Neoplasia 2010; 12:11 - 19
- Campos AC, Molognoni F, Melo FH, Galdieri LC, Carneiro CR, D'Almeida V, et al. Oxidative stress modulates DNA methylation by endogenous nitric oxide during cell substrate adhesion blockade. Neoplasia 2007; 9:1111 - 1121
- Robertson KD. DNA methylation, methyltransferases and cancer. Oncogene 2001; 20:3139 - 3155
- Guz J, Foksinski M, Olinski R. Global DNA hipomethylation: the meaning in carcinogenesis. Postepy Biochem 2010; 56:16 - 21
- McCabe MT, Davis JN, Day ML. Regulation of DNA methyltransferase 1 by the pRb/E2F1 pathway. Cancer Res 2005; 65:3624 - 3632
- Bogdanovic O, Veenstra GJ. DNA methylation and methyl-CpG binding proteins: developmental requirements and function. Chromosoma 2009; 118:549 - 565
- Tachibana M, Matsumura Y, Fukuda M, Kimura H, Shinkai Y. G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J 2008; 27:2681 - 2690
- Lehnertz B, Ueda Y, Derijck AA, Braunschweig U, Perez-Burgos L, Kubicek S, et al. Suv39 h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol 2003; 13:1192 - 1200
- Bapat SA, Jin V, Berry N, Balch C, Sharma N, Kurrey N, et al. Multivalent epigenetic marks confer microenvironment-responsive epigenetic plasticity to ovarian cancer cells. Epigenetics 2010; 5:716 - 712
- Pinskaya M, Morillon A. Histone H3 lysine 4 dimethylation: a novel mark for transcriptional fidelity?. Epigenetic 2009; 4:302 - 306
- Jenuwein T, Allis CD. Translating the histone code. Science 2001; 293:1074 - 1080
- Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res 2008; 647:21 - 29
- Yi J, Luo J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim Biophys Acta 2010; 1804:1684 - 1689
- Persson J, Ekwall K. Chd1 remodelers maintain open chromatin and regulate the epigenetics of differentiation. Exp Cell Res 2010; 316:1316 - 1323
- Bjornsson HT, Fallin MD, Feinberg AP. An integrated epigenetic and genetic approach to common human disease. Trends Genet 2004; 20:350 - 358
- Bode AM, Dong Z. Cancer prevention research—then and now. Nat Rev Cancer 2009; 9:508 - 516
- Islami F, Boffetta P, Ren JS, Pedoeim L, Khatib D, Kamangar F. High-temperature beverages and foods and esophageal cancer risk: a systematic. Int J Cancer 2009; 125:491 - 524
- Tucker MA. Melanoma epidemiology. Hematol Oncol Clin North Am 2009; 23:383 - 395
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 2009; 10:295 - 304
- Gallagher WM, Bergin OE, Rafferty M, Kelly ZD, Nolan IM, Fox EJ, et al. Multiple markers for melanoma progression regulated by DNA methylation: insights from transcriptomic studies. Carcinogenesis 2005; 26:1856 - 1867
- Muthusamy V, Duraisamy S, Bradbury CM, Hobbs C, Curley DP, Nelson B, et al. Epigenetic silencing of novel tumor suppressors in malignant melanoma. Cancer Res 2006; 66:11187 - 11193
- Klisovic DD, Katz SE, Effron D, Klisovic MI, Wickham J, Parthun MR, et al. Depsipeptide (FR901228) inhibits proliferation and induces apoptosis in primary and metastatic human uveal melanoma cell lines. Invest Ophthalmol Vis Sci 2003; 44:2390 - 2398
- van der Velden PA, Zuidervaart W, Hurks MH, Pavey S, Ksander BR, Krijgsman E, et al. Expression profiling reveals that methylation of TIMP3 is involved in uveal melanoma development. Int J Cancer 2003; 106:472 - 479
- Marini A, Mirmohammadsadegh A, Nambiar S, Gustrau A, Ruzicka T, Hengge UR. Epigenetic inactivation of tumor suppressor genes in serum of patients with cutaneous melanoma. J Invest Dermatol 2006; 126:422 - 431
- Liu S, Ren S, Howell P, Fodstad O, Riker AI. Identification of novel epigenetically modified genes in human melanoma via promoter methylation gene profiling. Pigment Cell Melanoma Res 2008; 21:545 - 558
- Thiery JP. Epithelial-mesenchymal transitions in cancer onset and progression. Bull Acad Natl Med 2009; 193:1969 - 1678
- Gaspar-Maia A, Alajem A, Polesso F, Sridharan R, Mason MJ, Heidersbach A, et al. Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature 2009; 13:863 - 868
- Sims RJ 3rd, Reinberg D. Stem cells: Escaping fates with open states. Nature 2009; 13:802 - 803
- Szutorisz H, Georgiou A, Tora L, Dillon N. The proteasome restricts permissive transcription at tissue-specific gene loci in embryonic stem cells. Cell 2006; 127:1375 - 1388
- Calvanese V, Lara E, Suárez-Alvarez B, Abu Dawud R, Vázquez-Chantada M, Martínez-Chantar ML, et al. Sirtuin 1 regulation of developmental genes during differentiation of stem cells. Proc Natl Acad Sci USA 2010; 107:13736 - 13741
- Bjelland S, Seeberg E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat Res 2003; 531:37 - 80
- Escargueil AE, Soares DG, Salvador M, Larsen AK, Henriques JA. What histone code for DNA repair?. Mutat Res 2008; 658:259 - 270
- Smerdon MJ. DNA repair and the role of chromatin structure. Curr Opin Cell Biol 1991; 3:422 - 428
- Amouroux R, Campalans A, Epe B, Radicella JP. Oxidative stress triggers the preferential assembly of base excision repair complexes on open chromatin regions. Nucleic Acids Res 2010; 38:2878 - 2890
- Costes SV, Chiolo I, Pluth JM, Barcellos-Hoff MH, Jakob B. Spatiotemporal characterization of ionizing radiation induced DNA damage foci and their relation to chromatin organization. Mutat Res 2010; 704:78 - 87
- Suter B, Livingstone-Zatchej M, Thoma F. Chromatin structure modulates DNA repair by photolyase in vivo. EMBO J 1997; 16:2150 - 2160
- O'Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet 2008; 4:1000155
- Bennett DC, Cooper PJ, Hart IR. A line of non-tumorigenic mouse melanocytes, syngeneic with the B16 melanoma and requiring a tumour promoter for growth. Int J Cancer 1987; 39:414 - 418
- Gonzalgo ML, Jones PA. Quantitative methylation analysis using methylation-sensitive single-nucleotide primer extension (Ms-SNuPE). Methods 2002; 27:128 - 133