Abstract
Oncogenic human papillomaviruses (HPV) are associated with nearly all cervical cancers and are increasingly important in the etiology of oropharyngeal tumors. HPV-associated head and neck squamous cell carcinomas (HNSCC) have distinct risk profiles and appreciate a prognostic advantage compared to HPV-negative HNSCC. Promoter hypermethylation is widely recognized as a mechanism in the progression of HNSCC, but the extent to which this mechanism is consistent between HPV(+) and HPV(-) tumors is unknown. To investigate the epigenetic regulation of gene expression in HPV-induced and carcinogen-induced cancers, we examined genome-wide DNA methylation and gene expression in HPV(+) and HPV(-) SCC cell lines. We used two platforms: the Illumina Infinium Methylation BeadArray and tiling arrays, and confirmed illustrative examples with pyrosequencing and quantitative PCR. These analyses indicate that HPV(+) cell lines have higher DNA methylation in genic and LINE-1 regions than HPV(-) cell lines. Differentially methylated loci between HPV(+) and HPV(-) cell lines significantly correlated with HPV-typed HNSCC primary tumor DNA methylation levels. Novel findings include higher promoter methylation of polycomb repressive complex 2 target genes in HPV(+) cells compared to HPV(-) cells and increased expression of DNMT3A in HPV(+) cells. Additionally, CDKN2A and KRT8 were identified as interaction hubs among genes with higher methylation and lower expression in HPV(-) cells. Conversely, RUNX2, IRS-1 and CCNA1 were major hubs with higher methylation and lower expression in HPV(+) cells. Distinct HPV(+) and HPV(-) epigenetic profiles should provide clues to novel targets for development of individualized therapeutic strategies.
Introduction
Squamous cell carcinomas (SCCs) have a complex etiology that includes life style behaviors, classical chemical carcinogenesis and infection with high risk human papillomaviruses (HPV). The role of HPV in the etiology of SCC of the uterine cervix has been known for many years, but increases in the incidence of oropharyngeal cancer1 are associated with high risk HPV.Citation2 Each year, there are an estimated 644,000 new cases of head and neck cancers diagnosed worldwide, with approximately 40,000 per year in the US.Citation3 HPV(+) oropharynx cancers differ dramatically from HPV(−) oropharyngeal and oral cancers that arise from excessive tobacco and alcohol exposure.Citation2,Citation4–Citation7 These differences include a predilection of HPV(+) tumors to have early metastasis to the lymph nodes of the neck,Citation8 a propensity for rapid growth and poor differentiation.Citation9 HPV(+) tumors are also generally more responsive to treatment and high cure rates can be achieved.Citation10 In contrast, HPV(−) SCC of the oral cavity and oropharynx tend to progress locally and exhibit greater resistance to treatment with chemotherapy and radiation. Additionally, these tumors have poor survival rates (∼50%) in spite of advances in surgical technology and sophisticated chemotherapy and radiation oncology methods.Citation11
Thus, it's not surprising that the development of head and neck cancer follows at least two distinct molecular pathways depending upon tumor HPV status. A striking example of this is the frequent loss of CDKN2A by chromosomal deletion, mutation or promoter hypermethylation in HPV(−) tumors, in contrast to high CDKN2A expression in HPV(+) tumors.Citation12 Although DNA methylation of the viral genome has been implicated both as a mechanism for masking the virus from the host cell and as a defense mechanism for the host cell, little is known regarding viral-induced changes in DNA methylation of the host genome as part of the carcinogenic pathway. Promoter hypermethylation studies have largely evaluated a limited number of candidate genes in HNSCC.Citation13–Citation17 Most were selected based on functional relevance in carcinogenesis in multiple tumor types, some of which are shown to be frequently methylated in HNSCC (p16, e-cadherin, RARβ, MGMT, DAP-kinase, DCC, GALR1, GALR2) and rarely methylated in HNSCC (RASSF1A, p73, p53, MLH1). Richards, et al. looked at global differences in CpG methylation in repetitive regions among HPV(+) and HPV(−) HNSCC tumors and found higher LINE-1 methylation and higher genomic stability among HPV(+) tumors.Citation18 Other epigenetic differences specific to HPV(+) tumors are less well described. Molecular studies comparing gene expression in HPV(−) and HPV(+) HNSCC tumors show distinct genomic signaturesCitation19–Citation21 reflecting underlying heterogeneous somatic genetic and epigenetic alterations. To date, nearly all studies have examined only one or two of the several possible genomic or epigenomic mechanisms leaving a major gap in our knowledge. To fully understand carcinogenic pathways it will be necessary to perform simultaneous assessment of the multiple mechanisms that can regulate gene expression.
Technological advances allow for a relatively unbiased interrogation of the entire genome to identify the constellation of genomic loci with altered epigenetic status. Recent studies have compared gene expression differences in HPV(+) and HPV(−) in HNSCC tumors,Citation19–Citation21 but there are no known studies of concurrent genome-wide methylation and gene expression in HPV(+) and HPV(−) tumors. Therefore, we compared the genome wide differences in DNA methylation (focusing on non-repetitive regions using whole genome tiling arrays and the Illumina Infinium HumanMethylation27 BeadArray) and gene expression in two HPV(+) and two HPV(−) squamous carcinoma cell lines. Using cell lines allowed us to identify the permanently affected gene expression profiles associated with the transformation mechanisms and avoid genetic changes that may be contributed by contaminating cells from tissue samples.
Results
Overall differences in methylation patterns between HPV(+) and HPV(−) SCC cells.
We evaluated overall distributional differences in DNA methylation between HPV(+) and HPV(−) cells in promoter regions (Illumina Infinium arrays) and genome-wide non-repetitive regions (MeDIP-tiling arrays). Overall, the HPV(+) cells showed greater levels of CpG methylation than the HPV(−) cells. This difference was exhibited by a strong trend in the Illumina methylation data covering mainly promoter regions, although in treating each sample as random, it did not reach statistical significance (p = 0.08, , B and Sup. Fig. 1). The MeDIP-tiling approach revealed that this trend is stronger in promoter regions than in other non-repetitive regions (). We estimate that approximately 10-times more genic regions are more highly methylated in HPV(+) than in HPV(−) cells ( and B). Unsupervised hierarchical clustering of the Illumina Infinium data, filtered based on three different levels of standard deviation of % methylation values across all four cell lines (see methods), was able to clearly separate the samples according to their HPV status (Sup. Fig. 2). Analysis of LINE-1 elements, repetitive elements that serve as a measure of global methylation, also revealed higher methylation in HPV(+) compared to HPV(−) cells (44% and 46% for HPV(−) vs. 76% and 78% for HPV(+) cell lines). Thus, HPV(+) cells had higher CpG methylation both in non-repetitive regions (genic and non-genic) and in repetitive regions.
Expression differences in HPV(+) and HPV(−) cells.
Of the 300 differentially expressed probe sets (248 unique genes) (2-fold with p-value < 0.01), 99 probe sets were upregulated in HPV(−) cells and 201 upregulated in HPV(+). We identified 183 concepts (biologically-related gene sets) as significantly upregulated in HPV(+) cells and 113 as significantly upregulated in HPV(−) cells (FDR < 0.05) using LRpath analysis (Sup. Table 3). Of these, the most significant chromosomal region we identified was 9p24 (p-value = 6.3 × 10−17; odds ratio = 129.0), which we observed to be lower expressed in HPV(−) cells. This region contains the genes UHRF2 (Nirf), C9orf123, C9orf46, CDC37I1, a cell division gene, RIN2, CIP150 and PPAPDC2. We also showed that the Pfam family of core histones was significantly upregulated in HPV(−) cells (p = 1.3 × 10−9).
Using ConceptGen software,Citation22 to test for publicly available gene expression datasets that have significant overlap of differentially expressed genes with our experiment, we found that genes downregulated in gefitinib-resistant cells (GEO identifier GSE10696) were the most significant group enriched with overexpressed genes in HPV(+) cells. This observation is consistent with the better response rate to anti-EGFR therapy in oropharynx cancers [many of which are HPV(+)] in the “Radiotherapy plus Cetuximab for Squamous-Cell Carcinoma of the Head and Neck” trial (ClinicalTrials.gov number NTC00004227).Citation23
Integrating methylation and expression data.
There is an inverse correlation between methylation and expression, i.e., genes with higher expression in HPV(+) cells tended to have less methylation in HPV(+) cells (, upper left petal/quadrant) and genes with decreased expression in HPV(+) tended to be more highly methylated in HPV(+) cell lines (Pearson correlation coefficient = −0.21, p-value < 10−100). Although the correlation may seem low, it must be interpreted in light of the fact that many genes are regulated by means other than promoter methylation. In general, genes more highly methylated in HPV(+) tumors tended to have low average expression levels across all four cell lines ( and D). In contrast, genes with higher promoter methylation in HPV(−) cells tended to be genes with mid to high expression level across all four cell lines ( and D). To prioritize candidate genes for being drivers of the carcinogenic process specific to HPV(+) or HPV(−) tumors, we characterized the sets of genes in the top left and bottom right quadrants of in terms of pathways and other biological concepts enriched with these genes. As described in the methods, 28 genes were chosen from the top left quadrant and 75 genes from the bottom right quadrant to characterize using functional (gene set) enrichment testing and interaction networks. Several of our findings are consistent with known differences between HPV-induced cellular changes and those induced by tobacco carcinogens.
Methylation patterns and expression of genes with higher methylation and lower expression in HPV(−) cells.
CDKN2A (p16ink4A) (chromosome 9p21) located in the top left quadrant of , is less expressed and more methylated in the HPV(−) tumors. An interaction network with the genes in this quadrant shows CDKN2A as an interaction hub among these genes, as is CK8 (Keratin 8); each of which has multiple interactions with other genes in the network (). Also notable in the candidate genes more highly expressed and less methylated in HPV(+) cells are PKCtheta, ESE3, RHOD, SOCS2, AnnexinIII and Annexin IX, as well as TSPAN1. As shown in (Illumina Infinium; MeDIP-tiling, Sup. Fig. 3), the promoter regions of CDKN2A and CDKN2B are more highly methylated in the HPV(−) cells (bottom half of fig.) and are known to be frequently silenced in HPV(−) head and neck cancers. The MeDIP-tiling data show that the entire region is more highly methylated in the HPV(−) than the HPV(+) cells, in contrast to most of the genome which is generally more highly methylated in the HPV(+) cells. In fact, only the downstream region of CDKN2A is more highly methylated in the HPV(+) cells (Sup. Fig. 3) and our expression data shows that this does not attenuate CDKN2A expression. Regulation of growth and cell size, as well as calcium-dependent phospholipid binding were found to be biological concepts enriched with these sets of genes ( and using ConceptGen).
Methylation patterns and expression of genes with higher methylation and lower expression in HPV(+) cells.
Among the 75 genes more highly methylated and less expressed in HPV(+) relative to HPV(−), vacuole was the most significantly enriched biological concept tested (p = 0.00076), involving the genes STS, ATP6V0C, HPS1, CTSL1, GNS, FUCA1 and VPS18. The PI3 kinase pathway (pantherdb.org) and regulation of kinase activity concept were also among the most enriched and included the genes IRS1, GNA11, GNAI2, EREG, CCNA1, RGS4 and PKIG (). The PI3 kinase signaling pathway has previously been identified as being enriched with genes overexpressed in HNSCC samples (which historically have been mainly HPV(−)) and in regions with increased copy number in oral premalignant lesions.Citation24 The major gene hubs were RUNX2, CD40 (TNFRSF5), IRS-1, Collagen III and Cyclin A1. p53 also interacted with several of the 75 candidate genes, specifically Mettl7a, Sestrin 3, FUCO, protein p8, PTPR-mu, MTS1(S100A4), IRS-1, Cyclin A1 and Lamin A/C.
Methylation patterns and expression of genes with both higher methylation and higher expression in HPV(+) cells.
There is a significant subset of genes that are both more highly methylated and more highly expressed in HPV(+) than in HPV(−) cells (top right quadrant of ). Although counter-intuitive, such a finding has multiple possible explanations shedding light onto the overall carcinogenic programs of HPV(+) versus HPV(−) tumors. In addition to DCC (deleted in colorectal carcinoma), this group of genes included KAL1, NF1, NGFR, GNAO1, SEMA3B and DCDC2. Predicted PRC2 target genes COL12a1, COL9a1, CYP2j2, GNAS, KAL1, MEST (previously identified as upregulated in HPV(+) HNSCC tumors versus normalCitation19), RASGRF1 and S1pr5 were also among the candidate set of more highly methylated and highly expressed genes in HPV(+) cells. An explanation for this observation is that chromosomal regions often lost in HPV(−) tumors are partially silenced by methylation in HPV(+) tumors. For example, chromosomal region 19q13 is rich in candidate tumor suppressor genesCitation25 and is frequently lost in head and neck tumors.Citation26 As shown in Supplemental Figure 4 this region is more highly methylated and exhibits greater gene expression in HPV(+) than in HPV(−) cells. To confirm this observation, we separately tested for significant differential methylation and expression for sites/genes in this chromosomal region and found significance in both cases (using LRpath: FDR < 0.05). This suggests the possibility that more chromosomal loss has occurred at 19q13 in HPV(−) tumor cells compared to HPV(+) cells, which exhibit more methylation. Genomic analysis will be necessary to test this hypothesis.
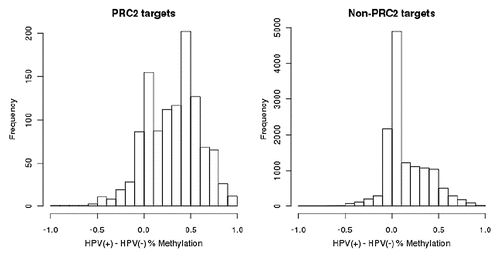
Polycomb repressive complex 2 (PRC2) targets tend to be more highly methylated in HPV(+) cells.
Polycomb repressive complex 2 (PRC2) proteins regulate genes involved in stem cell maintenance as well as growth and differentiation.Citation27 The promoters of PRC2 targets are more highly methylated in HPV(+) cells compared to non-PRC2 targets ( and B). The promoters of PRC2 targets are at median >19% more methylated in HPV(+) compared to HPV(−) cells, whereas the promoters of non-PRC2 targets are at median only 1% more methylated in HPV(+) compared to HPV(−) cells. Four PRC2 targets: CCNA1, PREX1, RUNX2 and SPON2 were among the 75 genes with higher methylation and lower expression in HPV(+) cells (lower right quadrant ).
Validation of selected targets: pyrosequencing, qPCR and primary tumors.
We chose to validate the methylation differences of two genes which had both higher methylation and higher expression in HPV(+): ESR1 (due to its significance and differential promoter methylation in other cancers),Citation28–Citation30 and DCC (identified as biologically important in head and neck cancer).Citation31 Illumina Infinium platform data showed ESR1 and DCC average methylation of 4% and 10% in HPV(−) tumors compared to 82% and 87% respectively in HPV(+) tumors. Pyrosequencing confirmed an average methylation of 9% in HPV(−) cells versus 94.5% methylation in HPV(+) cells for ESR1 (Sup. Fig. 5) and also confirmed higher DCC methylation in HPV(+) cells (average methylation 92.5%) compared to HPV(−) cells (average methylation 23.5%). We chose to validate the expression differences in two genes: CCNA1 due to its status as a protein-interaction hub among genes with higher methylation and lower expression in HPV(+) cells and because it has been shown to correlate with survival in HNSCCCitation32 and DNMT3a (as a possible explanation for the overall higher methylation observed in HPV(+) cells). qPCR showed that CCNA1 was expressed 7.3 times higher in HPV(−) cell lines compared to HPV(+), while DNMT3a was expressed 4.6 times higher in HPV(+) cell lines compared to HPV(−), which is consistent with the expression array data.
We analyzed 49 primary tumor DNAs (17 oral cavity and 32 oropharynx tumors, from paraffin embedded formalin fixed specimens) also assessed for HPV states (n = 21 HPV(+)), for CpG methylation using the Illumina Goldengate Cancer Panel. We compared our data from the four cell lines assessed with the Illumina Infinium platform to the Goldengate findings on the primary tumors. Of the CpG targets common to both platforms (n = 472), the top 12 most differentially methylated probes between HPV(+) and HPV(−) cell lines significantly correlated with tumor DNA methylation with correlation coefficients of 0.46. Overall, as observed when comparing HPV(+) and HPV(−) cell lines, the HPV(+) tumors showed greater levels of CpG methylation than the HPV(−) tumors (Sup. Fig.6). We also analyzed oral/oropharyngeal tumors (n = 28 HPV(−); n = 18 HPV(+)) for % methylation of LINE-1 elements. We observed significantly higher methylation in the HPV(+) samples (median HPV(+) = 68.7; median HPV(−) = 57.6; p = 0.004 by Mann-Whitney U test).
Discussion
Much of the heterogeneity of biological behavior and response to therapy in head and neck cancer can be explained by etiologic differences between traditional (tobacco/alcohol) and viral carcinogenesis. A few studies have attempted to evaluate the divergent pathways of gene expression in HPV(+) versus smoking related HNSCCCitation19–Citation21 using global repetitive DNA methylationCitation18 and somatic molecular differences.Citation33 Ours is the first study to use an integrated approach combining genome-wide epigenetic and gene expression changes to characterize the molecular differences between HPV(+) and HPV(−) tumors and allowing a preliminary assessment of the relationship between the epigentic changes and functionally relevant somatic expression. By using well-characterized HPV(+) and HPV(−) tumor cell lines, we eliminated the irrelevant information from contaminating stromal cells and controlled for the effects of cell culture. This allowed us to focus on tumor specific differences and eliminate noise, thus our analyses have provided insight into pathway differences between these two types of tumors.
To address generalizability of cell line epigenetic data, we compared the cell line epigenetic data to data from primary tumors. Both sample types were run on similar platforms (Illumina Infinium vs. Illumina Goldengate) as the Infinium platform is not recommended for DNA from formalin fixed paraffin embedded tumors. We found a strong correlation between differentially methylated targets between cell lines and primary tumors. Higher CpG methylation levels were noted in the HPV(+) compared to HPV(−) tumors, as observed in the cell line data. While the applicability of specific cell line epigenetic data remains to be seen, here we report general findings that indicate strong differences in carcinogenic pathways between HPV(+) and HPV(−) tumors.
Although availability of HPV(+) tumor cell lines are extremely limited, we were able to balance for gender and mechanism of tumor induction, i.e., high risk HPV or HPV(−) primary carcinogenic processes and age (47–53 years) at diagnosis. The inclusion of an HPV(+) cervical SCC allowed for an analysis of molecular pathways between the two types of HPV(+) cancers and the HPV(−) head and neck cancers. We found strong evidence that HPV(+) tumors have the same molecular pathways in carcinogenesisCitation34 as observed by others. There were many more similarities between the two HPV positive cell lines with respect to DNA methylation than between the two HPV(−) cell lines, as assessed by the Illumina Infinium platform (75th percentile for difference in % methylation between the two HPV(−) lines was 0.21 compared to 0.11 for HPV(+) lines). Thus, although the HPV(+) cell lines were from different sites, the methylation and expression signatures were similar.
As expected, the analysis showed relative overexpression of CDKN2A in HPV(+) compared to HPV(−) cells, with corresponding higher CpG methylation of CDKN2A in HPV(−) cells. This finding is consistent with the known mechanisms of tobacco-related carcinogenesis in HPV(−) SCC, where inactivation of p16 (CDKN2A) is the most common alteration.Citation35 The main mechanisms of p16 inactivation in HNSCC are homozygous deletions (40–60%), mutation (15–20%) and hypermethyation (5%).Citation36–Citation38 Conversely, in HPV(+) SCC, malignant transformation is attributed to the HPV E6 and E7 oncoproteins, which inhibit function of p53 and RB tumor suppressor genes respectively. HPV E7 sequesters Rb leading to E2F mediated transcription of CDKN2A and p16 protein overexpression, which feeds back to inhibit Rb activation under normal conditions. This is confirmed in our data by the predominant role these proteins play in our protein interaction analysis ().
Unsupervised clustering showed that the four cell lines are distinguishable by HPV status based solely on DNA methylation profiles. These differences were marked by overall greater DNA methylation across the genic and other non-repetitive genome in HPV(+) cells compared to HPV(−) cells, also observed in a sample of primary tumors. LINE-1 methylation, a measure of global methylation, was also higher in HPV(+) cell lines and in a larger population of tumor specimens, consistent with the findings of Richards et al. Thus the higher methylation in the HPV(+) tumor cells consists of both targeted genic methylation and global methylation that may or may not have functional consequences. The increased methylation in HPV(+) tumors may be partially explained by higher expression of DNMT3a in HPV(+) cells than in HPV(−) cells, although others have seen DNMT1 more highly expressed in HPV(+) tumors.Citation19 DNMT3a functions as a de novo methylator important in embryonic development, and has recently been seen to co-localize with EZH2, one of the polycomb repressive complex 2 (PRC2) proteins.Citation39 This finding supports our observation of a strong enrichment of highly methylated PRC2 targets in HPV(+) compared to HPV(−) cell lines (), and is consistent with the reported observation that the HPV E7 protein activates EZH2.Citation40 Although promoters of PRC2 target genes have previously been observed to be hypermethylated in cancer, these studies compared cancer versus normal cells.Citation41 To our knowledge, no studies have been performed on the relative difference in PRC2 target hypermethylation between cancer subtypes. PcG-targeted genes were more than twice as likely to be methylated in HPV(+) cell lines compared to HPV(−) cell lines, suggesting that PcG targets involved in differentiation may be silenced by promoter methylation. Because such a broad-reaching change in methylation affects a whole population of genes, rather than a more targeted set that would be indicative of natural selection within the tumor, we hypothesize that the higher methylation of PcG targets in HPV(+) tumors suggests that HPV directed genomic changes such as altered DNA methylation may occur very early in the carcinogenic process, leading to aberrant DNA methylation and creation of HNSCC stem cells.Citation42,Citation43 Thus, whether or not the specific epigenetic differences observed correspond to functional changes in gene expression is of critical importance to understanding if promoter methylation is serving as a driver of the carcinogenic process, or a passenger in response to overall aberrant methylation.
Together, these define a distinct mechanism in HPV-associated carcinogenesis. PcG target promoters are cell type specificCitation44 and differ between embryonal cells and adult tumors. It is intriguing to think that they may differ by tumor subtype, and thus could explain prognostic heterogeneity within tumor type. For example, prostate cancers with high expression of the PcG protein EZH2 have a poorer prognosis.Citation45 Our results indicate that PRC2-targeted genes are more highly methylated in HPV(+) tumor cells, identifying a key pathway with implications for our understanding of how oncogenic HPV may contribute to carcinogenesis. Of interest, for HPV(+) cancers, it has been shown that the HPV E7 protein regulates EZH2 through activation of E2F.Citation40 CK8, which was found to be an interaction hub in HPV(+) cells, is associated with simple or single-layered epithelia and thus may be related to the less differentiated nature of HPV(+) tumors.Citation46 This is consistent with observations that HPV(+) HNSCC are usually poorly differentiatedCitation2,Citation4–Citation7 in contrast to HPV(−) SCC which typically show greater evidence of squamous differentiation (i.e., variuos degrees of keratinization, dyskeratosis and keratin pearl formation).
A few studies have compared gene expression profiles in HPV(+) and HPV(−) HNSCC. Martinez et al. noted that annexin IX was under-expressed in both HPV(−) and HPV(+) versus normal oral epithelium.Citation19 We found annexin IX to have higher methylation and lower expression in HPV(−) cells. CCNA1 was found to be upregulated in HPV(−) over normal epithelium and we found it to be not only upregulated in HPV(−) but also that it has greater DNA methylation in HPV(+) tumor cells. Interestingly, CCNA1, along with DCC and CDKN2A, has been found to correlate with longer disease-free survival in HNSCC.Citation32 Genes in the 9p24 region were preferentially lower expressed in HPV(−) indicating a possible loss of this region. One of these genes was UHRF2, a ubiquitin-ligase involved in cell cycle regulation and proliferation, thought to be involved in histone modifications and regulation of the epigenetic code, and a possible drugable target for cancers.Citation47 Overexpression of UHRF2 leads to growth arrest, which is in concordance with its tumor suppressor properties supporting the better prognosis for HPV(+) patients.
The candidate methylation differences observed between HPV(+) and HPV(−) tumors and the pathways in which they take part provide several testable hypotheses for further research. Of particular interest will be the genes observed to have both higher promoter methylation and higher expression levels in HPV(+) cells compared to HPV(−) cells, as these serve as candidate genes that may be lost due to copy number changes or mutation on HPV(−) and partially lost due to promoter methylation in HPV(+). Validated differences may provide intriguing potential therapuetic targets and suggest novel approaches to cancer therapy. Altering gene expression in HPV(−) tumors to reflect that seen in the more treatment responsive HPV(+) tumors might prove to be a useful strategy. A novel approach to treating HNSCC would be to develop methods by which the methylation profiles in HPV(−) tumors would be altered to recreate the more therapeutically favorable HPV(+) profile. This strategy could potentially lead to a more responsive cancer phenotype and better outcomes.
Materials and Methods
Cell culture, nucleic acid isolation and bisulfite conversion.
HPV(+) and HPV(−) SCC cell lines were cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum (FBS) and prophylactic antibiotics. UM-SCC cell lines were established from University of Michigan (UM) Head and Neck cancer patients who gave written informed consent to use their tissue to create a cell line. UM-SCC-4 and UM-SCC-74A, established from a 47 year old female and 50 year old male respectively, both had HPV negative base of tongue cancer and UM-SCC-47, established from a 53 year old male with base of tongue cancer, is HPV-16 positive. CaSki, derived from an HPV-16 positive cervical carcinoma from a 40 year old female, was obtained in 1988 by Dr. Carey from Dr. Roland Pattillo, the originator of the cell line at the Medical College of Wisconsin, (Pattillo, RA).Citation48 All cell lines were genotyped by the UM DNA Core laboratory using Profiler Plus (Applied Biosystems, www.applied biosystems.com) in the earliest possible passage available in the UM Head and Neck Oncology cell line bank. The UM-SCC cell lines have unique genotypesCitation49 and the CaSki genotype is consistent with the ATCC catalog report. The cells are periodically retested for genotype and for mycoplasma contamination. Cells in log phase growth were harvested with Trypsin/EDTA, counted, washed and nucleic acids were isolated using modified protocols for the QiaAmp DNA Mini Kit for Blood and Body Fluids and RNEasy Mini Kit (Qiagen, Valencia, CA). A 20 ug aliquot of DNA was reserved for methylated DNA IP-on-Chip. Bisulfite conversion was carried out on 600 ng of DNA using the manufacturer's protocol (EZ DNA Methylation-Direct Kit, Zymo Research Corp., Orange, CA) for genome-wide methylation assessment using the Illumina Infinium HumanMethylation27 BeadChip platform (Illumina, San Diego, CA). For pyrosequencing validation 600 ng of DNA was bisulfite converted using the Qiagen EpiTect Bisulfite Conversion kit (Qiagen, Valencia, CA).
The commercially available Illumina Goldengate® Methylation Cancer Panel was utilized to detect DNA methylation patterns in 84 formalin-fixed paraffin-embedded (FFPE) head and neck tumor samples. DNA was isolated from FFPE tumor samples using the QIAamp DNA FFPE Tissue Kit (Qiagen, Valencia, CA) with a modification to the manufacturer's recommended lysis protocol (incubation overnight at 56°C in lysis buffer). We performed sodium bisulfite modification on 500 ng to 1 µg of extracted DNA using the EZ DNA Methylation kit (Zymo Research, Orange, CA) following the manufacturer's recommended protocol. Bead arrays were run at the University of Michigan DNA Sequencing Core Facility according to the manufacturer's protocol and bead arrays were imaged using Illumina BeadArray Reader software. Full details on these data are presented in Colacino et al. (submitted).
Genome wide DNA methylation determination with affymetrix tiling set.
Methylated DNA IP-on-Chip (MeDIP-tiling) was performed with GenPathway (San Diego, CA) using the GeneChip Human Tiling 2.0R Array Set (Affymetrix, Santa Clara, CA). Following preclearing with protein G agarose beads (Invitrogen), input DNA was denatured and immunoprecipitated using an anti-5-methyl-cytosine antibody (Abcam ab1884) and incubated overnight at 4°C, with protein G agarose beads. An aliquot of each precipitated sample was used for quantitaitive PCR (qPCR) to confirm the enrichment of methylated regions, using a GenPathway proprietary method. Immunoprecipitated and input DNA were amplified and qPCR again verified enrichment of methylated regions in IP'ed DNA. Amplified immunoprecipitated and input DNA were labeled according to Affymetrix procedures and hybridized to the gene chips. Most Infinium array probes are located in genic regions (Sup. Fig.7) while the Affymetrix tiling array represents a genome-wide measurement of CpG methylation, but not repetitive regions.
Gene expression.
RNA (5 ug) from each cell line, isolated from the same passage as the DNA used for methylation analysis, was analyzed on the Affymetrix U133Plus 2.0 array in the University of Michigan Affymetrix Core facility. cDNA was generated using Bio-Rad iScript cDNA Synthesis Kit. TaqMan Gene Expression Assays (Applied Biosystems) for CCNA1 and DNMT3a validation were performed using 25 ng of input cDNA. qPCR was run and analyzed on a Bio-Rad CFX96 Real-Time PCR Detection System.
Validation by pyrosequencing.
CpG methylation at specific loci was validated by pyrosequencing using bisulfite converted DNA. PCR and sequencing primers are listed in Supplemental Table 1. The PCR products were run on a 1.5% agarose gel to ensure PCR quality, correct product length and lack of contamination. Pyrosequencing was performed following the manufacturer's protocol on a PyroMark MD (Qiagen).
Statistical analysis.
The Illumina BeadStudio Software provides estimates, termed beta values, of the percent methylation for each probe site based on the Cy3 and Cy5 intensities. However, we developed and implemented a novel normalization strategy which improves the correlation among replicate samples and increases the levels of significance when the methylation levels of the groups are compared (Sup. Fig.8). The motivation and detailed normalization steps are provided in the supplement and diagrammed in Figure S9. We used an empirical Bayesian moderated t-test to assess significance between the percent methylation in HPV(+) versus HPV(−) samples, which improves results for studies of small sample size.Citation50 In follow-up analyses, both a p-value cutoff and % change in methylation cutoff were used.
The Affymetrix Human Tiling 2.0R Array Set is a set of 7 tiling arrays divided by chromosome and consisting of a total of more than 40 million 25-mer oligos. Raw data were read into R using the AffyTiling package and were analyzed as described in reference Citation51. Raw data were quantile normalized, then oligos were annotated to CpG islands and genes using the UCSC CpG islands and Known Genes tracks, respectively. An empirical Bayes approach with moderated variance in ANOVACitation52 was used to test for methylated and differentially methylated regions. This method takes into account the dependency of variance on intensity level and improves results in studies with small sample size. The estimates of fold change over input and between HPV(+) and HPV(−) cells were smoothed using a moving window average of 175 bp length.
Genome-wide expression differences between the HPV(+) and HPV(−) cells were evaluated with the Affymetrix Human Genome U133 Plus 2.0 Array platform. Raw data was preprocessed and normalized using RMA. Differential expression and significance was assessed using IBMT.Citation52
Bioinformatics analyses.
Beta values representing the % methylation levels of the four cell lines were clustered to determine whether the HPV(+) and HPV(−) cell lines could be correctly separated using unsupervised methods. CpG sites with low standard deviation for beta values across all four cell lines were filtered out, which is a common approach to remove genes that are not informative, and values for each site were centered by their mean. Hierarchical clustering with correlation similarity measures and average linkage clustering was performed using Gene Cluster and Java TreeView to visualize results.Citation53 To determine whether the results are robust, we used three different cutoffs for standard deviation resulting in three different lists of CpG sites to cluster: 0.01 (18,815 sites), 0.10 (11,205 sites) and 0.40 (2,457 sites). The range of standard deviations was 0.00017–0.57. Complete linkage and single linkage also resulted in the same conclusions.
Genes were chosen from the top left, bottom right and top right quadrants of using a combination of criteria: each gene chosen had a % change in methylation >20% with p-value < 0.10, and a p-value for change in expression < 0.10. We chose to use relaxed criteria since these sets of genes were chosen to use as input for additional bioinformatics analyses and meant only to assess candidates or perform gene set over-representation analysis which does not require the use of FDR criteria for individual genes.Citation54 For genes with higher methylation and lower expression in HPV(+), we used the additional criteria of an average background-adjusted and normalized log2-intensity ≥6.0 (a selected set is provided in Sup. Table 2). ConceptGenCitation22 was used to test for predefined gene sets (concepts) enriched with the chosen genes from the top left and bottom right quadrants of , which tests several types of gene sets, including Gene Ontology (GO), KEGG Pathways, literature-defined concepts from Medical Subject Headings and differentially expressed gene sets from publicly available microarray data. The same sets of genes were also uploaded to GeneGO Inc.,'s MetaCore software program, which we used to identify and visualize the protein interaction networks and identify potential hub proteins connecting the chosen genes in each set. The Dijkstra's shortest path algorithm was used with a two step maximum, including only high quality interactions. We defined a CpG island by the Illumina-provided annotation for the Infinium Methylation27 platform. To identify biologically defined gene sets or concepts, significantly enriched with genes up or downregulated in HPV(+) versus HPV(−) cells, we used LRpathCitation54 (see Sup. for more details). All array-based data are available in Gene Expression Omnibus (GEO), accession ID GSE24091.
Financial Support
This work was supported by the University of Michigan School of Public Health (SPH) and the SPH Department of Environmental Health Sciences. Support for J.A.C. was provided by an Institutional Training Grant from the National Institute of Environmental Health Sciences, T32 ES07062. Other grant support was from NIH-NIDCR R01 DE019126 (T.E.C.), NIH NCI NIDCR SPORE P50 CA097248 (T.E.C., L.S.R., M.E.P.P.) NIH-NCI Comprehensive Cancer Center Core Grant P30 CA46592, NIH NIDCD P30 DC05188.
Abbreviations
| FDR | = | false discovery rate |
| GEO | = | gene expression omnibus |
| HNSCC | = | head and neck squamous cell carcinomas |
| HPV | = | human papillomaviruses |
| IP | = | immunoprecipitate |
| PRC2 | = | polycomb repressive complex 2 |
| SCC | = | squamous cell carcinoma |
Figures and Tables
Figure 1 Global methylation in HPV(+) and HPV(−) cell lines. (A) Histogram of % methylation of CpG sites represented on the Illumina Infinium methylation array for each cell line. There is a shift to more sites with high methylation in HPV(+) cells (gray circles; see also Sup. Fig. 4). The number (%) of genomic sites with greater than 80% methylation in each cell line is: SCC-4: 6402 (23.2%); SCC-74A: 4138 (15.0%); SCC-47: 7707 (27.9%); CaSki: 9322 (33.8%). The Illumina array is designed to assess sites thought to be functionally relevant. (B) Scatterplot showing average percent methylation across all 4 cell lines (x-axis) versus the difference in methylation between HPV(+) and HPV(−) cells (y-axis) from Illumina array. Most of the differentially methylated sites have higher levels of methylation in HPV(+) cells (seen by the high density of points at the top of the plot), while very few have higher methylation in HPV(−) cells. (C) Histograms of log2 fold changes between HPV(+) and HPV(−) cells revealed by the Affymetrix MeDIP-tiling approach (global vs. promoter regions) confirms that overall methylation is higher in the HPV(+) cell lines. Sites greater than 0 have higher methylation in HPV(+) cells; sites less than 0 have higher methylation in HPV(−) cells. Fold change between the number of sites greater than 0 and less than 0 is provided in the plots. A significance cutoff of p < 0.0005 for the difference between HPV(+) and HPV(−) cells was used to filter the sites prior to the analysis. This trend for increased methylation in HPV(+) cells is stronger in promoter regions than in other non-repetitive regions (p = 1.9 × 10−26; odds ratio = 1.7, Fisher's Exact test).
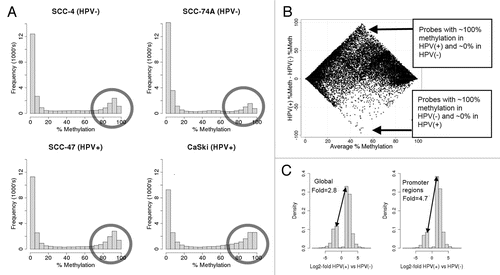
Figure 2 Integrating DNA methylation (Illumina Infinium platform) and gene expression data in HPV(+) and HPV(−) cell lines. (A) Histogram of average expression levels (across all 4 cell lines) for the 343 genes whose promoter regions are less methylated in HPV(+) than in HPV(−); (B) Histogram of average expression levels for the 4,525 genes whose promoter regions are more methylated in HPV(+) than in HPV(−). (A and B) together illustrate that genes with higher promoter methylation in HPV(+) cells are generally expressed at low levels. (C) Scatterplot showing the relationship between methylation and gene expression in HPV(+) and HPV(−) cell lines. The top half of the figure represents genes more highly expressed in HPV(+) cell lines and the right half of the figure shows genes more highly methylated in HPV(+) cell lines. Genes near the x-axis do not differ in expression even though many are differentially methylated in HPV(+) and HPV(−) cells. Genes within the loops are differentially expressed in HPV(+) and HPV(−) cells. Overall the expression negatively correlates with methylation, although not as strongly as expected due to the unexpected population of genes in the top right quadrant having higher methylation and expression in HPV(+) relative to HPV(−) cells (Pearson correlation coefficient = −0.21). Expression levels of genes near the y-axis are unrelated to the change in percent methylation. (D) Boxplot of HPV(+)/HPV(−) expression differences for genes with lower methylation in HPV(+) cells (left) and genes with higher methylation in HPV(+) cells (right).
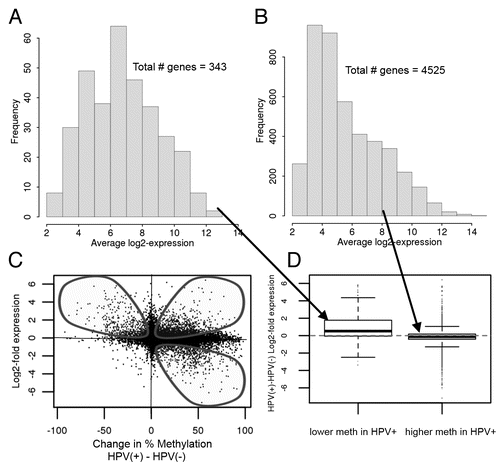
Figure 3 Characterization of genes with increased methylation and decreased expression level in HPV(−) compared to HPV(+) cell lines (A) CDKN2A and Keratin 8 are interaction hubs among genes with increased methylation and decreased expression level in HPV(−) relative to HPV(+) cell lines (, top left quadrant). Interactions among 28 input genes (circled in green/blue/red) with increased methylation and decreased expression level in HPV(−) compared to HPV(+) cell lines. The 28 genes were prioritized as being both downregulated and exhibiting higher promoter methylation in HPV(−) versus HPV(+). The network was created using GeneGO's MetaCore and the shortest path algorithm with a 2 step maximum. Interactions (all curated) are either based on binding (B), transcriptional regulation (TR), influence on expression (IE) or phosphorylation (+P) in humans and low trust interactions were discarded. Green arrows indicate activation, red arrows indicate inhibition and gray arrows are unspecified. Interaction types are indicated on the arrows. Additional genes in network (not circled) were included because they connected/interacted with two or more of the input genes. (The complete legend for GeneGO's MetaCore networks is provided as Sup. Fig.10). (B) Related biological concepts (p < 0.05) for genes with increased methylation and decreased expression level in HPV(−) relative to HPV(+) cell lines (, top left quadrant).
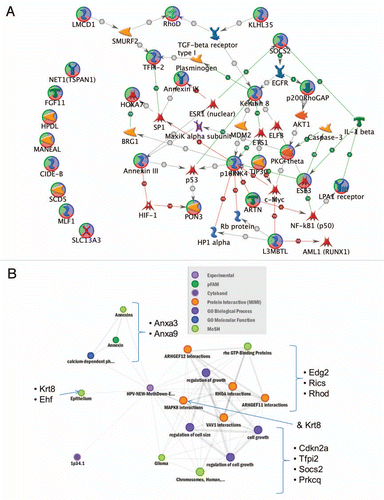
Figure 4 Network (A) and Heat Map (B) of enriched concepts (p < 0.05) for genes with increased methylation and decreased expression in HPV(+) relative to HPV(−) cells. Genes were limited to those that had mid-to-high overall levels of expression to avoid over-representation of passenger genes. Vacuole was the most significantly enriched concept, involving the genes STS, ATP6V0C, HPS1, CTSL1, GNS, FUCA1 and VPS18. The PI3 kinase Panther pathway and regulation of kinase activity concept included the genes IRS1, GNA11, GNAI2, EREG, CCNA1, RGS4 and PKIG. Plot generated from ConceptGen. (C) Runx2, IRS-1, CD40 and CCNA1 (Cyclin A1) are interaction hubs among genes with increased methylation and decreased expression level in HPV(+) relative to HPV(−) samples (-bottom right quadrant). Interactions among 75 input genes (circled in green/blue/red) with increased methylation and decreased expression level in HPV(+) compared to HPV(−) cell lines (Legend is provided as Sup. Fig. 10). The network was created using GeneGO's MetaCore and the shortest path algorithm with a 2 step maximum. Interactions (all curated) may be binding, cleavage, transcriptional regulation, influence on expression or phosphorylation in humans and low trust interactions were discarded. Green arrows indicate activation, red arrows indicate inhibition and gray arrows are unspecified. Additional genes in network (not circled) were included because they connected/interacted with two or more of the 75 input genes. To limit the complexity in we provide in Supplemental Figure 11 information in a zoomable format showing the interaction types between genes, which is provided.
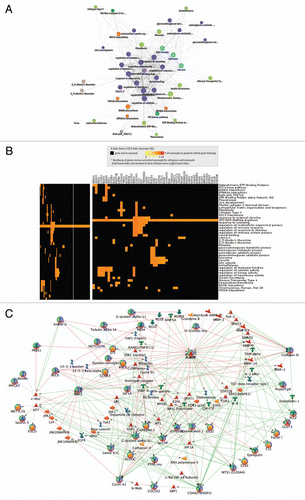
Figure 5 Promoter regions of polycomb repressive complex 2 (PRC2) targets tend to be much more highly methylated in HPV(+) cells than in HPV(−) cells, compared to promoters of non-PRC2 targets. Displayed are histograms of difference in percent methylation between HPV(+) and HPV(−) cells from the Illumina Infinium HumanMethylation27 BeadArray platform.
Additional material
Download Zip (6.1 MB)Acknowledgments
We thank Jung (Julie) Kim for submission of array data to Gene Expression Omnibus (GEO) and Martin Graham for preparing the cells used in this study.
References
- Shiboski CH, Schmidt BL, Jordan RC. Tongue and tonsil carcinoma: increasing trends in the USpopulation ages 20–44 years. Cancer 2005; 103:1843 - 1849
- Maxwell JH, Kumar B, Feng FY, Worden FP, Lee JS, Eisbruch A, et al. Tobacco use in human papillomavirus-positive advanced oropharynx cancer patients related to increased risk of distant metastases and tumor recurrence. Clin Cancer Res 2010; 16:1226 - 1235
- Marur S, Forastiere AA. Head and neck cancer: changing epidemiology, diagnosis and treatment. Mayo Clin Proc 2008; 83:489 - 501
- D'Souza G, Kreimer AR, Viscidi R, Pawlita M, Fakhry C, Koch WM, et al. Case-control study of human papillomavirus and oropharyngeal cancer. N Engl J Med 2007; 356:1944 - 1956
- Gillison ML, D'Souza G, Westra W, Sugar E, Xiao W, Begum S, et al. Distinct risk factor profiles for human papillomavirus type 16-positive and human papilloma-virus type 16-negative head and neck cancers. J Natl Cancer Inst 2008; 100:407 - 420
- Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst 2000; 92:709 - 720
- Worden FP, Kumar B, Lee JS, Wolf GT, Cordell KG, Taylor JM, et al. Chemoselection as a strategy for organ preservation in advanced oropharynx cancer: response and survival positively associated with HPV16 copy number. J Clin Oncol 2008; 26:3138 - 3146
- Hafkamp HC, Manni JJ, Haesevoets A, Voogd AC, Schepers M, Bot FJ, et al. Marked differences in survival rate between smokers and nonsmokers with HPV 16-associated tonsillar carcinomas. Int J Cancer 2008; 122:2656 - 2664
- Hafkamp HC, Manni JJ, Speel EJ. Role of human papillomavirus in the development of head and neck squamous cell carcinomas. Acta Otolaryngol 2004; 124:520 - 526
- Kumar B, Cordell KG, Lee JS, Worden FP, Prince ME, Tran HH, et al. EGFR, p16, HPV Titer, Bcl-xL and p53, sex and smoking as indicators of response to therapy and survival in oropharyngeal cancer. J Clin Oncol 2008; 26:3128 - 3137
- Shah JP, Gil Z. Current concepts in management of oral cancer surgery. Oral Oncol 2009; 45:394 - 401
- Hafkamp HC, Speel EJ, Haesevoets A, Bot FJ, Dinjens WN, Ramaekers FC, et al. A subset of head and neck squamous cell carcinomas exhibits integration of HPV 16/18 DNA and overexpression of p16INK4A and p53 in the absence of mutations in p53 exons 5–8. Int J Cancer 2003; 107:394 - 400
- Shaw R. The epigenetics of oral cancer. Int J Oral Maxillofac Surg 2006; 35:101 - 108
- Shaw RJ, Liloglou T, Rogers SN, Brown JS, Vaughan ED, Lowe D, et al. Promoter methylation of P16, RARbeta, E-cadherin, cyclin A1 and cytoglobin in oral cancer: quantitative evaluation using pyrosequencing. Br J Cancer 2006; 94:561 - 568
- Sanchez-Cespedes M, Esteller M, Wu L, Nawroz-Danish H, Yoo GH, Koch WM, et al. Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer Res 2000; 60:892 - 895
- Kato K, Hara A, Kuno T, Mori H, Yamashita T, Toida M, et al. Aberrant promoter hypermethylation of p16 and MGMT genes in oral squamous cell carcinomas and the surrounding normal mucosa. J Cancer Res Clin Oncol 2006; 132:735 - 743
- Viswanathan M, Tsuchida N, Shanmugam G. Promoter hypermethylation profile of tumor-associated genes p16, p15, hMLH1, MGMT and E-cadherin in oral squamous cell carcinoma. Int J Cancer 2003; 105:41 - 46
- Richards KL, Zhang B, Baggerly KA, Colella S, Lang JC, Schuller DE, et al. Genome-wide hypomethylation in head and neck cancer is more pronounced in HPV-negative tumors and is associated with genomic instability. PLoS One 2009; 4:4941
- Martinez I, Wang J, Hobson KF, Ferris RL, Khan SA. Identification of differentially expressed genes in HPV-positive and HPV-negative oropharyngeal squamous cell carcinomas. Eur J Cancer 2007; 43:415 - 432
- Schlecht NF, Burk RD, Adrien L, Dunne A, Kawachi N, Sarta C, et al. Gene expression profiles in HPV-infected head and neck cancer. J Pathol 2007; 213:283 - 293
- Slebos RJ, Yi Y, Ely K, Carter J, Evjen A, Zhang X, et al. Gene expression differences associated with human papillomavirus status in head and neck squamous cell carcinoma. Clin Cancer Res 2006; 12:701 - 709
- Sartor MA, Mahavisno V, Keshamouni VG, Cavalcoli J, Wright Z, Karnovsky A, et al. ConceptGen: a gene set enrichment and gene set relation mapping tool. Bioinformatics 2010; 26:456 - 463
- Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med 2006; 354:567 - 578
- Tsui IF, Poh CF, Garnis C, Rosin MP, Zhang L, Lam WL. Multiple pathways in the FGF signaling network are frequently deregulated by gene amplification in oral dysplasias. Int J Cancer 2009; 125:2219 - 2228
- Hartmann C, Johnk L, Kitange G, Wu Y, Ashworth LK, Jenkins RB, et al. Transcript map of the 3.7-Mb D19S112-D19S246 candidate tumor suppressor region on the long arm of chromosome 19. Cancer Res 2002; 62:4100 - 4108
- Hoque MO, Begum S, Sommer M, Lee T, Trink B, Ratovitski E, et al. PUMA in head and neck cancer. Cancer Lett 2003; 199:75 - 81
- Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 2006; 125:301 - 313
- Archer KJ, Mas VR, Maluf DG, Fisher RA. High-throughput assessment of CpG site methylation for distinguishing between HCV-cirrhosis and HCV-associated hepatocellular carcinoma. Mol Genet Genomics 2010; 283:341 - 349
- Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev 2007; 87:905 - 931
- Stephen JK, Chen KM, Raitanen M, Grenman S, Worsham MJ. DNA hypermethylation profiles in squamous cell carcinoma of the vulva. Int J Gynecol Pathol 2009; 28:63 - 75
- Carvalho AL, Chuang A, Jiang WW, Lee J, Begum S, Poeta L, et al. Deleted in colorectal cancer is a putative conditional tumor-suppressor gene inactivated by promoter hypermethylation in head and neck squamous cell carcinoma. Cancer Res 2006; 66:9401 - 9407
- Tan HK, Saulnier P, Auperin A, Lacroix L, Casiraghi O, Janot F, et al. Quantitative methylation analyses of resection margins predict local recurrences and disease-specific deaths in patients with head and neck squamous cell carcinomas. Br J Cancer 2008; 99:357 - 363
- Westra WH, Taube JM, Poeta ML, Begum S, Sidransky D, Koch WM. Inverse relationship between human papillomavirus-16 infection and disruptive p53 gene mutations in squamous cell carcinoma of the head and neck. Clin Cancer Res 2008; 14:366 - 369
- McLaughlin-Drubin ME, Munger K. Oncogenic activities of human papillomaviruses. Virus Res 2009; 143:195 - 208
- van der Riet P, Nawroz H, Hruban RH, Corio R, Tokino K, Koch W, et al. Frequent loss of chromosome 9p21-22 early in head and neck cancer progression. Cancer Res 1994; 54:1156 - 1158
- Nakahara Y, Shintani S, Mihara M, Ueyama Y, Matsumura T. High frequency of homozygous deletion and methylation of p16(INK4A) gene in oral squamous cell carcinomas. Cancer Lett 2001; 163:221 - 228
- Shintani S, Nakahara Y, Mihara M, Ueyama Y, Matsumura T. Inactivation of the p14(ARF), p15(INK4B) and p16(INK4A) genes is a frequent event in human oral squamous cell carcinomas. Oral Oncol 2001; 37:498 - 504
- Rocco JW, Sidransky D. p16(MTS-1/CDKN2/INK4a) in cancer progression. Exp Cell Res 2001; 264:42 - 55
- Rush M, Appanah R, Lee S, Lam LL, Goyal P, Lorincz MC. Targeting of EZH2 to a defined genomic site is sufficient for recruitment of Dnmt3a but not de novo DNA methylation. Epigenetics 2009; 4:404 - 414
- Holland D, Hoppe-Seyler K, Schuller B, Lohrey C, Maroldt J, Durst M, et al. Activation of the enhancer of zeste homologue 2 gene by the human papillomavirus E7 oncoprotein. Cancer Res 2008; 68:9964 - 9972
- Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, et al. Epigenetic stem cell signature in cancer. Nat Genet 2007; 39:157 - 158
- Rajasekhar VK, Begemann M. Concise review: roles of polycomb group proteins in development and disease: a stem cell perspective. Stem Cells 2007; 25:2498 - 2510
- Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci USA 2007; 104:973 - 978
- Squazzo SL, O'Geen H, Komashko VM, Krig SR, Jin VX, Jang SW, et al. Suz12 binds to silenced regions of the genome in a cell-type-specific manner. Genome Res 2006; 16:890 - 900
- Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002; 419:624 - 629
- Inada H, Izawa I, Nishizawa M, Fujita E, Kiyono T, Takahashi T, et al. Keratin attenuates tumor necrosis factor-induced cytotoxicity through association with TRADD. J Cell Biol 2001; 155:415 - 426
- Bronner C, Achour M, Arima Y, Chataigneau T, Saya H, Schini-Kerth VB. The UHRF family: oncogenes that are drugable targets for cancer therapy in the near future?. Pharmacol Ther 2007; 115:419 - 434
- Pattillo RA, Hussa RO, Story MT, Ruckert AC, Shalaby MR, Mattingly RF. Tumor antigen and human chorionic gonadotropin in CaSki cells: a new epidermoid cervical cancer cell line. Science 1977; 196:1456 - 1458
- Brenner JC, Graham MP, Kumar B, Saunders LM, Kupfer R, Lyons RH, et al. Genotyping of 73 UM-SCC head and neck squamous cell carcinoma cell lines. Head Neck 2010; 32:417 - 426
- Smyth GK. Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Statistical Applications in Genetics and Molecular Biology 2004; 3:3
- Sartor MA, Schnekenburger M, Marlowe JL, Reichard JF, Wang Y, Fan Y, et al. Genome-wide analysis of aryl hydrocarbon receptor binding targets reveals an extensive array of gene clusters that control morphogenetic and developmental programs. Environmental Health Perspectives 2009;
- Sartor MA, Tomlinson CR, Wesselkamper SC, Sivaganesan S, Leikauf GD, Medvedovic M. Intensity-based hierarchical Bayes method improves testing for differentially expressed genes in microarray experiments. BMC Bioinformatics 2006; 7:538
- de Hoon MJ, Imot S, Nolan J, Miyano S. Open source clustering software. Bioinformatics 2004; 20:1453 - 1454
- Sartor MA, Leikauf GD, Medvedovic M. LRpath: a logistic regression approach for identifying enriched biological groups in gene expression data. Bioinformatics 2009; 25:211 - 217