Abstract
Aberrant TGFβ signaling pathway may alter the expression of down-stream targets and promotes ovarian carcinogenesis. However, the mechanism of this impairment is not fully understood. Our previous study has identified RunX1T1 as a putative SMAD4 target in an immortalized ovarian surface epithelial cell line, IOSE. In this study, we report that transcription of RunX1T1 was confirmed to be positively regulated by SMAD4 in IOSE cells and epigenetically silenced in a panel of ovarian cancer cell lines by promoter hypermethylation and histone methylation at H3 lysine 9. SMAD4 depletion increased repressive histone modifications of RunX1T1 promoter without affecting promoter methylation in IOSE cells. Epigenetic treatment can restore RunX1T1 expression by reversing its epigenetic status in MCP3 ovarian cancer cells. When transiently treated with a demethylating agent, the expression of RunX1T1 was partially restored in MCP3 cells, but gradual re-silencing through promoter re-methylation was observed after the treatment. Interestingly, SMAD4 knockdown accelerated this re-silencing process, suggesting that normal TGF-beta signaling is essential for the maintenance of RunX1T1 expression. In vivo analysis confirmed that hypermethylation of RunX1T1 was detected in 35.7% (34/95) of ovarian tumors with high clinical stages (P=0.035) and in 83% (5/6) of primary ovarian cancer-initiating cells. Additionally, concurrent methylation of RunX1T1 and another SMAD4 target, FBXO32 which was previously found to be hypermethylated in ovarian cancer was observed in this same sample cohort (P<0.05). Restoration of RunX1T1 inhibited cancer cell growth. Taken together, dysregulated TGFβ/SMAD4 signaling may lead to epigenetic silencing of a putative tumor suppressor, RunX1T1, during ovarian carcinogenesis.
Introduction
Transforming growth factorβ (TGFβ) is a potent cytokine capable of various physiological processes including cell growth and differentiation in mammals.Citation1 Upon binding to its receptor, TGFβ initiates a signal transduction cascade, resulting in the activation of R-SMAD transcription factors. The activated R-SMAD then associates with SMAD4 and other cofactors which together translocate into the nucleus to regulate gene transcription.Citation2
The TGFβ signaling pathway plays a central role in the growth control of ovarian surface epithelium (OSE) during ovulation.Citation3 It is suggested that TGFβ may prevent over-proliferation of OSE during normal ovulatory cycles.Citation3,Citation4 Thus loss of such signaling may lead to transformation and subsequent ovarian tumorigenesis.Citation5 In this regard, resistance to TGFβ-mediated growth inhibition is frequently observed in the early stages of ovarian cancer.Citation6,Citation7 On the contrary, in more advanced stages of development and disease progression, increased networking between TGFβ and other growth-factor transduction pathways has been shown to further promote tumor proliferation and invasion.Citation8,Citation9 Although, the mechanism underlying this phenomenon is not fully understood, epigenetic modifications have been demonstrated to participate in this processes.Citation10
Epigenetic modifications play an important role in regulating gene transcription.Citation11 Methylation of DNA which occurs at the cytosine residues is one of the most common mechanisms in controlling epigenetic silencing in mammalian cell.Citation12 On the other hand, post-translational modifications of chromatin including acetylation and methylation at the lysine residue of histones are also known to control gene activity.Citation13 In cancer cells, such epigenetic components act in concert to aberrantly repress gene transcription responsible for growth suppression and genomic stability. However, how these epigenetic modifications are established is not fully understood. Recently, we have identified a TGFβ/SMAD4 target, ADAM19 which is expressed in normal OSE cells but is epigenetically silenced in ovarian cancer cells showing dysregulated TGFβ signaling.Citation14 This and several other resultsCitation15,Citation16 suggest that dysregulation of a signaling pathway may lead to epigenetic silencing of the downstream targets.
In the present study, we provide more direct evidences to demonstrate that aberrant TGFβ/SMAD4 signaling may lead to epigenetic silencing of its target gene in ovarian cancer. Our results showed that epigenetic silencing of another TGFβ/SMAD4 target, RunX1T1 (also known as ETO or MTG8) is mediated through DNA methylation as well as histone modification in ovarian cancer. Knockdown of SMAD4 accelerated re-silencing of RunX1T1 through faster re-methylation of the promoter in demethylated MCP3 ovarian cancer cells. Interestingly, methylation of RunX1T1 was detected in ovarian cancer patients and their corresponding ovarian cancer initiating cells (OCIC) thus suggesting that methylation of RunX1T1 is crucial for ovarian carcinogenesis.
Results
RunX1T1 is a SMAD 4 regulated gene in ovarian cancer.
Our previous study using integrative ChIP-Chip and expression arrays have identified RunX1T1 (also known as ETO or MTG8) as a SMAD4 target gene in IOSE.Citation17 After TGFβ treatment, IOSE cells showed a progressive increase of RunX1T1 expression in a time-dependent manner (Fig. S2). To further confirm the role of SMAD4 in regulating transcription of RunX1T1, we performed a knockdown experiment in IOSE and SKOV3 cells using siRNA against SMAD4. IOSE showed a 50.5% and 40% downregulation of SMAD4 and RunX1T1 expression, respectively. Similarly, 86% and 66% downregulation of SMAD4 and RunX1T1 expression was also observed in SKOV3 cells ( and C). Downregulation of RunX1T1 correlated with decreased expression of SMAD4 in both cells. These results confirmed that RunX1T1 is a SMAD4 regulated gene in IOSE cells as well as in ovarian cancer cell line.
RunX1T1 promoter contains SMAD 4 binding element.
Bioinformatic analysis has identified three potential SMAD4 binding elements (SBE) from −500 bp to +200 bp in the RunX1T1 putative promoter region ().Citation17 This region exhibited SMAD4 recruitment upon TGFβ activation in IOSE cells as demonstrated previously by ChIP-chip (Fig. S3) and ChIP-PCR.Citation17 To evaluate if this region contains any promoter activity, we performed reporter assay using a luciferase plasmid construct containing this potential RunX1T1 promoter. Robust luciferase activity was observed in SKOV3 ovarian cancer cells transfected with plasmid containing this putative promoter () thus suggesting that this region contains RunX1T1 promoter activity.
RunX1T1 is epigenetically silenced in ovarian cancer cell lines.
Our previous study revealed that downregulation of TGFβ target genes are observed in a panel of ovarian cancer cell lines.Citation14,Citation18 To examine if a similar phenomenon can be observed for RunX1T1, mRNA levels of RunX1T1 were examined by quantitative real time RT-PCR in the same panel of cancer cell lines (namely, HeyC2, SKOV3, MCP3, MCP2, A2780 and CP70). Lower or no expression of RunX1T1 expression was found in all ovarian cancer cell lines as compared with IOSE cells ().
To investigate if epigenetic mechanisms contribute to the silencing of RunX1T1 in ovarian cancer cell lines, we first examined the proximal promoter region of RunX1T1 for aberrant epigenetic modifications in the ovarian cancer cell lines. Bisulphite pyrosequencing (), COBRA () and MSP () revealed that a 334 bp CpG island covering around the TSS (−236 bp to +98 bp) of the RunX1T1 promoter was methylated to varying degrees in ovarian cancer cell lines but was almost completely unmethylated in IOSE cells. Traditional bisulphite sequencing confirmed these result in three selected ovarian cancer cell lines in addition to IOSE cells (Fig. S4). To further demonstrate that methylation of this region suppressed transcription, in vitro methylation assay using the above-mentioned RunX1T1 luciferase construct was performed. As expected, in vitro methylation inhibited RunX1T1 promoter activity (). Taken together, these results suggested that DNA methylation plays a significant role in the suppression of RunX1T1 in ovarian cancer cell lines.
Histone modification on RunX1T1 promoter correlates with its expression in cancer cell lines.
Beside DNA methylation, histone modifications also control transcription by affecting chromatin structure. We therefore analyzed the histone status in respect to active and repressive chromatin marks at the RunX1T1 promoter. Chromatin immunoprecipitation (ChIP) experiments were performed using antibodies against dimethylated lysine 4 of histone H3 (H3K4me2), dimethylated lysine 9 of histone H3 (H3K9me2) and trimethylated lysine 27 of histone H3 (H3K27me3). Levels of H3K4me2 at region closed to the RunX1T1 transcriptional start site were more predominantly enriched in IOSE cells than in other cancer cell lines (). On the contrary, higher levels of the repressive histone mark H3K9me2 were found in these cancer cell lines (). However, no correlation between the level of H3K27me3 and expression of RunX1T1 was found in these cell lines (data not shown). Apparently, enrichment of dimethyl-H3K4 and dimethyl-H3K9 at the RunX1T1 promoter coincided with the expression of RunX1T1 in ovarian cell lines. Hence, histone modifications are also involved in the gene silencing of RunX1T1 in ovarian cancer cell lines.
Epigenetic treatments restore RunX1T1 expression by reversing epigenetic status.
After showing that epigenetic modifications may be involved in the silencing of RunX1T1 in ovarian cancer cell lines, we wanted to further clarify that the expression of RunX1T1 is dependent upon its epigenetic status by reversing it and restoring its expression. We therefore treated the cells with the DNMT inhibitor, 5-aza-2′deoxycytidine (5aza) and/or the HDAC inhibitor, trichostatin A (TSA) and examined the epigenetic status of the RunX1T1 promoter. Treatment with 5aza resulted in partial reactivation of RunX1T1 in these cells (). This reactivation was due to partial promoter demethylation as demonstrated in MCP3 ovarian cancer cells (). It is also interesting to note that with the exception of SKOV3, cells with lower DNA methylation (MCP3, MCP2) exhibited higher RunX1T1 reactivation. Co-treatment with 5aza and TSA resulted in a more prominent effect in these cell and a similar promoter demethylation in MCP3 cells ( and B). Surprisingly, treatment with TSA alone resulted in a higher level of reactivation than 5aza alone without affecting the methylation of the promoter ( and B) suggesting that histone modifications may be more important in regulating RunX1T1 transcription, at least in MCP3 cells. As expected, treatment with 5aza and/or TSA also resulted in a relaxation of chromatin in MCP3 cells as demonstrated by about 2-fold increase in the ratio of H3K4me2 versus H3K9me2 (K4/K9, ). However, it is also important to point out that SKOV3 cells which has partial promoter methylation and yet high H3K9me2 did not respond to the epigenetic treatment. Taken together, these results suggested that RunX1T1 is repressed by epigenetic mechanisms in ovarian cancer cell lines.
Epigenetic status of RunX1T1 promoter was controlled by SMAD 4.
As RunX1T1 is a SMAD4 targets in IOSE cells, we sought evidence that SMAD4 plays a role in controlling the epigenetic status of RunX1T1. We examined DNA methylation as well as histone modifications at the RunX1T1 promoter in the aforementioned SMAD4 knockdown experiments in IOSE and SKOV3 cells ( and C). In IOSE cells expressing RunX1T1, knockdown of SMAD4 resulted in a 2-fold decrease in the K4/K9 ratio () without affecting the DNA methylation status () at the RunX1T1 promoter. In SKOV3 cells expressing a low level of RunX1T1, on the contrary, knockdown of SMAD4 only resulted in a slight decrease of the K4/K9 ratio (). However, a slight increase in DNA methylation (∼5%) was observed (). This may be due to the fact that the promoter regions of SKOV3 cells are already in a compact chromatin state () but remain only partially methylated at the promoter CpG sites (). Taken together, these result suggested that SMAD4 plays a significant role in controlling epigenetic status of RunX1T1 expression via epigenetic modifications.
Downregulation of SMAD 4 accelerated epigenetic silencing of RunX1T1.
Our previous study suggested that aberrant TGFβ/SMAD may contribute to epigenetic silencing of its downstream target ADAM19 in ovarian cancer.Citation14 To provide more direct evidence of this signaling-mediated epigenetic silencing, we choose the ovarian cancer cell line MCP3 as it showed the lowest expression of RunX1T1 and highest re-expression of RunX1T1 upon epigenetic drug treatment to perform this experiment ( and ). Cells were first treated with the DNMT inhibitor, 5aza to restore RunX1T1 expression followed by SMAD4 knockdown to observe the changes of RunX1T1 expression as well as DNA methylation in the process of RunX1T1 re-silencing (). As expected, expression of RunX1T1 was restored after treatment of 5aza. This re-expression is associated with de-methylation of RunX1T1 promoter ( and D). The expression of RunX1T1 gradually decreased concomitant with gradual increased in RunX1T1 DNA methylation after demethylation treatment (). Interestingly, SMAD4 knockdown accelerated this re-silencing process as observed by accelerated RunX1T1 mRNA downregulation (). Importantly, this phenomenon is due to faster re-methylation of the RunxX1T1 promoter which is more evident at day 6 of the experiment ( and D). These results provided more direct evidence that disruption of the TGFβ/SMAD4 signaling pathways can lead to epigenetic silencing of its target gene.
Methylation of RunX1T1 associates with tumor stage in ovarian cancer patients.
We next sought to determine the association between methylation of RunX1T1 and clinical-pathological parameters in ovarian cancer patients. MSP was performed to analyze the promoter methylation status of RunX1T1 in 95 ovarian epithelial cancer patient samples (Sup. Table 1). Representative MSP results were shown in . There are a total of 35.7% (34/95) of samples showing methylation of RunX1T1 (). Methylation was able to be confirmed by bisulphite pyrosequencing of selected cancer samples (LA557 and 588) () showing that our MSP assay can detect methylation levels as low as 30% at the CpG sites where the MSP primers are located thus suggesting that it is a sensitive assay for methylation detection.Citation19 Furthermore, significant association between RunX1T1 methylation and higher stage tumor was observed in this sample cohort (p < 0.05, ). Although not statistically significant, patients with methylation of RunX1T1 tended to have shorter progression-free survival than patients without methylation (Fig. S5 and p = 0.09). These results suggested that methylation of RunX1T1 may be associated with the tumor progression. However, the role of RunX1T1 methylation as a prognostic indicator may need further investigation.
Concurrent hypermethylation of RunX1T1 and FBXO32 in ovarian cancer patients.
We have recently demonstrated epigenetic silencing of a newly identified TGFβ/Smad4 target gene, FBXO32 in ovarian cancer.Citation18 Promoter hypermethylation of FBXO32 was observed in HeyC2 ovarian cancer cell line with aberrant TGFβ signaling. In the current study, promoter hypermethylation of RunX1T1, another TGFβ/Smad4 target, is also observed in HeyC2 cells thus suggesting that dysregulation of TGFβ signaling may contribute to concurrent methylation of these two genes in ovarian cancer. In this regard, it is of interest to investigate if there is any correlation between methylation of RunX1T1 and FBXO32 in this cohort of ovarian cancer patients. As expected, methylation of RunX1T1 coincided with that of FBXO32 in this samples cohort (p < 0.05, ). However, further investigations will need to be performed to examine if TGFβ signaling is dysregulated in these cancer patients.
Methylation of RunX1T1 in ovarian cancer initiating cells (OCIC).
Our previous study demonstrated that ovarian cancer may be originated from ovarian cancer initiating cells (OCIC). These OCICs displaying stem cell phenotype exist as suspension spheres in culture and have the ability to self-renew and differentiate to a phenotypically diverse tumor cell population.Citation20 Therefore, we thought to examine if promoter methylation of RunX1T1 occurred in these OCICs. Methylation of RunX1T1 can be found in five out of six (83%) OCIC spheres obtained from ovarian cancer patients as examined by MSP assay ( and B). These result suggested that methylation of RunX1T1 may be crucial for ovarian carcinogenesis.
RunX1T1 suppresses tumor growth in vitro by colony formation assay and growth assay.
Previous studies demonstrated that RunX1T1 may function as a transcriptional repressors.Citation21,Citation22 To investigate the role of RunX1T1 in ovarian cancer cell lines, colony formation assays was performed in SKOV3 and CP70 ovarian cancer cells transfected either with a RunX1T1-expressing plasmid or control vector. Overexpression of RunX1T1 showed a dramatic decrease in colony numbers as compared to control cells ( and B). Additionally, SKOV3 cells stable-expressing RunX1T1 also demonstrated a growth reduction (). Interestingly, this growth reduction correlated with the expression of RunX1T1 in the cells (). Taken together, these results suggested that RunX1T1 may be a potential tumor suppressor in ovarian cancer.
Discussion
Dyrsregulation of the TGFβ/SMAD4 signaling pathway is a common event leading to loss of growth inhibition in ovarian cancer.Citation14,Citation23,Citation24 However the mechanism leading to this dysregulation is not fully understood. Several groups have demonstrated that dysregulation of a signaling pathway may lead to establishment of epigenetic silencing of their downstream targets in various cancers.Citation15,Citation16,Citation25 We recently proposed a model to explain the establishment of epigenetic modifications in ovarian cancer with dysregulated TGFβ signaling.Citation26 In this model, transcriptional inactivation due to transient deprivation of transcription factors can be reversed by signal restoration. Under long-term signal disruption, recruitment of repressive factors causes progressive accumulation of DNA methylation of the targeted promoter which eventually establishes an epigenetic memory for the generation of an inactive heterochromatin state. This hypothesis is further supported by the observations from our current study: first, expression of RunX1T1 which is upregulated by TGFβ in IOSE cells was found to be epigenetically silenced in ovarian cancer cell line with dysregulated TGFβ/SMAD4 signaling;Citation14 second, SMAD4 depletion resulted in chromatin condensation in IOSE cells and accelerated promoter re-methylation of RunX1T1 in demethylated MCP3 cells; third, concurrent methylation of RunX1T1 and another TGFβ target gene, FBXO32 was found in the same cohort of ovarian cancer patient samples. Taken together, these results reinforce the hypothesis that aberrant TGFβ/SMAD4 signaling pathway can lead to epigenetic silencing of its downstream target, RunX1T1 in ovarian cancer.
However, it is noteworthy to point out that histone modifications may be more important in controlling epigenetic silencing of RunX1T1 as treatment of TSA resulted in a similar level of gene reactivation with 5aza without loss of DNA methylation. Additionally, in demethylated MCP3 cells, SMAD4 knockdown resulted in RunX1T1 suppression after 2 days but maximum re-methylation were observed at day 6 of siRNA treatment ( and D) suggesting that histone modifications may precede DNA methylation in the silencing of RunX1T1. However, due to sample limitation, the histone chromatin status cannot be examined. On the other hand, additional factors may still be involved in the epigenetic silencing of RunX1T1. For example, SKOV3 cells showing enhanced SMAD4 expression exhibited reduced RunX1T1 levels as compared to IOSE cells. Further, epigenetic treatments cannot restore RunX1T1 expression in SKOV3 cells. As the transcriptional regulatory module of RunX1T1 was predicted to be controlled by both SMAD4 and p53,Citation17 the lower expression of RunX1T1 in SKOV3 cells may be caused by the fact that the transcriptional module was disrupted in SKOV3 cells which bear a p53 mutation.Citation27,Citation28 This may also explain why epigenetic treatment cannot restore RunX1T1 expression in SKOV3 cells. Accordingly, a similar phenomenon may also occur in patients as p53 is frequently mutated in ovarian cancer.Citation29,Citation30 Further experiments are underway to reconstruct these epigenetic events in IOSE cells.
In this study, restoration of RunX1T1 caused a marked reduction in cancer growth in vitro. Although the role of RunX1T1 in ovarian cancer is not clearly understood, studies in acute myelogenous leukaemia (AML) found that RunX1-RunX1T1 fusion protein which is one of the most common molecular abnormalities in AML can lead to transcriptional repression of its target genes.Citation31 Further investigation of RunX1T1 revealed that it may function as a transcriptional corepressor. For example, RunX1-RunX1T1 fusion protein can associate with DNMT to downregulate RunX1 target gene through DNA methylation.Citation32 Furthermore, RunX1T1 has been shown to be associated with corepressor N-CoR/Sin3A and HDAC in mediating transcriptional repression.Citation21,Citation22 Thus it is reasonable to believe that RunX1T1 may participate in TGFβ growth inhibition in normal ovarian epithelial cells through similar mechanisms and that loss of RunX1T1 may confer tumor growth in ovarian cancer.
The TGFβ signaling pathway is important in regulating cell differentiation as well as stem-cell maintenance.Citation33 However, recent reports have demonstrated that disruption of the TGFβ signaling facilitates the formation of cancer stem/progenitor cells in liver and breast cancer.Citation34–Citation36 In our current study using primary human ovarian tumors, we demonstrated that methylation of RunX1T1 existed in the cancer stem cell component, ovarian cancer initiating cells (OCIC). The existence of methylation of RunX1T1 in both primary tumor and OCIC suggested that disruption of TGFβ signaling is important in maintaining stem cell population in ovarian cancer. However, the absence of RunX1T1 methylation in several primary cancer samples suggest that methylation of RunX1T1 may be crucial for carcinogenesis in a sub-group of ovarian cancer which ultimately developed into a higher stage tumor. Alternatively, disruption of RunX1T1 may be regulated by a mechanism other than DNA methylation in that group of patients.
Investigators have searched for independent prognostic factors in ovarian cancer for a long time.Citation37 Clinico-pathological factors such as age at diagnosis, tumor stage, grade and tumor volume are being searched for correlative effect with patient survival. Oftentimes, these parameters proved to be non ideal or not sensitive enough.Citation38 The use of DNA methylation as prognostic marker in ovarian cancer has been widely described in reference Citation39 and Citation40. In this study, we found that methylation of RunX1T1 was observed in 35.7% of ovarian cancer patients and significantly associated with higher tumor stage. Addition of RunX1T1 methylation to existing marker panels may be able to improve the prognosis of ovarian cancer. However, further studies may be required to validate RunX1T1 methylation in a larger sample cohort for its clinical application.
In conclusion, we have demonstrated that silencing of a TGFβ/SMAD4 target gene, RunX1T1 is mediated through DNA methylation as well as histone modifications in ovarian cancer. This epigenetic silencing may be established by aberrant TGFβ/SMAD4 signaling in ovarian cancer. The tumor suppressor function of RunX1T1 in ovarian cancer and its cancer stem/progenitor cells deserves further investigation.
Materials and Methods
Patient samples.
Ninety-five ovarian cancer samples were obtained from Tri-Service General Hospital, Taipei, Taiwan (Sup. Table 1). Four normal ovarian surface epithelial (NOSE) cells were acquired from patients during surgery for benign gynecological disease at Indiana University as previously described in reference Citation41 and Citation42. For culture of ovarian cancer initiating cells, five ovarian cancer patient samples were obtained from either the Ohio State University Cancer Center, Columbus, OH or Changhua Christian Hospital, Changhua, Taiwan. All studies involving human ovarian cancer samples were approved by the Institutional Review Boards of Indiana University, Ohio State University, Tri-Service General hospital and Changhua Christian hospital.
Cell culture and epigenetic treatment.
Immortalized ovarian surface epithelia cells (IOSE) were maintained in a 1:1 mixture of medium 199 (Sigma, St. Louis, MO) and 105 (Sigma) supplemented with 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA), 400 ng/ml hydrocortisone (Sigma), 10 ng/ml EGF and 50 units/ml of penicillin/streptomycin (Invitrogen).Citation43 Ovarian cancer cell lines A2780, CP70, MCP2, MCP3 were propagated with RPMI 1640 (Invitrogen) containing 10% fetal bovine serum. HeyC2 cell was cultured with DMEM containing 5% FBS, 1% NEAA, 1% Gln and 1% HEPES. SKOV3 cell was cultured with McCoy's 5A containing 10% FBS, 1% NEAA, 1% Gln and 1% HEPES. For epigenetic treatment, 1 × 106 cells were seeded onto 90-mm plates and treated with 0.5 µM 5′-aza-2′-deoxycytidine (5-azaDC; Sigma) for 72 h or trichostatin A (TSA, 0.5 µM; Sigma) for 12 h. For 5-azaDC treatment, media was changed and new drug was added every 24 h. For TGFβ treatment, a total 5 × 104 of IOSE cells were seeded into 35-mm plate. After an overnight culture, cells were treated with 10 ng/ml of TGFβ1 (Sigma). Cells were then harvested at designated time for expression analysis.
Culture of ovarian cancer initiating cells (OCIC).
OCIC was cultured as described previously in reference Citation20. In brief, fresh tumors were minced, suspended in DMEM/F12 medium (Invitrogen), and mixed with 300 units/mL of collagenase (Invitrogen) and hyaluronidase (Sigma), followed by overnight incubation at 37°C incubator with 5% CO2. Enzymatically disaggregated suspensions were washed twice with DMEM/F12, the resulting tumor cells were cultured using serum-free DMEM/F12 supplemented with 5 ug/mL insulin (Sigma), 20 ng/mL human recombinant epidermal growth factor (EGF; Invitrogen), 10 ng/mL basic fibroblast growth factor (bFGF; Invitrogen) and 0.4% bovine serum albumin (BSA; Sigma), in ultra low attachment plates (Corning). Cells were continuously cultured until spheres were formed. Spheres were trypzined every week to obtain single cells for propagation of spheres.
RNA interference studies.
IOSE or SKOV3 cells (3 × 105) were seeded in a 60 mm dish overnight and then transfected with predesigned siRNA against Smad4 (siRNA ID: s8405; Ambion, Austin, TX), or negative control (Ambion) at a concentration of 100 nM using Lipofectamine 2000 (Invitrogen). After 48 h, total RNA was harvested for quantitative RT-PCR analysis.
Bisulphite conversion and combined bisulphite restriction analysis (COBRA).
Genomic DNA (0.5 µg) was bisulphite-modified by EZ DNA Methylation Kit (Zymo Research, Orange, CA) according to manufacturer's protocol. For COBRA analysis, bisulphite-modified DNA was first amplified using RunX1T1 specific primer followed by digestion with 20 U of AciI (GGCG) or Hinp1I (GCGC) (New England Biolabs, Ipswich, MA). Multiple AciI and Hinp1I recognition sites were introduced into the bisulphite converted sequence where the original genomic sequence had been methylated while unmethylated locations were unable to introduce any. The digested PCR products were separated by gel electrophoresis using 3% GenePure high-resolution agarose (ISC BioExpress, Kaysville, UT) and stained with ethidium bromide. All primer sequences were listed in Supplemental Table 2.
Bisulphite sequencing and methylation specific PCR (MSP).
The bisulphite-modified DNA was subjected to bisulphite sequencing and MSP as previously described in reference Citation18. For bisulphite sequencing, bisulphite PCR products were cloned into the TOPO TA cloning kit (Invitrogen) according to manufacturer's protocol. Five randomly selected clones were sequenced by Genomics Biotechnology Company (Taiwan). Sequencing results were analyzed by BiQ analyzer.Citation44 All primer sequences were listed in Supplemental Table 2.
Bisulphite pyrosequencing.
Bisulphite-modified DNA was subjected to PCR amplification strategy using a tailed reverse primer in combination with a biotin-labeled universal primer as described in reference Citation45. PCR and sequencing primers were designed using PyroMark Assay Design 2.0 (Qiagen GmbH, Hilden, Germany). RunX1T1 promoter (−339 to −75) was PCR-amplified in a 25 µl reaction containing 12.5 µl of 2× RBC Sensizyme Hotstart Taq premix (RBC Bioscience, Taiwan) 0.5 µM of each primer and 6.8 µl (around 35 ng) of bisulphite-modified DNA for 95°C for 5 min followed by 50 cycles of 95°C for 30 s, 62°C for 1 min and 72°C for 45 s and a final extension at 72°C for 7 min. 1.5 µl of each PCR reaction were analysed on a 1% agarose gel before pyrosequencing. Pyrosequencing was performed on the PyroMark Q24 (Qiagen) using the Pyro Gold Reagents (Qiagen) according to the manufacturer's protocol. The methylation level of eleven CpG sites from −187 to −104 with respective to transcriptional start site (TSS) was measured. The methylation percentage of each cytosine was determined by the fluorescence intensity of cytosines divided by the sum of fluorescence intensity of cytosines and thymines at each CpG site. In-vitro methylated DNA (Millipore) was included as positive control for pyrosequencing. Representative pyrograms were shown in Figure S1. All primer sequences were listed in Supplemental Table 2.
RNA extraction and quantitative reverse transcription-PCR.
Total RNA from cell lines was extracted using Trizol (Invitrogen) as previously described in reference Citation18. In brief, 1 µg of total RNA was treated with DNase I (amplification grade, Invitrogen) before first-strand cDNA synthesis using reverse transcriptase (Superscript II RT, Invitrogen). PCR reactions were carried out using ABI Stepone real time PCR system (Applied Biosystems, Foster City, CA). Primers used to amplify RunX1T1 and GAPDH cDNA can be found in Supplemental Table 2. Relative expression of RunX1T1 was calculated using the comparative Ct method.
Quantitative ChIP-PCR.
Chromatin immunoprecipitation (ChIP)-PCR was performed as described previously in reference Citation17. In brief, cultured cells (2 × 106) were cross-linked with 1% formaldehyde and then washed with phosphate buffered saline (PBS) in the presence of protease inhibitors. Cells were homogenized and their chromatin was subjected to ChIP pull down using magnetic Dynal beads (Invitrogen) and antibodies against dimethyl-H3K9, trimethyl-H3K27 or dimethyl-H3K4 (Millipore, Temecula, CA), respectively. Fold-enrichment of amplified DNAs by ChIP was assessed using previously described protocols.Citation46 Specific primers for amplification are listed in Supplemental Table 2.
Plasmid construct.
The full length human RunX1T1 cDNA sequence was PCR amplified using specific primers (Sup. Table 2) in IOSE cells. The PCR fragment was inserted into NheI-XhoI sites of mammalian expression vector pIRESII EGFP (Promega, Madison, WI). The putative promoter region containing SMAD4 binding element (SBE) of RunX1T1 was PCR amplified using genomic DNA from IOSE cells with specific primers (Sup. Table 2), which covered −557 to +170 bp of putative promoter region. The PCR products were inserted into XhoI and BglII sites of pGL3-Basic (Promega), which was predigested with Xho I and Bgl II. All plasmid constructs were verified by sequencing.
Promoter reporter assay.
SKOV3 cells (1 × 105) were transiently transfected with 1 µg of pGL3-RunX1T1 promoter construct or pGL3 basic vector along with 100 ng pRL-TK using lipofectamine 2000 reagents in a 6 well plate. After 48 h, the luciferase activities were measured using a Dual Luciferase Reporter Assay System (Promega) on a luminometer (Turner Designs, Sunnyvale, CA). Renilla luciferase expression plasmid (pRL-TK) was used to normalize for transfection efficiency. For In vitro DNA methylation, RunX1T1 promoter was excised from the pGL3-RunX1T1 plasmid with XhoI and BglII, isolated by gel elution and in vitro methylated using a recombinant CpG methyltransferase (M.SssI, New England Biolabs, Beverly, MA). Excised fragment without SssI methylase treatment was acted as control. DNA methylation was verified using methylation-sensitive restriction enzyme, BstUI. The methylated and unmethylated promoter was re-liagted into the pGL3 promoter, and 50 ng of ligated DNA was co-transfected with 25 ng renilla luciferase vectors into SKOV3 cells in a 24-well plate and luciferase activities was monitored as described above.
Re-silencing of RunX1T1.
MCP3 cells (2.5 × 105) were plated in 60 mm plate and treated with 0.5 µM 5′-aza-2′-deoxycytidine for 10 h. After demethylation treatment, cells were replaced with fresh medium and cultured for 7 days. Cells were then seeded onto 60 mm plate and then transfected with predesigned siRNA against Smad4 (siRNA ID: s8405; Ambion), or negative control (Ambion) at a concentration of 100 nM. Total RNA and DNA were harvested after 48 hour (day 2) and 144 hour (day 6) for quantitative RT-PCR analysis and COBRA methylation analysis.
Colony formation assay and cell growth assay.
Approximately 1.5 µg of RunX1T1 expression vector or empty vector were transfected into SKOV3 or CP70 cells (1 × 106 cells) in a 10-cm plate using Lipofectamine 2000 (Invitrogen) according to manufacturer's protocol. Twenty four hours after transfection, cells were counted and replated in triplicate and cultured for 3 weeks in RPMI 1640 containing 10% fetal bovine serum supplemented with 400 µg/ml G418 (Invitrogen, Paisley, UK). The surviving colonies were stained with 0.4% crystal violet (Sigma) in 50% methanol, and visible colonies were counted. Experiments were repeated twice and the average number of colonies from 6 plates for each plasmid was obtained. For cell growth assay, SKOV3 cells were transfected with RunX1T1 expression vector or empty vector. Forty eight hours after the experiment, transfected cells (around 1,000 cells) were replated in a 10-cm plate and selected for stable clones using RPMI 1640 containing 10% fetal bovine serum supplemented with 400 µg/ml G418 for at least 1 month. Stable transfectants were subcloned using cloning cylinder and harvested for RNA extraction to confirm RunX1T1 expression. RunX1T1-expressing SKOV3 stable cells or mock-transfected cells were plated at 2 × 104 cells per well in a 6-well plate. Cells were collected by trypsin-EDTA (Invitrogen) and cell numbers were counted by hematcytometer at day 2, 4 and 6 after transfection. Data was expressed as mean values ± standard deviations of the triplicates against time.
Figures and Tables
Figure 1 RunX1T1 is a SMAD4 target gene. (A) Schematic diagram depicting the positions of putative SMAD4-bining elements (SBE) with respect to the transcriptional start site (TSS) of RunX1T1. Arrow-head indicates the location of the primers for promoter cloning. Quantitative RT-PCR analysis of the mRNA levels of SMAD4 (B) and RunX1T1 (C) in siRNA knockdown experiments. IOSE and SKOV3 cells transfected with predesigned siRNAs against SMAD4 resulted in reduction in SMAD4 and RunX1T1 mRNA. Error bar represents standard deviation calculated from triplicates. (D) RunX1T1 promoter contains promoter activity. SKOV3 cells transfected with control vector (pGL3, empty vector) or vector containing RunX1T1 promoter (RunX1T1) was assayed for luciferase activity 48 hours after transfection. (E) In vitro methylation inhibited RunX1T1 promoter activity. SKOV3 cells transfected either with plasmid containing unmethylated or in vitro methylated RunX1T1 promoter was assayed for luciferase activity 48 hours after transfection. Error bars indicates standard deviation from triplicates.
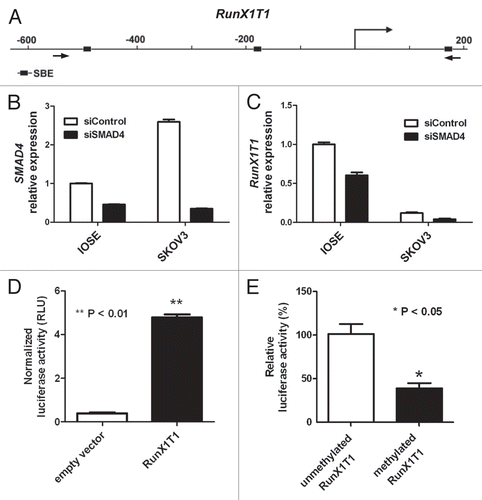
Figure 2 RunX1T1 is epigenetically silenced by promoter methylation in ovarian cancer cell lines. (A) RunX1T1 expression in IOSE and ovarian cancer cell lines. Total RNA were isolated from various cells and converted into cDNA for amplification with specific primers for RunX1T1. Relative level of expression after quantitative RT-PCR analysis were calculated and compared with IOSE (set as 1). (B) Bisulphite pyrosequencing analysis of CpG sites at RunX1T1 promoter from −187 to −104 in IOSE and ovarian cancer cell lines. Upper part indicates the genome map of RunX1T1 promoter with corresponding locations of all CpG sites from −236 to +98 and AciI cut sites in this region. Lower part illustrates DNA methylation at each CpG site (circle) of different samples with intensity of blue color indicating methylation level. The CpG sites interrogated by bisulphite prosequencing are indicated by blue bar underneath. IVD (in vitro methylated DNA) was included as positive control of bisulphite pyrosequencing. The Methylation status of the RunX1T1 promoter region was analyzed by COBRA assay (C) and MSP (D). In COBRA assay, bisulphite-modified DNA was PCR amplified by and digested with AciI. C, digested by restriction enzyme; U, undigested control. In MSP reaction, M and U indicates the presence of methylated and unmethylated alleles respectively. For both reaction, IVD (in vitro methylated DNA) was used as positive control for methylation and water (H2O) was used as a negative control for PCR. The locations of the primers for COBRA and MSP reaction are indicated by arrows.
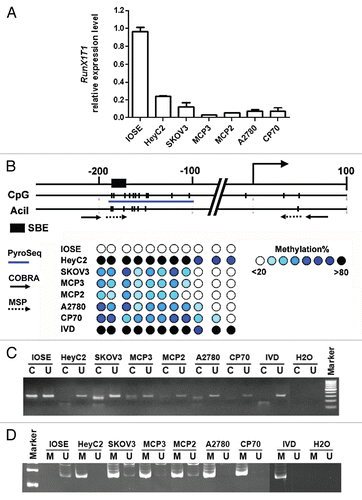
Figure 3 RunX1T1 is epigenetically silenced by histone modifications in ovarian cancer cell lines. ChIP assays were performed with antibodies directed against dimethyl-H3-K4 (H3K4me2) and dimethyl-H3-K9 (H3K9me2) in RunX1T1 promoter in various ovarian cancer cell lines. Regions of RunX1T1 ChIP-PCR analysis are indicated in the schematic diagram above. The relative binding of each antibody to the corresponding region was measured by the amount ChIP DNA against a standard curve generated from input DNA. Each error bar represents standard deviation calculated from triplicates.
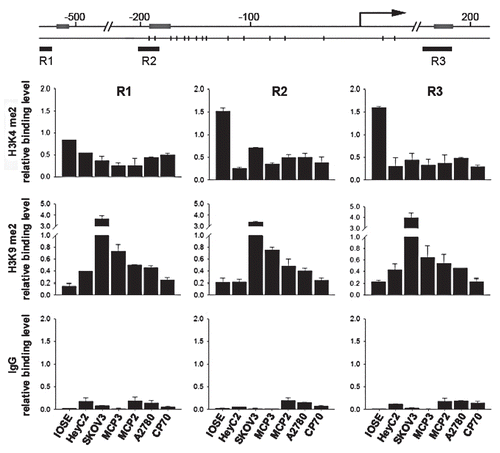
Figure 4 Epigenetic treatments restore RunX1T1 expression by reversing epigenetic status of RunX1T1 promoter. (A) RunX1T1 expression in ovarian cancer cell lines after epigenetic treament. IOSE cell and various ovarian cancer cell lines treated with 5-aza-2′-deoxycytidine (5aza) and/or trichostatin A (TSA) were examined for RunX1T1 expression by RT-PCR. Each error bar represents standard deviation calculated from triplicates. (B) Bisulphite pyrosequencing analysis of CpG sites from −187 to −104 () in MCP 3 cells treated with 5aza and/or TSA. (C) ChIP PCR were performed with antibodies directed against dimethyl-H3-K4 (H3K4me2) and dimethyl-H3-K9 (H3K9me2) in region R2 () of RunX1T1 promoter in MCP 3 cells treated with 5aza and/or TSA. The relative binding of each antibody to the corresponding region was measured by the amount ChIP DNA against a standard curve generated from input DNA. Each error bar represents standard deviation calculated from triplicates. The ratio between H3K4me2 versus H3K9me2 (K4/K9) of each treatment are shown (lower part).
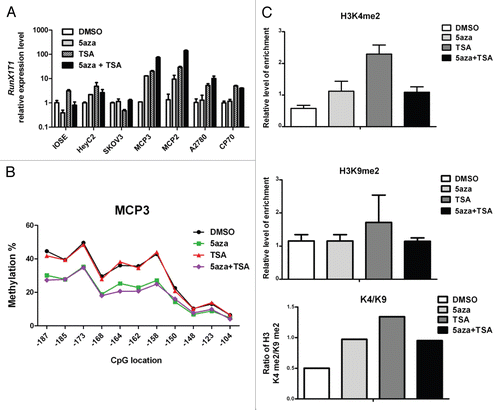
Figure 5 Knockdown of SMAD4 changes the epigenetic status of RunX1T1 promoter. IOSE cells or SKOV3 cells were transfected with siRNA against either SMAD4 (siSMAD4) or control (siControl). Histone modifications of the indicated histone marks in region R2 () of RunX1T1 promoter were analyzed in (A) IOSE cells or (C) SKOV3 cells. The ratio between H3K4me2 versus H3K9me2 (K4/K9) of each treatment are shown (lower part). Bisulphite pyrosequencing analysis of CpG sites from −187 to −104 in (B) IOSE cells or (D) SKOV3 cells.
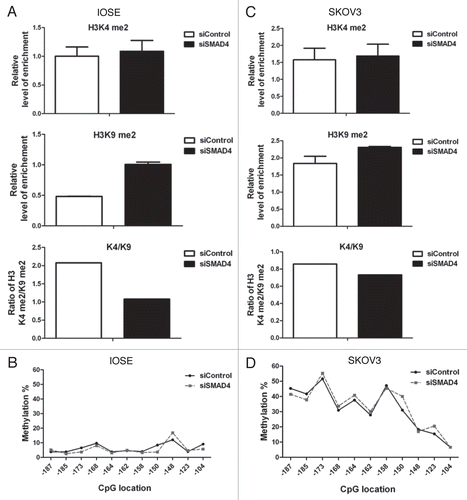
Figure 6 Knockdown of Smad4 accelerates re-silencing of RunX1T1 in MCP3 cells. (A) MCP3 cells were treated with 0.5 mM of 5-aza for 10 hrs. After demethylation treatment, cells were replaced with fresh medium and cultured for another 7 days before transfection with siRNA against SMAD4 (siSMAD4) or scramble control (sicontrol), DNA and RNA were harvested for methylation and expression experiment on day 0, 2 and 6 after siRNA transfection. (B) Quantitative RT-PCR analysis of mRNA levels of RunX1T1 after various treatments at the indicated time. Error bars indicate SD calculated from triplicates. (C) Methylation analysis of RunX1T1 promoter by COBRA assay on day 0, 2 and 6 after siRNA transfection. Knock down of SMAD4 accelerated re-methylation of RunX1T1. This phenomenon is more evident on day 6 of the experiment as indicated by stronger AciI digestion in siRNA transfection group (red arrow head) than in control group (white arrow head). (D) Bisulphite pyrosequencing analysis of CpG sites from −187 to −104 on day 6 (D6) of the experiment. Methylation level of DMSO-treated MCP 3 cells were included as reference.
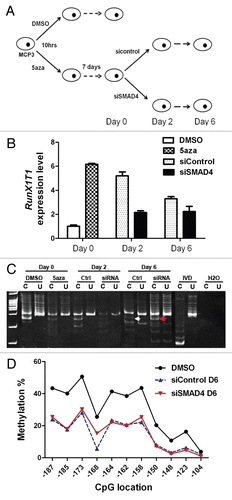
Figure 7 Methylation analysis of RunX1T1 promoter in 95 ovarian cancer patients. (A) Representative gel image of MSP analysis of RunX1T1 promoter in six ovarian cancer patients (LA345, LA540, LA557, 588, A881, B049). The MSP result is summarized in . Primer sets used is indicated in . M and U indicates the presence of methylated and unmethylated alleles respectively. IVD was used as positive control for methylation and water (H2O) was used as a negative control for PCR. (B) Selected samples with or without RunX1T1 promoter methylation (LA557 and 588) were selected for confirmation by bisulphite pyrosequencing. (C) Dot plot showing the association between methylation of RunX1T1 and another TGFβ/SMAD4 target gene, FBXO32 in the same cohort of ovarian cancer patients. Methylation of FBXO32 in this cohort of ovarian cancer patients was demonstrated previously by qMSP.Citation18 Current hypermethylation of RunX1T1 and FBOX32 was observed in this ovarian cancer patients cohort. *p < 0.05.
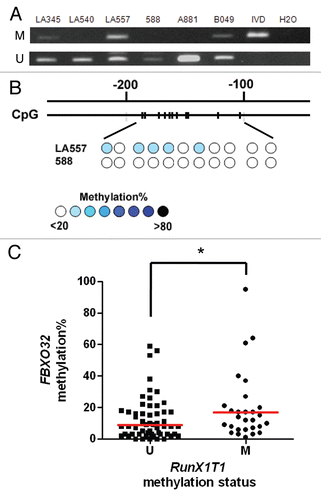
Figure 8 Methylation analysis of RunX1T1 in ovarian cancer initiating cells OCIC. (A) Typical spheroids form after about ten passages of repeating suspension culture from ovarian patient samples. Magnification, 100×. (B) MSP gel image of six OCIC samples isolated from primary ovarian tumor patients. M and U indicates the presence of methylated and unmethylated alleles respectively. The sample names are indicated on the top part. For both reaction, IVD (in vitro methylated DNA) was used as positive control for methylation and water (H2O) was used as a negative control for PCR.
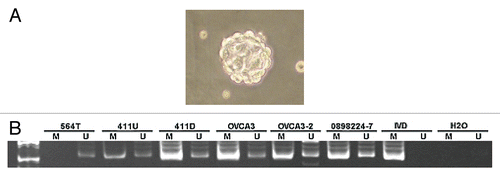
Figure 9 RunX1T1 exhibits tumor suppressor activity in ovarian cancer in vitro. (A) Expression of RunX1T1 inhibited tumor growth as demonstrated in colony forming assay. SKOV3 and CP70 cells transfected with RunX1T1 expressing vector showed a significant reduction in the number of colonies as compared with control after G418 selection. (B) Quantitative analysis of the number of colonies formed by colony forming assay. Each error bar represents standard error calculated from triplicates. (C) Stable transfectants of SKOV3 cells transfected either with RunX1T1 expression vector (RunX1T1_#3. RunX1T1_#8) or empty vector were selected for growth analysis. The number of cell was determined by hematcytometer at the designated day. (D) Expression level of the RunX1T1 stable transfectant and empty vector control was detected by quantitative RT-PCR. RunX1T1 restoration showed marked reduction in tumor growth. **p < 0.01.
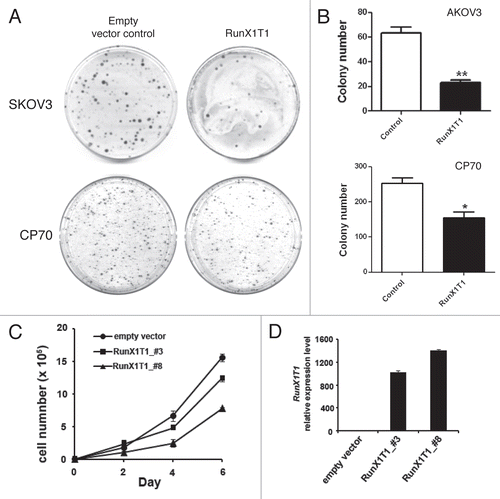
Table 1 Association between methylation of RunX1T1 and tumor stage in 95 ovarian cancer patients
Additional material
Download Zip (2.3 MB)Acknowledgments
This work was supported by research grant from Changhua Christian Hospital and National Science Council, Republic of China (NSC97-2320-B-194-002-MY3).
References
- Waite KA, Eng C. From developmental disorder to heritable cancer: it's all in the BMP/TGFbeta family. Nat Rev Genet 2003; 4:763 - 773
- Feng XH, Derynck R. Specificity and versatility in TGFβeta signaling through Smads. Annu Rev Cell Dev Biol 2005; 21:659 - 693
- Wong AS, Leung PC. Role of endocrine and growth factors on the ovarian surface epithelium. J Obstet Gynaecol Res 2007; 33:3 - 16
- Berchuck A, Rodriguez G, Olt G, Whitaker R, Boente MP, Arrick BA, et al. Regulation of growth of normal ovarian epithelial cells and ovarian cancer cell lines by transforming growth factor-beta. Am J Obstet Gynecol 1992; 166:676 - 684
- Nilsson EE, Skinner MK. Role of transforming growth factor beta in ovarian surface epithelium biology and ovarian cancer. Reprod Biomed Online 2002; 5:254 - 258
- Yamada SD, Baldwin RL, Karlan BY. Ovarian carcinoma cell cultures are resistant to TGFbeta1-mediated growth inhibition despite expression of functional receptors. Gynecol Oncol 1999; 75:72 - 77
- Baldwin RL, Tran H, Karlan BY. Loss of c-myc repression coincides with ovarian cancer resistance to transforming growth factorbeta growth arrest independent of transforming growth factor beta/Smad signaling. Cancer Res 2003; 63:1413 - 1419
- Tanaka Y, Kobayashi H, Suzuki M, Kanayama N, Terao T. Transforming growth factor-beta1-dependent urokinase upregulation and promotion of invasion are involved in Src-MAPK-dependent signaling in human ovarian cancer cells. J Biol Chem 2004; 279:8567 - 8576
- Rodriguez GC, Haisley C, Hurteau J, Moser TL, Whitaker R, Bast RC Jr, et al. Regulation of invasion of epithelial ovarian cancer by transforming growth factorbeta. Gynecol Oncol 2001; 80:245 - 253
- Balch C, Fang F, Matei DE, Huang TH, Nephew KP. Minireview: epigenetic changes in ovarian cancer. Endocrinology 2009; 150:4003 - 4011
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3:415 - 428
- Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007; 128:683 - 692
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell 2007; 128:707 - 719
- Chan MW, Huang YW, Hartman-Frey C, Kuo CT, Deatherage D, Qin H, et al. Aberrant transforming growth factor beta1 signaling and SMAD4 nuclear translocation confer epigenetic repression of ADAM19 in ovarian cancer. Neoplasia 2008; 10:908 - 919
- Leu YW, Yan PS, Fan M, Jin VX, Liu JC, Curran EM, et al. Loss of estrogen receptor signaling triggers epigenetic silencing of downstream targets in breast cancer. Cancer Res 2004; 64:8184 - 8192
- Ren M, Pozzi S, Bistulfi G, Somenzi G, Rossetti S, Sacchi N. Impaired retinoic acid (RA) signal leads to RARbeta2 epigenetic silencing and RA resistance. Mol Cell Biol 2005; 25:10591 - 10603
- Qin H, Chan MW, Liyanarachchi S, Balch C, Potter D, Souriraj IJ, et al. An integrative ChIP-chip and gene expression profiling to model SMAD regulatory modules. BMC Syst Biol 2009; 3:73
- Chou JL, Su HY, Chen LY, Liao YP, Hartman-Frey C, Lai YH, et al. Promoter hypermethylation of FBXO32, a novel TGFbeta/SMAD4 target gene and tumor suppressor, is associated with poor prognosis in human ovarian cancer. Lab Invest 2010; 90:414 - 425
- Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996; 93:9821 - 9826
- Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res 2008; 68:4311 - 4320
- Davis JN, McGhee L, Meyers S. The ETO (MTG8) gene family. Gene 2003; 303:1 - 10
- Guo C, Hu Q, Yan C, Zhang J. Multivalent binding of the ETO corepressor to E proteins facilitates dual repression controls targeting chromatin and the basal transcription machinery. Mol Cell Biol 2009; 29:2644 - 2657
- Sunde JS, Donninger H, Wu K, Johnson ME, Pestell RG, Rose GS, et al. Expression profiling identifies altered expression of genes that contribute to the inhibition of transforming growth factor-beta signaling in ovarian cancer. Cancer Res 2006; 66:8404 - 8412
- Evangelou A, Letarte M, Jurisica I, Sultan M, Murphy KJ, Rosen B, et al. Loss of coordinated androgen regulation in nonmalignant ovarian epithelial cells with BRCA1/2 mutations and ovarian cancer cells. Cancer Res 2003; 63:2416 - 2424
- Dumont N, Wilson MB, Crawford YG, Reynolds PA, Sigaroudinia M, Tlsty TD. Sustained induction of epithelial to mesenchymal transition activates DNA methylation of genes silenced in basal-like breast cancers. Proc Natl Acad Sci USA 2008; 105:14867 - 14872
- Chou JL, Chen LY, Lai HC, Chan MW. TGFbeta: friend or foe? The role of TGFbeta/SMAD signaling in epigenetic silencing of ovarian cancer and its implication in epigenetic therapy. Expert Opin Ther Targets 2010; 14:1213 - 1223
- Vikhanskaya F, Erba E, D'Incalci M, Broggini M. Introduction of wild-type p53 in a human ovarian cancer cell line not expressing endogenous p53. Nucleic Acids Res 1994; 22:1012 - 1017
- Shin KS, Sullenger BA, Lee SW. Ribozyme-mediated induction of apoptosis in human cancer cells by targeted repair of mutant p53 RNA. Mol Ther 2004; 10:365 - 372
- Milner BJ, Allan LA, Eccles DM, Kitchener HC, Leonard RC, Kelly KF, et al. p53 mutation is a common genetic event in ovarian carcinoma. Cancer Res 1993; 53:2128 - 2132
- Okamoto A, Sameshima Y, Yokoyama S, Terashima Y, Sugimura T, Terada M, et al. Frequent allelic losses and mutations of the p53 gene in human ovarian cancer. Cancer Res 1991; 51:5171 - 5176
- Tonks A, Pearn L, Musson M, Gilkes A, Mills KI, Burnett AK, et al. Transcriptional dysregulation mediated by RUNX1-RUNX1T1 in normal human progenitor cells and in acute myeloid leukaemia. Leukemia 2007; 21:2495 - 2505
- Liu S, Shen T, Huynh L, Klisovic MI, Rush LJ, Ford JL, et al. Interplay of RUNX1/MTG8 and DNA methyltransferase 1 in acute myeloid leukemia. Cancer Res 2005; 65:1277 - 1284
- Watabe T, Miyazono K. Roles of TGFbeta family signaling in stem cell renewal and differentiation. Cell Res 2009; 19:103 - 115
- Bierie B, Stover DG, Abel TW, Chytil A, Gorska AE, Aakre M, et al. Transforming growth factor-beta regulates mammary carcinoma cell survival and interaction with the adjacent microenvironment. Cancer Res 2008; 68:1809 - 1819
- Tang Y, Kitisin K, Jogunoori W, Li C, Deng CX, Mueller SC, et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGFbeta and IL-6 signaling. Proc Natl Acad Sci USA 2008; 105:2445 - 2450
- Tang B, Yoo N, Vu M, Mamura M, Nam JS, Ooshima A, et al. Transforming growth factorbeta can suppress tumorigenesis through effects on the putative cancer stem or early progenitor cell and committed progeny in a breast cancer xenograft model. Cancer Res 2007; 67:8643 - 8652
- Kulasingam V, Pavlou MP, Diamandis EP. Integrating high-throughput technologies in the quest for effective biomarkers for ovarian cancer. Nat Rev Cancer 2010; 10:371 - 378
- Bertelsen K, Holund B, Andersen JE, Nielsen K, Stroyer I, Ladehoff P. Prognostic factors and adjuvant treatment in early epithelial ovarian cancer. Int J Gynecol Cancer 1993; 3:211 - 218
- Chan MW, Wei SH, Wen P, Wang Z, Matei DE, Liu JC, et al. Hypermethylation of 18S and 28S ribosomal DNAs predicts progression-free survival in patients with ovarian cancer. Clin Cancer Res 2005; 11:7376 - 7383
- Wei SH, Balch C, Paik HH, Kim YS, Baldwin RL, Liyanarachchi S, et al. Prognostic DNA methylation biomarkers in ovarian cancer. Clin Cancer Res 2006; 12:2788 - 2794
- Ahluwalia A, Yan P, Hurteau JA, Bigsby RM, Jung SH, Huang TH, et al. DNA methylation and ovarian cancer. I. Analysis of CpG island hypermethylation in human ovarian cancer using differential methylation hybridization. Gynecol Oncol 2001; 82:261 - 268
- Wei SH, Chen CM, Strathdee G, Harnsomburana J, Shyu CR, Rahmatpanah F, et al. Methylation microarray analysis of late-stage ovarian carcinomas distinguishes progression-free survival in patients and identifies candidate epigenetic markers. Clin Cancer Res 2002; 8:2246 - 2252
- Gillan L, Matei D, Fishman DA, Gerbin CS, Karlan BY, Chang DD. Periostin secreted by epithelial ovarian carcinoma is a ligand for alpha(V)beta(3) and alpha(V) beta(5) integrins and promotes cell motility. Cancer Res 2002; 62:5358 - 5364
- Bock C, Reither S, Mikeska T, Paulsen M, Walter J, Lengauer T. BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics 2005; 21:4067 - 4068
- Colella S, Shen L, Baggerly KA, Issa JP, Krahe R. Sensitive and quantitative universal Pyrosequencing methylation analysis of CpG sites. Biotechniques 2003; 35:146 - 150
- Cheng AS, Jin VX, Fan M, Smith LT, Liyanarachchi S, Yan PS, et al. Combinatorial analysis of transcription factor partners reveals recruitment of c-MYC to estrogen receptor-alpha responsive promoters. Mol Cell 2006; 21:393 - 404