Abstract
The placenta acts not only as a conduit of nutrient and waste exchange between mother and developing fetus, but also functions as a regulator of the intrauterine environment. Recent work has identified changes in the expression of candidate genes, often through epigenetic alteration, which alter the placenta's function and impact fetal growth. In this study, we used the Illumina Infinium HumanMethylation27 BeadChip array to examine genome-wide DNA methylation patterns in 206 term human placentas. Semi-supervised recursively partitioned mixture modeling was implemented to identify specific patterns of placental DNA methylation that could differentially classify intrauterine growth restriction (IUGR) and small for gestational age (SGA) placentas from appropriate for gestational age (AGA) placentas, and these associations were validated in a masked testing series of samples. Our work demonstrates that patterns of DNA methylation in human placenta are reliably and significantly associated with infant growth and serve as a proof of principle that methylation status in the human term placenta can function as a marker for the intrauterine environment, and could potentially play a critical functional role in fetal development.
Introduction
Fetal growth and development are complex processes that can be influenced by a myriad of environmental conditions and exposures, including xenobiotics,Citation1 maternal stress and psychology,Citation2 maternal nutrition and activityCitation3,Citation4 and sociodemographic factors.Citation5 This intrauterine environment is critical in “programming” the fetus for various health and disease outcomes throughout life.Citation6 The placenta effectively mediates the appropriate response to and integration of such environmental signals, allowing for healthy growth and development. Adverse intrauterine conditions can alter placenta gene expression and subsequent functions resulting in alteration of infant growth,Citation7,Citation8 which in turn has been linked to an increased incidence of metabolic and cardiovascular disease.Citation9
The changes in placental gene expression resulting from the intrauterine environment can be attributed in part to epigenetic alterations. Numerous links have been made between infant growth restriction and specific epigenetic alterations, including changes to gene imprinting status and to DNA methylation, thereby implicating such regulation in appropriate growth and development.Citation10–Citation12 DNA methylation has been associated with the regulation of genes regulating trophoblast migration and invasion,Citation13 as well as to endometrial receptivity to implantation.Citation14 Animal models have suggested that methylation plays a critical role in placenta development, and alterations to its methylation pattern can lead to adverse placenta morphology and birth outcome.Citation15 In humans, normal hypermethylation of the vitamin D 24-hydroxylase gene is thought to allow for greater availability of vitamin D at the maternal-fetal interface,Citation16 while altered DNA methylation of specific genes in imprinting control regions, including one controlling H19/IGF2 in the placenta, has been linked to preeclampsia and intrauterine growth restriction (IUGR).Citation17,Citation18
As the placenta plays a critical role in responding to the intrauterine environment and in controlling infant growth, and as the environment can influence the pattern of DNA methylation, we sought to examine the association between infant growth and profiles of genome-wide, gene-specific DNA methylation in human placenta. These data will allow us to identify general epigenomic alterations in the placenta related to an adverse intrauterine environment, represented by poor fetal growth.
Results
DNA methylation patterning in the human placenta.
DNA methylation profiles were obtained on 206 term human placenta samples using the Human-Methylation-27 BeadChip array. describes the maternal and infant characteristics of the randomly generated training and testing datasets. As these profiles represent term births and were oversampled for growth restricted infants, the mean gestational age was 38 weeks for both groups, and growth restricted infants (SGA or IUGR) made up 43% of the population. There were no significant differences in the clinical characteristics between training and testing sets as demonstrated by the associated p values.
Platform variability across BeadChips was assessed by profiling a reference sample derived from peripheral blood DNA on each array. The correlation coefficients calculated between each combination of control blood arrays had a median value of 0.98. As an additional measure of array reproducibility, 12 placenta samples were repeated across arrays. A median correlation coefficient of 0.95 was found between the 12 technical replicates (Sup. Fig. 1), indicating agreement within each sample.
To identify those genomic loci most significantly associated with SGA or IUGR diagnoses, a linear mixed effects model was fit using the training data only for each of the 26,486 autosomal loci in the dataset. These linear effects models had as their response, arc-sine square-root transformed methylation β value, a fixed-effect term indicating growth status (i.e., SGA/IUGR or AGA), and a random-effect term for BeadChip. depicts the volcano plot of the fixed effect estimates from the linear mixed effects models, representing the magnitude of the association between DNA methylation extent at each locus and growth status on the x-axis and the log p value of the test of the association on the y-axis. The greater number of points on the left-hand side of the volcano plot indicates that there are a larger number of loci whose methylation is inversely associated with SGA or IUGR status.
DNA methylation profiles are significantly associated with SGA or IUGR status.
Using the nested cross-validation procedure described in Koestler et al.,Citation19 we determined that the most appropriate number of loci to include in the RPMM was 22. Those 22 loci along with the estimates of association of growth status and corresponding genomic information are listed in Supplemental Table 1. Subsequently, fitting RPMM to the training data using these 22 loci resulted in 5 methylation classes (). Classes 1 and 2, the left-most branches of the dendrogram, contained 56% of all the samples, with 40 observations and 37 observations, respectively (). The remaining samples were predicted to be within the right branches of the dendrogram, with Class 3 containing 43 observations, and Classes 4 and 5 both having 9 observations. There was a highly significant difference in the prevalence of SGA or IUGR placentas across all classes (p = 0.0007, Permutation Chi Square test). Notably, the left classes (Classes 1 and 2) contained a greater proportion of samples from growth restricted infants while the right-most classes (3–5) showed reduced prevalence of growth restricted samples ().
Using the RPMM solution based on the training data and a naïve Bayes procedure, we predicted methylation class for the observations in the testing data (). The samples were predicted to be members of the defined RPMM classes with a distribution within the classes similar to that observed in the training data (). SGA or IUGR diagnosis was again significantly different by class (p = 0.0087, Permutation Chi Square test), with the left branch classes (1 and 2) containing 70% of all SGA and IUGR samples. No samples in the testing dataset were predicted to be in Class 5. There were no significant associations between predicted methylation class and infant gender, delivery method, maternal age or maternal BMI (Sup. Table 2). To examine the validity of the methylation at these points, we specifically examined the methylation of these 22 loci in the 12 placenta samples with replicate arrays. The mean difference between the replicates for the 22 loci selected did not exceed 0.17 (Sup. Table 3), which is the reported sensitivity of the Infinium Golden Gate Methylation array,Citation20 suggesting that differences between placentas located on different BeadChip arrays were related to biological variance and not chip to chip platform variance.
To estimate the association between predicted methylation class and SGA or IUGR diagnosis while controlling for potential confounders, a multivariable logistic regression model was constructed () using the testing data. Controlling for infant gender, delivery method, maternal age and maternal BMI, infants whose placenta methylation profile was predicted to be in Classes 1 and 2 had nearly 3 times the odds of being SGA or IUGR compared to infants predicted to be in Classes 3–5 (OR 2.94; 95% CI 1.05, 7.38). There were no significant associations observed between SGA or IUGR diagnosis and infant gender, delivery method, maternal age, nor maternal BMI.
Of the 22 genes present in our profile, fourteen demonstrated reduced methylation associated with SGA or IUGR diagnosis, while only OMG, PDC, RPE65, SERPINA5, APBA2, CHML, SLC25A18 and MEP1A showed increased methylation (Sup. Table 1). To examine the potential biological relevance of genes targeted for alteration and to explore which biological systems are most affected, a gene-set enrichment analysis (GSEA) based on KEGG-defined pathways was performed. Using the combined training and testing data, we compared pathways that were over-represented among loci associated with altered growth status. Pathways with a nominal p < 0.05 for both a GSEA statistic and independent Wilcoxon rank sum test are listed in and include pathways important in neurodegenerative diseases such as Alzheimer and Huntington disease, as well as basic cellular functions including protein export, transcription and DNA repair.
In addition to examining the functional consequences of differential methylation in placentas of growth restricted and normal infants, we hypothesized that the observed differential methylation profiles may represent an altered epigenetic process related to the intrauterine environment and growth. This process may result from cell signaling leading to epigenetic alterations at specific regions targeted by certain transcription factors. Therefore, we examined over-representation of transcription factor binding sites (TFBS) within 1 kb of loci exhibiting differential methylation by growth status. The over-represented TFBS, again with a nominal p < 0.05 for both a GSEA statistic and independent Wilcoxon rank sum tes, are listed in and include NFE2, C/EBPβ and FOXO4.
Discussion
A number of studies have begun to examine the association between growth restriction and epigenetic regulation in fetal tissues including placenta.Citation16–Citation18,Citation21–Citation23 Much of the recent research on placental epigenetics has focused on the role of genomic imprinting and new technologies have been developed to more efficiently identify imprinted genes.Citation24 From this we have learned much about the timing of epigenetic remodelingCitation25 and the role of specific genes, including imprinted tumor-suppressor genes in the human placenta.Citation26 Specific genes, such as H19/IGF2, have been studied in the context of growth restriction, demonstrating differential methylation of the imprinting control regions in placentas from growth restricted infants.Citation17,Citation18 A number of other imprinted loci, such as PHLDA2, ILK2, NNAT, CCDC86, PEG10, PLAGL1, DHCR24, ZNF331 and CDKAL1 have been shown to demonstrate differential expression between growth restricted and non-restricted infant placentas.Citation10 Animal models have described the functional significance of alterations in imprinting; for example, loss of the maternal allele of Grb10 limits placental size and efficiency.Citation27 Our goal in this study was to build on this knowledge and to expand the examination of epigenetic alterations to encompass a larger genome-wide profile of genes, beyond those genes subjected to regulation by genomic imprinting, and thus to identify if the intrauterine environment can be represented by gross alteration to the epigenetic landscape of the placenta.
We focused our investigation on methylation patterning due to the highly stable nature of DNA methylation marks. Previous studies have shown that labor induces altered expression of genes in the human placentaCitation28 and, while the methylation status is stable, the corresponding gene expression may not be.Citation29 Therefore, we believe that examination of methylation may reflect changes occurring over the course of in utero development and not only at the final moments of pregnancy and delivery. We used a relatively unbiased, genome-wide method to identify loci whose methylation status in the term placenta was most associated with infant birth weight as a marker of growth. The genome-wide analysis was driven by our hypothesis that the complex and multifactorial outcome of infant growth in the relatively non-pathologic context from which our population is drawn, results from the interplay of various genes, and thus, profiling their regulation by DNA methylation can provide insight into the global regulation of the genome. In order to avoid the confounding effects of premature birth, we only included placentas from patients with gestational age >36 weeks. Our analysis, using a novel validated statistical strategy aimed not at identifying a single locus whose methylation is associated with growth, but more importantly, a pattern of alterations, identified that the pattern of methylation of 22 critical loci is highly predictive of SGA or IUGR diagnosis. The identified pattern is potentially indicative of altered cellular processes leading to targeted DNA methylation alterations that are linked to infant growth restriction. The GSEA examining transcription factor binding sites is aimed at better characterizing genomic similarities amongst loci whose methylation was associated with growth status. The CCAAT/enhancer binding protein β (C/EBPβ) is a downstream effector of estrogen-mediated implantation and decidualization,Citation30 and controls target genes such as PLAC1, which are involved in the maternal-placental interface.Citation31 Thus, DNA methylation of CEBPβ target genes may affect the maternal-fetal interface, resulting in growth restriction. The GSEA also identified over-representation of FOXO4 TFBS as targets of methylation alteration. FOXO4, a homeobox transcription factor, has been localized to differentiated syncytiotrophoblastsCitation32 and has been shown to be involved in cellular stress responses.Citation33 It may likewise play a role in integrating environmental signals, resulting in altered placenta function and infant growth.
The GSEA of KEGG pathways revealed two neurological disease pathways: Huntington disease and Alzheimer disease. While it is unlikely that the methylation patterns we identified in the placenta are directly linked to the development of these diseases later in life, it is known that the placenta plays a critical role in neuropeptide homeostasis for the developing fetus.Citation34–Citation36 The placenta has been postulated to represent the “third brain” that links the developed (maternal) and developing (fetal) brains,Citation35 playing a critical role in the pathophysiology of intrauterine insults on the developing nervous system.Citation37 Placental production of the corticotropin-releasing hormone (CRH) and thyrotropin-releasing hormone (TRH) subserves intrauterine hypothalamic control of fetal pituitary development throughout most of gestation.Citation38,Citation39 Thus, our findings may represent alterations to such pathways related to growth restriction and potentially linking growth restriction with neurodevelopmental and mental health outcomes later in life. Additional studies are warranted to expand on these examinations and these later life endpoints to clarify the biological mechanisms at play.
It is important to note that we cannot definitively determine if these altered profiles of DNA methylation are a response of the placenta to the intrauterine environment and/or growth restriction, or are extant, such that they have led to the growth phenotypes observed. Nonetheless, by oversampling for infants who were small for gestational age, we generated a robust interrogation of the effects of intrauterine growth on placental DNA methylation and identified strong, significant and independent associations between these specific profiles of DNA methylation and infant growth. An additional limitation of this study lies in the overrepresentation of CpG island-associated loci found on the Illumina Infinium Human Methylation27 BeadArray. Ongoing research is now revealing that gene regulatory methylation events may occur in regions outside of CpG islands, such as on CpG shores found up to 2 kb upstream of the regulated gene.Citation40 More comprehensive approaches including more inclusive arrays and genome-wide sequencing will be needed to fully identify all regions contributing to the regulation of infant growth.
The biological basis and implications of these altered profiles remains unclear. One possibility is that these different profiles represent changes in the population of cells present in the placenta. It is clear that individual tissues and cells demonstrate unique patterns of DNA methylationCitation41 and that altered DNA methylation can identify specific sub-populations of cells in a highly sensitive manner.Citation42 Thus, within our placenta tissues we may be detecting, changes in the distribution of mature trophoblasts, immune cells and stromal cells, or even changes to sub-populations of these cells within these placenta tissue samples. Alternatively, these profiles may reflect phenotypic differences in the maturity or differentiation of cells in the placenta, as it is clear that epigenetic mechanisms play critical roles in cellular differentiation.Citation43 The difficulty in obtaining placental tissue from uncomplicated pregnancies at various time points throughout pregnancy limits available data that could be used to examine this hypothesis more definitively.
In summary, we have demonstrated the methylation profile of 22 genes from human term placentas yielded five different classes, and that these classes differed significantly by SGA or IUGR diagnosis. This work serves as a proof of principle that variation in the DNA methylation profile of human term placenta can serve as a marker of growth. Further analysis is warranted to elucidate additional covariates, including environmental factors and exposures that may be affecting the methylation profiles that distinguish these classes. As prospective associations have already been demonstrated for peripheral blood-based methylation profiles and diseases such as acute myeloid leukemia, longitudinal follow-up on these subjects may demonstrate the prospective utility of these placenta-specific methylation profiles.Citation44 This would strengthen our hypothesis that epigenetic alterations in the placenta are acting functionally to program the health of an individual far beyond the intrauterine environment.
Methods
Study design.
From September 2008 through September 2009, 206 residual placenta samples, oversampled for SGA infants (<10th percentile of birth weight) or with a diagnosis of IUGR, were collected from Women and Infants Hospital in Providence, RI in accordance with protocols approved by the Institutional Review Boards of both Women and Infants Hospital and Brown University. The diagnosis of IUGR was made through sonographic measurements demonstrating an estimated fetal weight less than the 10th percentile for gestational age. Measurements also included abdominal circumference, biparietal diameter, femur length and head circumference.Citation45 Samples were collected from women between the ages of 18–42, considered at term (≥36 weeks) with no history of preeclampsia, gestational diabetes, psychological disorders, and whose infants were viable, with no known genetic disorders. Placenta samples were collected within 2 h of parturition by excising 6–8 small pieces (massing approximately 1 g each), free of maternal decidua, from the maternal side of the placenta 2 cm from the umbilical cord insertion site. The sample was immediately placed in RNAlater™ (Applied Biosystems, Inc., Foster City, CA) and stored at 4°C. At least 72 h later, placenta samples were blotted dry of RNAlater™ and stored at −80°C until processed. Data on the infant's birth weight, length, gender, IUGR status, maternal demographics and delivery history were recorded from the medical chart. The birth weight percentile was calculated from the infant's birth weight and gestational age using the Fenton growth chart.Citation46
DNA extraction and modification.
DNA was extracted from the placenta samples using the QIAamp DNA Mini Kit (Qiagen, Inc., Valencia, CA) following manufacturer's protocols. Purified DNA was quantified with the NanoDrop ND1000 spectrophotometer, and 1 µg of placental DNA was bisulfite modified using the EZ DNA Methylation Kit D5008 (Zymo Research, Irvine, CA). In addition, a single peripheral blood sample from an adult not associated with the study was extracted and bisulfite modified in the same manner as the placenta samples and was included on each BeadChip to allow for inter-array normalization.
Infinium DNA methylation microarray.
Methylation profiling was performed using the Illumina Infinium Human Methylation27 BeadArray (Illumina, San Diego, CA) at the UCSF Institute for Human Genetics Genomic Core Facility following standardized protocols at the facility. The methylation status of a specific CpG site was calculated from the intensity of the methylated (M) and unmethylated (U) alleles, as the ratio of fluorescent signals β = Max(M,0)/[Max(M,0) + Max(U,0) + 100]. On this scale, 0 < β < 1, with β values close to 1 indicating complete methylation and β values close to 0 indicating no methylation. Quality assurance was assessed by detection p values, and no locus had a sizable fraction (>25%) of p values above a predetermined threshold (10−5). In addition, the multivariate characteristics (e.g., Cholesky residualsCitation47 or Mahalanobis distance based on fitted mean vector and variance-covariance matrix) of array control probes supplied by Illumina were used to diagnose problems such as poor bisulfite conversion or color-specific problems; none were noted. Finally, only autosomal loci were considered, thus our analysis was performed on 26,486 loci. We have previously demonstrated that DNA methylation detected using this array-based approach can be verified using alternative strategies including bisulfite sequencing approaches.Citation48,Citation49
Statistical methods.
The statistical workflow is illustrated in . We used Semi-Supervised Recursively Partitioned Mixture Models (SS-RPMM),Citation19 to identify methylation profiles associated with SGA or IUGR diagnosis. This method uses a Recursively Partitioned Mixture Model (RPMM),Citation50 for clustering methylation data. RPMM is an unsupervised model-based method for clustering data that has been demonstrated to perform effectively and efficiently for methylation data derived from the Illumina array technologies.Citation41,Citation49,Citation51–Citation53 Such an approach allows for inference in addressing the associations between the methylation-based clusters and covariates.
Training and testing sets were obtained by randomly sampling from the total population within infant gender and a β-distributed RPMM was fit to the training data using the M CpG loci most associated with SGA or IUGR diagnosis, where M was determined as described in Koestler et al.Citation19 The resulting model provides a latent class structure on the pre-selected loci, which was then used in conjunction with naïve Bayes procedure, to predict methylation class for the observations in the masked testing data. To ensure that the predicted methylation classes for the testing data were not due to factors other than SGA or IUGR diagnosis, the association between potential confounders and the predicted methylation classes were examined. Chi-Square permutation tests were performed on categorical variables and a Kruskal Wallis permutation test was performed on continuous variables. To further examine the association between the predicted methylation classes and SGA or IUGR diagnosis controlling for potential confounders, we fit a multivariable logistic regression model to the testing data, with SGA or IUGR diagnosis as the dependent variable predicted methylation class as the independent variable, and controlled for infant gender, mode of delivery (vaginal vs. Caesarean section) and maternal age.
GSEA,Citation54 was used to explore the biologic relevance of alterations in DNA methylation in the human placenta in distinguishing SGA or IUGR diagnoses from samples derived from AGA placentas.
Financial Support
Supported by grants P20RR018728 from the NIH-NCRR and grants P42ES013660 and T32ES007272 from the NIH-NIEHS.
Figures and Tables
Figure 1 Data analysis schematic. Placenta samples were split into training and testing datasets matched with equal proportions of SGA or IUGR samples in each group. Semi-Supervised Recursively Partitioned Mixture Model data analysis was used to rank the methylation of each locus as associated with SGA or IUGR diagnosis.
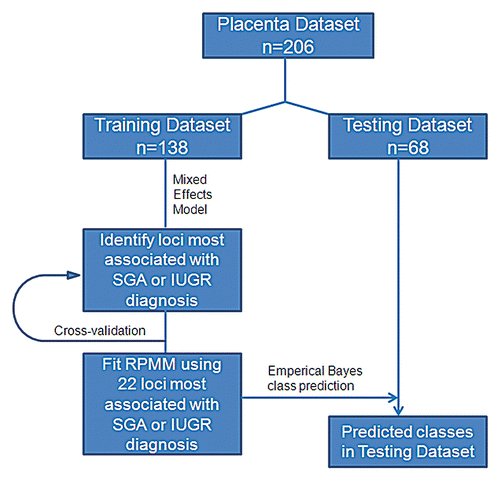
Figure 2 Association between methylation and birth weight. Volcano plot examining the association between SGA or IUGR diagnosis and methylation extent across all 26,486 autosomal loci examined. Negative log-transformed p values generated from the linear mixed effects model are plotted against the model coefficient (adjusting for gestational age). The area above the solid blue line indicates a p value < 0.05. This coefficient represents the magnitude of the effect of SGA or IUGR status on methylation.
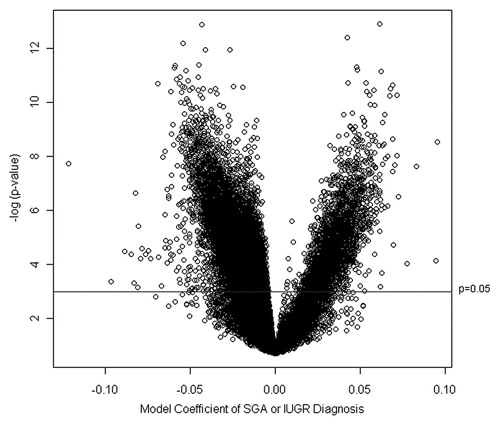
Figure 3 Methylation profiles defined by 22 loci are associated with infant growth status. The Recursively Partitioned Mixture Model-based classification of placenta samples (columns) based on 22 loci (rows) is depicted on the heatmap, with the five classes separated by red lines in the (A) training and (B) testing series. The prevalence of growth restriction or normal births within each of the classes is shown below the heatmap. The Chi-square p value listed below the tables indicates a significant difference in the proportion of SGA participants between classes for both training and testing datasets.
TABLE 1 Characteristics of the subjects involved in the study
Table 2 Multivariable analysis of the association between predicted SSRPMM class for the observations in the test data and SGA or IUGR classification; controlled for infant gender, maternal age, delivery method, parity, maternal BMI at time of delivery and maternal smoking during pregnancy
Table 3 Gene set enrichment analysis (GSEA) of KEGG pathways over-represented amongst loci associated with SGA or IUGR status
Table 4 Gene set enrichment analysis (GSEA) of transcription factor binding sites over-represented within 1 kb of loci associated with SGA or IUGR status
Additional material
Download Zip (2.1 MB)Acknowledgments
The authors would like to thank Keila Vega, Joyce Lee and Gilda Ferro for collecting the placenta samples and patient information, and to Andrew Rainey and Alison Migliori for processing the samples in the laboratory.
References
- Crinnion WJ. Maternal levels of xenobiotics that affect fetal development and childhood health. Altern Med Rev 2009; 14:212 - 222
- Kinsella MT, Monk C. Impact of maternal stress, depression and anxiety on fetal neurobehavioral development. Clin Obstet Gynecol 2009; 52:425 - 440
- Christian P, Stewart CP. Maternal micronutrient deficiency, fetal development and the risk of chronic disease. J Nutr 2010; 140:437 - 445
- Perkins CC, Pivarnik JM, Paneth N, Stein AD. Physical activity and fetal growth during pregnancy. Obstet Gynecol 2007; 109:81 - 87
- Fogleman KA, Herring AH, Kaczor D, Pusek SN, Jo H, Thorp JM. Factors that influence the timing of spontaneous labor at term. J Matern Fetal Neonatal Med 2007; 20:813 - 817
- Silveira PP, Portella AK, Goldani MZ, Barbieri MA. Developmental origins of health and disease (DOHaD). J Pediatr (Rio J) 2007; 83:494 - 504
- Maulik D, Frances Evans J, Ragolia L. Fetal growth restriction: pathogenic mechanisms. Clin Obstet Gynecol 2006; 49:219 - 227
- Burton GJ, Jauniaux E, Charnock-Jones DS. The influence of the intrauterine environment on human placental development. Int J Dev Biol 2010; 54:303 - 312
- Lumey LH, Stein AD, Kahn HS, van der Pal-de Bruin KM, Blauw GJ, Zybert PA, et al. Cohort profile: the Dutch Hunger Winter families study. Int J Epidemiol 2007; 36:1196 - 1204
- Diplas AI, Lambertini L, Lee MJ, Sperling R, Lee YL, Wetmur J, et al. Differential expression of imprinted genes in normal and IUGR human placentas. Epigenetics 2009; 4:235 - 240
- McMinn J, Wei M, Schupf N, Cusmai J, Johnson EB, Smith AC, et al. Unbalanced placental expression of imprinted genes in human intrauterine growth restriction. Placenta 2006; 27:540 - 549
- Maccani MA, Marsit CJ. Epigenetics in the placenta. Am J Reprod Immunol 2009; 62:78 - 89
- Rahnama F, Shafiei F, Gluckman PD, Mitchell MD, Lobie PE. Epigenetic regulation of human trophoblastic cell migration and invasion. Endocrinology 2006; 147:5275 - 5283
- Rahnama F, Thompson B, Steiner M, Shafiei F, Lobie PE, Mitchell MD. Epigenetic regulation of E-cadherin controls endometrial receptivity. Endocrinology 2009; 150:1466 - 1472
- Serman L, Vlahovic M, Sijan M, Bulic-Jakus F, Serman A, Sincic N, et al. The impact of 5-azacytidine on placental weight, glycoprotein pattern and proliferating cell nuclear antigen expression in rat placenta. Placenta 2007; 28:803 - 811
- Novakovic B, Sibson M, Ng HK, Manuelpillai U, Rakyan V, Down T, et al. Placenta-specific methylation of the vitamin D 24-hydroxylase gene: implications for feedback autoregulation of active vitamin D levels at the fetomaternal interface. J Biol Chem 2009; 284:14838 - 14848
- Bourque DK, Avila L, Penaherrera M, von Dadelszen P, Robinson WP. Decreased placental methylation at the H19/IGF2 imprinting control region is associated with normotensive intrauterine growth restriction but not preeclampsia. Placenta 2010; 31:197 - 202
- Tabano S, Colapietro P, Cetin I, Grati FR, Zanutto S, Mando C, et al. Epigenetic modulation of the IGF2/H19 imprinted domain in human embryonic and extra-embryonic compartments and its possible role in fetal growth restriction. Epigenetics 2010; 5:313 - 324
- Koestler DC, Marsit CJ, Christensen BC, Karagas MR, Bueno R, Sugarbaker DJ, et al. Semi-supervised recursively partitioned mixture models for identifying cancer subtypes. Bioinformatics 2010; 26:2578 - 2585
- Bibikova M, Fan JB. GoldenGate assay for DNA methylation profiling. Methods Mol Biol 2009; 507:149 - 163
- Einstein F, Thompson RF, Bhagat TD, Fazzari MJ, Verma A, Barzilai N, et al. Cytosine methylation dysregulation in neonates following intrauterine growth restriction. PLoS One 2010; 5:8887
- Guillomot M, Taghouti G, Constant F, Degrelle S, Hue I, Chavatte-Palmer P, et al. Abnormal expression of the imprinted gene Phlda2 in cloned bovine placenta. Placenta 2010; 31:482 - 490
- Breton CV, Byun HM, Wenten M, Pan F, Yang A, Gilliland FD. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am J Respir Crit Care Med 2009; 180:462 - 467
- Daelemans C, Ritchie ME, Smits G, Abu-Amero S, Sudbery IM, Forrest MS, et al. High-throughput analysis of candidate imprinted genes and allele-specific gene expression in the human term placenta. BMC Genet 2010; 11:25
- Pozharny Y, Lambertini L, Ma Y, Ferrara L, Litton CG, Diplas A, et al. Genomic loss of imprinting in first-trimester human placenta. Am J Obstet Gynecol 2010; 202:391
- Guilleret I, Osterheld MC, Braunschweig R, Gastineau V, Taillens S, Benhattar J. Imprinting of tumor-suppressor genes in human placenta. Epigenetics 2009; 4:62 - 68
- Charalambous M, Cowley M, Geoghegan F, Smith FM, Radford EJ, Marlow BP, et al. Maternally-inherited Grb10 reduces placental size and efficiency. Dev Biol 2010; 337:1 - 8
- Lee K, Shim S, Kang K, Kang J, Park D, Kim S, et al. Global gene expression changes induced in the human placenta during labor. Placenta 2010; 31:698 - 704
- Avila L, Yuen RK, Diego-Alvarez D, Penaherrera MS, Jiang R, Robinson WP. Evaluating DNA methylation and gene expression variability in the human term placenta. Placenta 2010; 31:1070 - 1077
- Liang XH, Zhao ZA, Deng WB, Tian Z, Lei W, Xu X, et al. Estrogen regulates amiloride-binding protein 1 through CCAAT/enhancer-binding protein-beta in mouse uterus during embryo implantation and decidualization. Endocrinology 2010; 151:5007 - 5016
- Fant M, Weisoly DL, Cocchia M, Huber R, Khan S, Lunt T, et al. PLAC1, a trophoblast-specific gene, is expressed throughout pregnancy in the human placenta and modulated by keratinocyte growth factor. Mol Reprod Dev 2002; 63:430 - 436
- Lappas M, Lim R, Riley C, Menon R, Permezel M. Expression and localisation of FoxO3 and FoxO4 in human placenta and fetal membranes. Placenta 2010; 31:1043 - 1050
- Brenkman AB, van den Broek NJ, de Keizer PL, van Gent DC, Burgering BM. The DNA damage repair protein Ku70 interacts with FOXO4 to coordinate a conserved cellular stress response. FASEB J 2010; 24:4271 - 4280
- Petraglia F, Coukos G, Volpe A, Genazzani AR, Vale W. Involvement of placental neurohormones in human parturition. Ann NY Acad Sci 1991; 622:331 - 340
- Yen SS. The placenta as the third brain. J Reprod Med 1994; 39:277 - 280
- Yen SS. Yen SS, Jaffe RB. Endocrine-metabolic adaptions in pregnancy. Reproductive Endocrinology 1991; Philadelphia WB Saunders 936 - 970
- Lester BM, Padbury JF. Third pathophysiology of prenatal cocaine exposure. Dev Neurosci 2009; 31:23 - 35
- Power ML, Schulkin J. Functions of corticotropin-releasing hormone in anthropoid primates: from brain to placenta. Am J Hum Biol 2006; 18:431 - 447
- Bajoria R, Babawale M. Ontogeny of endogenous secretion of immunoreactive-thyrotropin releasing hormone by the human placenta. J Clin Endocrinol Metab 1998; 83:4148 - 4155
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009; 41:178 - 186
- Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet 2009; 5:1000602
- Wang HY, Wang RF. Regulatory T cells and cancer. Curr Opin Immunol 2007; 19:217 - 223
- Mulero-Navarro S, Esteller M. Epigenetic biomarkers for human cancer: the time is now. Crit Rev Oncol Hematol 2008; 68:1 - 11
- Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 2010; 17:13 - 27
- Zimmer EZ, Divon MY. Sonographic diagnosis of IUGR-macrosomia. Clin Obstet Gynecol 1992; 35:172 - 184
- Fenton TR. A new growth chart for preterm babies: Babson and Benda's chart updated with recent data and a new format. BMC Pediatr 2003; 3:13
- Houseman EA, Ryan LM, Coull BA. Cholesky residuals for assessing normal errors in a linear model with correlated outcomes. J Amer Stat Assoc 2004; 99:383 - 394
- Marsit CJ, Houseman EA, Christensen BC, Gagne L, Wrensch MR, Nelson HH, et al. Identification of methylated genes associated with aggressive bladder cancer. PLoS One 5:12334
- Poage GM, Christensen BC, Houseman EA, McClean MD, Wiencke JK, Posner MR, et al. Genetic and epigenetic somatic alterations in head and neck squamous cell carcinomas are globally coordinated but not locally targeted. PLoS One 2010; 5:9651
- Houseman EA, Christensen BC, Yeh RF, Marsit CJ, Karagas MR, Wrensch M, et al. Model-based clustering of DNA methylation array data: a recursive-partitioning algorithm for high-dimensional data arising as a mixture of beta distributions. BMC Bioinformatics 2008; 9:365
- Marsit CJ, Christensen BC, Houseman EA, Karagas MR, Wrensch MR, Yeh RF, et al. Epigenetic profiling reveals etiologically distinct patterns of DNA methylation in head and neck squamous cell carcinoma. Carcinogenesis 2009; 30:416 - 422
- Christensen BC, Houseman EA, Godleski JJ, Marsit CJ, Longacker JL, Roelofs CR, et al. Epigenetic profiles distinguish pleural mesothelioma from normal pleura and predict lung asbestos burden and clinical outcome. Cancer Res 2009; 69:227 - 234
- Christensen BC, Marsit CJ, Houseman EA, Godleski JJ, Longacker JL, Zheng S, et al. Differentiation of lung adenocarcinoma, pleural mesothelioma and nonmalignant pulmonary tissues using DNA methylation profiles. Cancer Res 2009; 69:6315 - 6321
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005; 102:15545 - 15550