Abstract
Alterations in DNA methylation patterns, both at specific loci and overall in the genome, have been associated with many different health outcomes. In cancer and other diseases, most of these changes have been observed at the tissue level. Data on whether DNA methylation changes in white blood cells (WBC) can serve as a useful biomarker for different health outcomes are much more limited, but rapidly emerging. Epidemiologic studies have reported associations between global WBC methylation and several different cancers including cancers of the colon, bladder, stomach, breast and head and neck, as well as schizophrenia and myelodysplastic syndrome. Evidence for WBC methylation at specific loci and disease risk is more limited, but increasing. Differences in WBC DNA methylation by selected risk factors including demographic (age, gender, race), environmental exposures (benzene, persistent organic pollutants, lead, arsenic, and air pollution), and other risk factors (cigarette smoke, alcohol drinking, body size, physical activity and diet) have been observed in epidemiologic studies though the patterns are far from consistent. Challenges in inferences from the existing data are primarily due to the cross-sectional and small size of most studies to date as well as the differences in results across assay type and source of DNA. Large, prospective studies will be needed to understand whether changes in risk factors are associated with changes in DNA methylation patterns, and if changes in DNA methylation patterns are associated with changes in disease endpoints.
DNA methylation may play an important role in causing disease by silencing genes through hypermethylation or activating genes through hypomethylation.Citation1–Citation7 In addition to gene-specific DNA methylation, lower genome-wide aberrant DNA methylation (also referred to as global methylation) in regions that are normally methylated, such as repeats or transposable elements can lead to genomic instability and altered gene transcription impacting normal growth and development in target tissues. Although a large literature exists on DNA methylation changes at the tissue level, less is known about whether DNA methylation measured in WBC can be used as an intermediate biomarker for different health outcomes. As blood biomarkers can be measured repeatedly over time in epidemiologic studies, they can be very useful to understand how disease risk changes over the life course. Several requirements need to be met when evaluating whether a potential biomarker is useful for over time monitoring. First, the intermediate marker needs to be associated with the endpoint of interest. Second, exposures that logically precede the measurement of the biomarker should be correlated with the marker. Third, changes in the exposure levels should result in changes in the intermediate biomarker. Fourth, changes in the intermediate biomarker should be associated with change in the endpoint. Here, we review whether WBC DNA methylation fulfills the requirements to be used as a potential biomarker in human epidemiology studies, specifically focusing on whether the studies to date suggest that risk factors are associated with WBC DNA methylation.
A number of methods are available for the analysis of CpG methylation levels.Citation8 Overall, levels of 5-methyl-cytosine (5-mC) can be quantified by enzymatic hydrolysis of the DNA, chromatographic separation of the nucleosides, and analysis by HPLC or MS. A methyl acceptor assay that is dependent upon the ability of a bacterial enzyme to specifically methylate unmethylated Cs in CpG sites using [3H] S-adenosylmethionine has also been used extensively (referred to here as the [3H]-methyl acceptor assay). The Luminometric Methylation Assay (LUMA) targets all CCGG sequences in the genome by using methylation-sensitive restriction enzymes to discriminate methylated and unmethylated DNA followed by pyrosequencing. Repetitive element sequences, such as Alu, LINE-1 and Sat2, have also been analyzed in a number of studies, as a surrogate for genome-wide methylation levels; such analysis requires bisulfite conversion of cytosine to uracil followed by PCR. In the pyrosequencing analysis of repetitive elements, the PCR step amplifies a given region using primers outside the target sequence containing CpG sites so that both methylated and unmethylated sequences are amplified. This is followed by pyrosequencing of the region of interest. For methylation specific assays such as MethyLight, a Taqman quantitative real-time PCR assay is performed, using primers and probes that are methylation specific and require all CpG sites in the region to be methylated for amplification to occur. To assay gene specific methylation, MethyLight and pyrosequencing are the most commonly used methods in large human population studies. However, studies employing array methodologies are beginning to emerge as well.Citation9–Citation11
WBC Methylation and Disease
Alterations in DNA methylation patterns, both at specific loci and overall genomic level, have been associated with many different cancers.Citation12 These alterations include hypermethylation of tumor suppressor genes, hypomethylation of oncogenes, and overall lower levels of global methylation, and have primarily been observed at the tissue level. More recently, DNA methylation changes have also been observed in plasma as a marker of circulating tumor DNACitation13. Less is known about changes in WBC DNA methylation levels and cancer risk, although studies are rapidly emerging. Specifically, at least eight studies measuring overall WBC global DNA methylation in different cancer types including colon,Citation14,Citation15 bladder,Citation16–Citation18 stomach,Citation19 breast,Citation20 and head and neck cancerCitation21 have found an elevated risk for cancer between those in the lowest quantile of global DNA methylation compared to those in the highest quantile of risk; the association was statistically significant in six of the studies (see for summary). The few studies that exist investigating gene-specific methylation in WBC and cancer risk have mostly focused on selected genes for breast cancer and colon cancer but also support the potential for gene-specific methylation measured in WBC as a biomarker of riskCitation22–Citation27 (see for summary). However, data are still very limited and systematic and unbiased approaches to the selection of genes have not been routinely employed. The first paper using Illumina Infinium HumanMethylation27 arrays that interrogate 27,000 CpG sites to differentiate ovarian cancer cases from controls suggests the potential of this approach as a tool for detecting or predicting certain cancers.Citation11
While research in this field is at an early stage, increasing evidence is accumulating on the role of epigenetics, including DNA methylation (genome-wide hypomethylation and gene-specific hypo- and hypermethylation) as well as histone modifications, in numerous other diseases in addition to cancer, including autoimmunity, asthma and neurologic diseases. For example, alterations in gene expression impacted through epigenetic mechanisms can lead to the development of autoreactive lymphocytes in systemic lupus erythematosus, synoviocyte proliferation in rheumatoid arthritis or neural demyelination in multiple sclerosis (reviewed in ref. Citation28). Studies in asthma are also leading to a new understanding of the role of environmental exposures in altering DNA methylation as well as histone modifications (reviewed in ref. Citation29). Twin studies in Alzheimer disease support the notion that epigenetic mechanisms mediate risk (reviewed in ref. Citation30). However, since many studies of gene-specific methylation are performed on brain autopsy samples, it is not clear whether the observed epigenetic changes actually represent a cause or a consequence of the disease. Studies on WBC DNA methylation and other disease points are more limited but are starting to emerge with respect to Alzheimers's disease, schizophrenia and myelodysplastic syndrome.Citation30–Citation32
Associations Between WBC DNA Methylation and Epidemiologic Risk Factors
Epidemiologic studies have investigated the association between demographic, environmental and behavioral risk factors with both WBC global and gene-specific DNA methylation. These studies are summarized in and , respectively. In this review we searched for all articles published to date on each respective risk factor and WBC DNA methylation measured in human populations. We did not include studies that investigated DNA methylation in tissue.
Demographic factors.
Age. Growing evidence supports that WBC global DNA methylation, particularly in blood, changes with age.Citation9,Citation33,Citation34 In the two studies directly assessed the percentage of 5-mC content, a statistically significant decrease of 5-mC with older age was observed in one,Citation33 but not in the other.Citation18 Age was also significantly associated with lower global methylation measured by the [3H]-methyl acceptor assay.Citation35 Studies have correlated age with two of the repetitive elements, LINE-1 and Alu. Although one study correlated lower levels of LINE-1 with increasing age,Citation36 most of the studies did not find an age-dependent effect on blood LINE-1 methylation.Citation17,Citation21,Citation37–Citation41 In contrast, all of the studies investigating age and Alu methylation supported a lower level of DNA methylation of this repetitive element with increasing age.Citation34,Citation36,Citation37,Citation41 Gene-specific methylation has been correlated with age in some studies ().Citation34,Citation42 However, studies looking across the genome have found infrequent age-dependent changes in CpG island methylation.Citation43–Citation45
The overall impact of age may account for a small proportion of inter-individual variation.Citation33,Citation36,Citation37 Inter-individual variability in methylation of repetitive elements has been reported to be in the range 5–25%.Citation46 Using information from monozygotic twin pairs, significant differences in total 5-mC content in WBC was found among older twins, suggesting environmental exposure throughout the life course might have a large impact on the decline in global DNA methylation over time.Citation34 Measurement of within-individual changes in WBC DNA methylation by LUMA on 111 individuals after 10 years found both increased and decreased methylation, with over half exhibiting >5% change.Citation9 The CpG array data on a subset of the same subjects suggested that most sites lost methylation over time.Citation9 This study also supported familial clustering in changes in DNA methylation, which indicates that both environmental and genetic factors are determinants of DNA methylation patterns.Citation9 Evidence supporting a change in DNA methylation with time has led some to suggest that smoking, diet, and other epidemiologic risk factors may influence these epigenetic changes.Citation47–Citation50
Gender. Many studies have found that global DNA methylation was higher in males than in females.Citation17,Citation21,Citation33,Citation37,Citation38,Citation39,Citation46 For example, total 5-mC content in WBC was higher in males than females in one study,Citation33 but this result was not replicated in a larger study measuring 5-mC.Citation18 Of the repetitive elements, LINE-1 has been investigated more frequently and was associated with lower levels in females compared to males in mostCitation17,Citation21,Citation37–Citation39,Citation46 but not all studies.Citation41 In contrast, there has been no consistent pattern associated with Alu methylation.Citation37,Citation41,Citation46
A genome-wide study of DNA methylation profile among twins and healthy singletons independently found that all CpG sites on the X chromosome were highly methylated in blood DNA from females, consistent with X-inactivation.Citation44,Citation51 A gender-dependent difference in gene-specific methylation in blood has also been reported for CALCA, MGMT, MTHFR, MAOA, DRD4, SERT and F8.Citation42,Citation46,Citation52 Folate is essential for erythrocyte formation and development and, because menstruation regularly depletes their supply of erythrocytes, females may tend to have lower levels of circulating folate,Citation21 potentially explaining some differences in gender levels of DNA methylation.
Race and ethnicity. Only a few studies have examined the association between race/ethnicity and DNA methylation in WBC.Citation21,Citation39,Citation53,Citation54 Among the repetitive elements, only LINE-1 has been investigated, with whites having higher levels of LINE-1 methylation compared with Hispanic and non-Hispanic black groups in one study,Citation39 and the opposite finding in another study.Citation21
Environmental factors.
Several studies in humans have established an association between DNA methylation and environmental factors, including benzene, persistent organic pollutants, lead, arsenic and air contaminants.
Benzene. Low-dose benzene exposure was associated with decreased methylation of LINE-1 and Alu sequences, and with hypermethylation of p15 and hypomethylation of MAGE-1.Citation55 In 11 patients with benzene poisoning, the average methylation level of p16, but not of p15, was higher than in a control group.Citation56
Persistent organic pollutants. Two studies evaluated the relation between plasma persistent organic pollutant (POP) concentrations and WBC global DNA methylation.Citation41,Citation57 In 70 Greenlandic Inuit, a population presenting some of the highest reported levels of POP worldwide, a significant inverse linear relationship was found for DDT, DDE, β-hexachlorocyclohexane, oxychlordane, α-chlordane, mirex, several PCBs and the sum of all persistent organic pollutants with Alu methylation.Citation57 However, as the exposure levels in Greenlandic Inuit are much higher than those in the US general population, it is unclear whether the findings apply to other populations with lower exposures. A study in KoreaCitation41 found that most organochlorine (OC) pesticides were inversely and significantly associated with Alu methylation. The strongest OC pesticide associations with methylation were observed with oxychlordane, trans-nonachlor and DDE. Most PCBs and PBDEs showed non-significant inverse trends with Alu methylation. In this same study, POPs were not associated with LINE-1 methylation.Citation41
Lead. Prenatal maternal lead exposure, measured as maternal tibia and patella lead, was inversely associated with genomic WBC methylation (Alu and LINE-1) in 103 umbilical cord samples from a birth cohort in Mexico City.Citation58 Patella, tibia and blood lead were measured in 517 male participants of the Normative Aging Study and an inverse association was found between patella lead levels and LINE-1 methylation, but not with Alu methylation.Citation59
Arsenic. Using the methyl acceptor assay, an unexpected dose-dependent hypermethylation of WBC DNA was found in Bangladeshi adults with chronic arsenic exposure.Citation35 This effect was modified by folate, suggesting that arsenic-induced increases in DNA methylation were dependent upon methyl availability. Arsenic has been associated with gene-specific hypermethylation of p53 and p16 promoter regions in blood DNA of subjects exposed to toxic level of arsenic compared to controls.Citation60 Hypermethylation of the p16 promoter was also observed in WBC DNA from 103 patients with arseniasis compared to 110 healthy subjects.Citation61
Air pollution. The effects of particulate matter (PM) exposure on Alu, LINE-1 and gene-specific methylation was examined in steel plant workers with well-characterized exposure to PM with aerodynamic diameters <10 µm (PM10).Citation62 Long-term exposure to PM10 was negatively associated with methylation in both Alu and LINE-1.Citation62 Exposure to black carbon (BC), a marker of traffic particles, was also associated with decreased DNA methylation in LINE-1.Citation63 Another study in the same cohort looked at the association for six months of exposure to air pollution (PM2.5 and two components, BC and sulfate) and DNA methylation. Prolonged exposure to BC and sulfate particles was associated with hypomethylation of both LINE-1 and Alu repetitive elements.Citation64NOS2 promoter methylation was significantly lower in post-exposure blood samples compared to baseline.Citation62
Behavioral factors.
Smoking. The cellular modifications resulting from exposures to the chemicals present in cigarette smoke have been widely investigated and include DNA adducts, gene mutations, micronuclei, chromosome aberrations, sister chromatin exchanges and DNA strand breaks (reviewed in ref. Citation65). It has also been proposed that DNA methylation changes could result from this exposure. The mechanisms through which smoking might lead to epigenetic changes are not clearly understood. Some research has shown that smokers have lower levels of blood folate than non-smokers.Citation66 Additionally, DNA methyltransferases have been found to bind DNA at sites of DNA damage, which results in altered methylation patterns on these regions, suggesting another molecular mechanism for the generation of aberrant DNA methylation by exposures to chemicals such as those present in cigarettes.Citation67 Studies looking at genomic DNA methylation levels have not found an association between cigarette smoking and variations in different global DNA methylation measurements. Only a few studies have investigated overall 5-mC content and smoking and found no difference by cigarette smoking patterns.Citation18,Citation20,Citation68 More research has focused on the effect of smoking on the methylation levels of repetitive elements. LINE-1, Alu and AluYb8 methylation did not differ between current and never/former smokers.Citation17,Citation21,Citation37,Citation41,Citation57,Citation69 Some studies recruited small number of subjects; however, the overall consistency of the results suggests that genomic DNA methylation levels are not affected by active smoking exposure in adulthood.
Research focusing on DNA methylation at specific genomic regions has found differential effects of smoking status on these epigenetic marks. A discovery study looked at blood DNA of smokers and non-smokers using the Illumina Infinium HumanMethylation27 array, allowing for the identification of specific loci that showed significant differences between smokers and non-smokers.Citation10 Using two different analytical methods, the investigators isolated F2RL3, a gene that encodes thrombin protease-activated receptor-4 (PAR-4), as the most relevant epigenetic modification resulting from smoking exposure. In the same study, they were able to replicate this finding in a different population. Along with PAR-1, PAR-4 is essential in thrombin-mediated platelet aggregation and overall maintenance of vascular integrity.Citation70 This locus had not been previously studied in relation to smoking and its discovery can be a useful step in identifying proteins involved in smoking induced molecular changes. In a study looking at differences between male and female smokers, analysis of methylation of the MAOA 5′ untranslated region in lymphocytic cell lines (LCL) and blood from smokers and non-smokers found that while males had lower levels of LCL methylation at this locus, females only showed a difference in blood.Citation71 In a study of African-Americans, COMT promoter methylation levels did not differ by smoking status. However, in a more detailed analysis statistically significant differences were uncovered for position -193 of this promoter. To further understand this association, the authors replicated the study in another set of individuals finding consistent results.Citation72
It has been suggested that chemicals affecting epigenetic marks could have a larger impact when exposure occurs in utero or earlier in life, a time when epigenetic modifications are being established. Overall levels of genomic DNA methylation have been investigated in several studies in children and adults. Prenatal exposure to cigarette smoke has been associated with an increase in the overall blood levels of DNA methylation in adulthood.Citation53 Other studies have focused on individual repetitive element methylation. LINE-1, Sat2 and Alu methylation was measured in adults and children prenatally exposed to smoking. LINE-1 and Sat2 levels were lower in exposed individuals when compared to unexposed ones.Citation73 However, other elements such as LINE-1 do not seem to be affected by exposure to cigarette smoke. Specific sequences have also been identified in association with in utero smoking exposure. The 5′ untranslated region of the BDNF promoter was hypermethylated in a group of exposed young adults.Citation74,Citation75 Several genes were investigated in blood samples from adults in a cohort of individuals born preterm that were exposed to smoking, with lower levels found for IGF2 methylation. However, no associations were found for GNASAS, INSIGF and LEP.Citation75 Similarly, environmental tobacco smoke (ETS) exposure in childhood has been associated with methylation changes in adult life. ETS exposure was found to be related to an increased methylation in Sat2.Citation73 It is important however to consider that in utero and childhood ETS exposure information is collected retrospectively in most studies, which can limit the validity of these findings.
Alcohol. Alcohol consumption may decrease DNA methylation in hepatic tissue by disrupting folate metabolism and/or methionine synthesis, which might decrease levels of the universal methyl donor S-adenosylmethionine (SAM).Citation76 Excessive alcohol consumption has been shown to alter DNA methylation in humans. Compared with controls, patients suffering from alcoholism had 8–10% higher methylation in CCGG sequences.Citation77,Citation78 Associations between alcohol drinking and repetitive element DNA methylation levels have shown inverse associations between Alu and alcohol drinking.Citation37 However, several studies did not find an association between alcohol consumption and LINE1 methylation.Citation21,Citation37,Citation41,Citation79 Gene-specific DNA methylation has not been widely investigated in association with alcohol consumption. One study found a significant increase in HERP (homocysteine-induced ER protein) promoter methylation in patients with alcohol dependence.Citation78
Body mass index. Studies investigating the association between DNA methylation and body mass index (BMI) are limited to measures of repetitive element methylation. A large study carried out by Zhu et al. found that in WBC DNA, LINE-1 and Alu methylation levels were not correlated with BMI.Citation37 Similar results have been reported in two other studies, one investigating LINE-1 and Alu levels and another one looking at LINE-1 only.Citation41,Citation79 In contrast, in a study of women of child bearing age, Piyathilake et al. found that higher BMI was associated with lower LINE-1 methylation.Citation80
Physical activity. Only one study has investigated the association between physical activity and DNA methylation. Using a cross-sectional design, Zhang and colleagues reported a trend of higher levels of LINE-1 methylation with higher levels of physical activity.Citation79
Dietary folate. Folate is required for the synthesis of methionine, a precursor of the universal methyl donor SAM. SAM provides methyl groups in a variety of biochemical reactions, including methylation of DNA. One would expect that a deficiency of methyl groups in the diet might lead to hypomethylation of DNA in several tissues, including blood. However, associations between global DNA methylation in blood and dietary folate and plasma levels of folate have not been generally detected in observational studies. Serum folate, red blood cell 5-methyltetrahydrofolate, red blood cell non-methylfolate and red blood cell total folate were not associated with 5-mC content in blood.Citation81 Another study did not observe any association between 5-mC content in blood and nutrients including folate, vitamin B1, B2, B3, B12 and total protein.Citation18 Individuals with lower dietary folate showed no difference in LINE-1 methylation compared with individuals with higher dietary folate intake.Citation21,Citation79 Although there is little data to date investigating dietary factors and gene-specific WBC methylation, there was an intriguing report studying maternal diet, as measured by exposure to famine, and gene-specific methylation patterns in IGF2.Citation82 Although this study investigated overall maternal undernutrition, the association was robustly tested using both time and sibling controls and provides evidence that it may be important to consider the timing of exposures when examining DNA methylation patterns.
Studies Examining Changes in WBC DNA Methylation after Risk Factor Changes
Given the limitations arising from drawing inferences from cross-sectional studies, reports that investigate whether changes in exposures are related to changes in DNA methylation patterns are particularly compelling. For example, a randomized estrogen replacement therapy trial in postmenopausal women found that conjugated equine estrogen was associated with a statistically significant increase in percentage of 5-mC in peripheral mononuclear cells compared with women provided with placebo,Citation83 though there was no difference in promoter methylation in the ERα, ERβ and p16.Citation83
Changes in global WBC methylation from folate intake have been more frequently investigated in controlled feeding or randomized placebo-controlled intervention trials with seven studies reported, primarily using the [3H]-methyl acceptor assay.Citation54,Citation84–Citation89 The controlled feeding studies start with a folate depletion diet of around 120 µg/day for, most frequently, 7 weeks. This is then followed by high doses ranging up to 800 µg/day. In the five controlled feeding studies, only one found a significant decrease in WBC methylation during the depletion phase (See for summary). During supplementation, two studies found an increase in methylation.Citation84,Citation87 Two double-blind randomized trials used doses as high as 2,000 µg/day, but no significant change was observed.Citation85,Citation88 These studies are limited by the relatively small number of subjects, which ranged from 8 to 61, and the relatively short time frames of the trials.
Methodological Challenges
The main challenge in making inferences from studies examining risk factor associations and DNA methylation is that most of the studies are cross-sectional in nature and mainly focus on risk factors later in life. With cross-sectional studies it is difficult to understand the direction of the association and causality cannot be determined. Studies focusing on early life factors such as prenatal smoke exposures and prenatal famine exposure are few but intriguing, in that they suggest that some early life factors may have persistent associations with global and gene-specific methylation patterns in adulthood.Citation53,Citation73,Citation74,Citation75,Citation82 Prospective studies, like the folate intervention studies described above, which can investigate a contrast or change in exposure and its association with a change in DNA methylation levels, can provide particularly compelling data. Another challenge with most of the existing studies is that many of them are small in sample size, which limits the ability to examine effect modification with any precision. For example, MTHFR 677TT has been investigated as a potential modifier between folate and DNA methylation, but the results were not consistent.Citation87,Citation89 These studies point to the importance of considering potential modifying effects of genes in one-carbon metabolic pathways when investigating the effect of folate on DNA methylation, but also reveal the need for studies of sufficient sample size as well as a more systematic approach to the evaluation of gene-environment interactions when considering risk factor associations with DNA methylation levels.
Source of WBC
Using data from multiple DNA sources from the same individuals, we have observed variation in genomic DNA methylation within specific WBC types.Citation90 Given the variation in function and gene expression levels of specific WBC types, it is not surprising the both gene-specific and global methylation levels vary by type. This complicates investigating DNA methylation in WBC. It is well known that there are differences in total WBC counts in healthy individuals, with a range of 5,000–10,000/µl, but also differences in cell populations. No association was found between the total number of WBC and either Alu or LINE-1 methylation, or between the differential blood count and Alu methylation.Citation37 Percent neutrophils and lymphocytes exhibited a positive and negative association with LINE-1 methylation, respectively.Citation37 It is also clear that during acute infections or inflammation and in cancer, an abnormally large increase in the number of WBCs in blood (leukocytosis) is observed. Other illnesses, such as neutrophilia, lymphocytosis and granulocytosis, target specific types of WBC. Drugs such as NSAIDs can also cause leukocytosis. Exercise has impacts on neutrophilia,Citation91 while a meta-analysis found that depression and stress (e.g., a standard life event scale for naturalistic stressors or an experimental stressor) affects cell numbers and ratios.Citation92 Cancer patients also demonstrate alterations in specific cell types. For example, neutrophil and lymphocyte counts were elevated and reduced respectively in advanced stage uterine cervical cancer.Citation93 MartinCitation94 suggested that definitive analyses require specific methodologies to account for shifts in cellular population heterogeneity.
Most studies of DNA methylation in blood have been carried out using total WBC isolated from the simple centrifugation of blood and collection of the “buffy coat”, since it is the easiest process and provides the greatest amount of DNA. Centrifugation of blood over a density gradient allows the separation of the more abundant granulocytes (∼85% of total WBC) from the mononuclear cells, which are comprised of monocytes and lymphocytes. Many publications refer to the mononuclear cell fraction as lymphocytes even though they contain ∼15% monocytes. The isolation of pure lymphocytes or specific lymphocyte subpopulations, requires additional steps, such as flow cytometric separation using cell surface receptor antibody binding, and is thus rarely, if ever, done in epidemiologic studies. Given the lack of cell purification in most studies and the variation in cell populations in individual samples, care should be taken in interpreting results.
Overall Summary
Epidemiologic studies investigating associations between WBC DNA methylation and different disease endpoints suggest the potential for measuring WBC DNA methylation as a biomarker of risk. Nevertheless, many studies have been retrospective in nature and require confirmation in prospective designs to ensure that the biomarker is not influenced by the disease process. A number of risk factors have been associated with WBC DNA methylation both globally and gene-specifically, although few consistent patterns have emerged. Interestingly, when patterns exist with a given risk factor and DNA methylation levels, they appear to differ across markers (e.g., lower Alu methylation with increasing age and lower LINE-1 methylation in females). Findings that suggest correlations between early life exposure to environmental factors and DNA methylation are intriguing but need to be replicated in larger, prospective studies. Inconsistent patterns arise due to the challenges of interpreting results from different assays and from different sources of DNA. Intervention studies examining whether changes in exposures within individuals are associated with changes in DNA methylation offer a robust way to test specific hypotheses. More data needed to support the use of monitoring biomarkers; there are still no epidemiologic studies examining whether changes in WBC methylation over time are associated with changes in disease endpoints. Large, prospective studies that focus on specific windows of susceptibility will be needed to understand whether DNA methylation patterns measured in WBC represent a useful biomarker on the pathway linking environmental exposures to disease endpoints.
Figures and Tables
Figure 1 Epidemiologic studies investigating the association between WBC global DNA methylation and cancer risk (comparing lowest quantile to highest quantile). Tertiles used are from references Citation14–Citation16 and Citation19–Citation21. Quartiles used are from reference Citation18. Deciles used are from reference Citation17. +, upper 95% confidence limit exceeds 10.
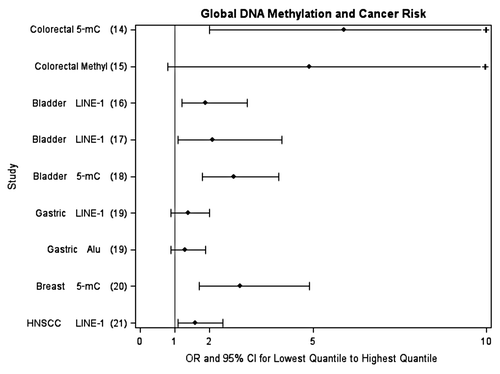
Figure 2 Epidemiologic studies investigating the association between WBC gene-specific DNA methylation and cancer risk. Methylation cutpoints specific to each study, for details see references Citation22–Citation27.
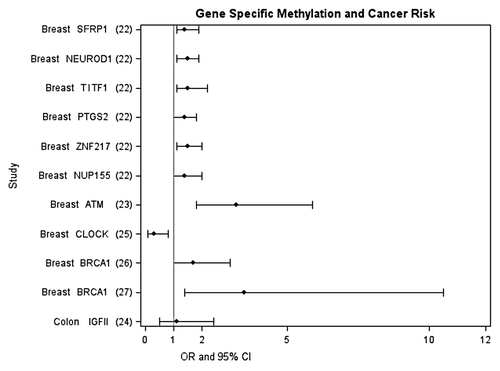
Table 1 Risk factors investigated in studies of WBC DNA global methylation by assay type
Table 2 Risk factors investigated in studies of gene-specific DNA methylation
Table 3 Folate intervention studies
Acknowledgements
We would like to acknowledge research support from the Breast Cancer Research Foundation and NIH grants U01 CA69398, P30 CA13696 and P30 ES009089.
References
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3:415 - 428.
- Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer 2004; 4:143 - 153.
- Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983; 301:89 - 92.
- Tyko B. Genetic and epigenetic mosaicism in cancer precursor tissues. Ann NY Acad Sci 2003; 983:43 - 54
- Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene 2002; 21:5400 - 5413
- Feinberg AP. The epigenetics of cancer etiology. Semin Cancer Biol 2004; 14:427 - 432
- Feinberg AP, Gehrke CW, Kuo KC, Ehrlich M. Reduced genomic 5-methylcytosine content in human colonic neoplasia. Cancer Res 1988; 48:1159 - 1161
- Fraga MF, Esteller M. DNA methylation: a profile of methods and applications. Biotechniques 2002; 33:636 - 649
- Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA 2008; 299:2877 - 2883
- Breitling Lutz P, Yang R, Korn B, Burwinkel B, Brenner H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet 2011; 88:450 - 457
- Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Gayther SA, Apostolidou S, et al. An epigenetic signature in peripheral blood predicts active ovarian Ccancer. PLoS One 2009; 4:8274
- Taby R, Issa JPJ. Cancer epigenetics. CA Cancer J Clin 2010; 60:376 - 392
- Zhang YJ, Wu HC, Shen J, Ahsan H, Tsai WY, Yang HI, et al. Predicting hepatocellular carcinoma by detection of aberrant promoter methylation in serum DNA. Clin Cancer Res 2007; 13:2378 - 2384
- Lim U, Flood A, Choi SW, Albanes D, Cross AJ, Schatzkin A, et al. Genomic methylation of leukocyte DNA in relation to colorectal adenoma among asymptomatic women. Gastroenterology 2008; 134:47 - 55
- Pufulete M, Al-Ghnaniem R, Leather AJM, Appleby P, Gout S, Terry C, et al. Folate status, genomic DNA hypomethylation and risk of colorectal adenoma and cancer: a case control study. Gastroenterology 2003; 124:1240 - 1248
- Cash HL, Tao L, Yuan JM, Marsit CJ, Houseman EA, Xiang YB, et al. LINE-1 hypomethylation is associated with bladder cancer risk among non-smoking Chinese. Int J Cancer 2011;
- Wilhelm CS, Kelsey KT, Butler R, Plaza S, Gagne L, Zens MS, et al. Implications of LINE1 methylation for bladder cancer risk in women. Clin Cancer Res 2010; 16:1682 - 1689
- Moore LE, Pfeiffer RM, Poscablo C, Real FX, Kogevinas M, Silverman D, et al. Genomic DNA hypomethylation as a biomarker for bladder cancer susceptibility in the Spanish Bladder Cancer Study: a case-control study. Lancet Oncol 2008; 9:359 - 366
- Hou L, Wang H, Sartori S, Gawron A, Lissowska J, Bollati V, et al. Blood leukocyte DNA hypomethylation and gastric cancer risk in a high-risk Polish population. Int J Cancer 2010; 127:1866 - 1874
- Choi JY, James SR, Link PA, McCann SE, Hong CC, Davis W, et al. Association between global DNA hypomethylation in leukocytes and risk of breast cancer. Carcinogenesis 2009; 30:1889 - 1897
- Hsiung D, Marsit C, Houseman E, Eddy K, Furniss C, McClean M, et al. Global DNA methylation level in whole blood as a biomarker in head and neck squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev 2007; 16:108 - 114
- Widschwendter M, Apostolidou S, Raum E, Rothenbacher D, Fiegl H, Menon U, et al. Epigenotyping in peripheral blood cell DNA and breast cancer risk: A proof of principle study. PLoS One 2008; 3:2656
- Flanagan JM, Munoz-Alegre M, Henderson S, Tang T, Sun P, Johnson N, et al. Gene-body hypermethylation of ATM in peripheral blood DNA of bilateral breast cancer patients. Hum Mol Genet 2009; 18:1332 - 1342
- Kaaks R, Stattin P, Villar S, Poetsch AR, Dossus L, Nieters A, et al. Insulin-like growth factor-II methylation status in lymphocyte DNA and colon cancer risk in the northern Sweden Health and Disease Cohort. Cancer Res 2009; 69:5400 - 5405
- Hoffman AE, Yi CH, Zheng T, Stevens RG, Leaderer D, Zhang Y, et al. CLOCK in breast tumorigenesis: Genetic, epigenetic and transcriptional profiling analyses. Cancer Res 2010; 70:1459 - 1468
- Iwamoto T, Yamamoto N, Taguchi T, Tamaki Y, Noguchi S. BRCA1 promoter methylation in peripheral blood cells is associated with increased risk of breast cancer with BRCA1 promoter methylation. Breast Cancer Res Treat 2010; 1 - 9
- Wong EM, Southey MC, Fox SB, Brown MA, Dowty JG, Jenkins MA, et al. Constitutional methylation of the BRCA1 promoter is specifically associated with BRCA1 mutation-associated pathology in early-onset breast cancer. Cancer Prev Res 2011; 4:23 - 33
- Brooks WH, Le Dantec C, Pers JO, Youinou P, Renaudineau Y. Epigenetics and autoimmunity. J Autoimmun 2010; 34:207 - 219
- Ho SM. Environmental epigenetics of asthma: An update. J Allergy Clin Immunol 2010; 126:453 - 465
- Chouliaras L, Rutten BPF, Kenis G, Peerbooms O, Visser PJ, Verhey F, et al. Epigenetic regulation in the pathophysiology of Alzheimer's disease. Prog Neurobiol 2010; 90:498 - 510
- Shimabukuro M, Sasaki T, Imamura A, Tsujita T, Fuke C, Umekage T, et al. Global hypomethylation of peripheral leukocyte DNA in male patients with schizophrenia: A potential link between epigenetics and schizophrenia. J Psychiatr Res 2007; 41:1042 - 1046
- Romermann D, Hasemeier B, Metzig K, Gohring G, Schlegelberger B, Langer F, et al. Global increase in DNA methylation in patients with myelodysplastic syndrome. Leukemia 2008; 22:1954 - 1956
- Fuke C, Shimabukuro M, Petronis A, Sugimoto J, Oda T, Miura K, et al. Age related changes in 5-methylcytosine content in human peripheral leukocytes and placentas: An HPLC-based study. Ann Hum Genet 2004; 68:196 - 204
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Nat Acad Sci USA 2005; 102:10604 - 10609
- Pilsner JR, Liu X, Ahsan H, Ilievski V, Slavkovich V, Levy D, et al. Genomic methylation of peripheral blood leukocyte DNA: influences of arsenic and folate in Bangladeshi adults. Am J Clin Nutr 2007; 86:1179 - 1186
- Bollati V, Schwartz J, Wright R, Litonjua A, Tarantini L, Suh H, et al. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev 2009; 130:234 - 239
- Zhu ZZ, Hou L, Bollati V, Tarantini L, Marinelli B, Cantone L, et al. Predictors of global methylation levels in blood DNA of healthy subjects: a combined analysis. Int J Epidemiol 2010; 1 - 10
- El-Maarri O, Walier M, Behne F, van uum J, Singer H, Diaz-Lacava A, et al. Methylation at global LINE-1 repeats in human blood are affected by gender but not by age or natural hormone cycles. PLoS One 2011; 6:16252
- Zhang FF, Cardarelli R, Carroll J, Fulda KG, Kaur M, Gonzalez K, et al. Significant differences in global genomic DNA methylation by gender and race/ethnicity in peripheral blood. Epigenetics 2011; 6
- Chalitchagorn K, Shuangshoti S, Hourpai N, Kongruttanachok N, Tangkijvanich P, Thong-ngam D, et al. Distinctive pattern of LINE-1 methylation level in normal tissues and the association with carcinogenesis. Oncogene 2004; 23:8841 - 8846
- Kim KY, Kim DS, Lee SK, Lee IK, Kang JH, Chang YS, et al. Association of low-dose exposure to persistent organic pollutants with global DNA hypomethylation in healthy Koreans. Environ Health Perspect 2009; 118
- Sarter B, Long TI, Tsong WH, Koh WP, Yu MC, Laird PW. Sex differential in methylation patterns of selected genes in Singapore Chinese. Hum Genet 2005; 117:402 - 403
- Tra J, Kondo T, Lu Q, Kuick R, Hanash S, Richardson B. Infrequent occurrence of age-dependent changes in CpG island methylation as detected by restriction landmark genome scanning. Mech Ageing Dev 2002; 123:1487 - 1503
- Boks MP, Derks EM, Weisenberger DJ, Strengman E, Janson E, Sommer IE, et al. The relationship of DNA methylation with age, gender and genotype in twins and healthy controls. PLoS One 2009; 4:6767
- Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet 2006; 38:1378 - 1385
- El-Maarri O, Becker T, Junen J, Manzoor S, Diaz-Lacava A, Schwaab R, et al. Gender specific differences in levels of DNA methylation at selected loci from human total blood: a tendency toward higher methylation levels in males. Hum Genet 2007; 122:505 - 514
- Foley DL, Craig JM, Morley R, Olsson CJ, Dwyer T, Smith K, et al. Prospects for epigenetic epidemiology. Am J Epidemiol 2009; 169:389 - 400
- Jaenisch R, Hochedlinger K, Blelloch R, Yamada Y, Baldwin K, Eggan K. Nuclear cloning, epigenetic reprogramming and cellular differentiation. Cold Spring Harb Symp Quant Biol 2004; 69:19 - 27
- Bjornsson HT, Fallin MD, Feinberg AP. An integrated epigenetic and genetic approach to common human disease. Trends Genet 2004; 20:350 - 358
- Herceg Z. Epigenetics and cancer: towards an evaluation of the impact of environmental and dietary factors. Mutagenesis 2007; 22:91 - 103
- Shen Y, Matsuno Y, Fouse SD, Rao N, Root S, Xu R, et al. X-inactivation in female human embryonic stem cells is in a nonrandom pattern and prone to epigenetic alterations. Proc Natl Acad Sci USA 2008; 105:4709 - 4714
- Wong CC, Caspi A, Williams B, Craig IW, Houts R, Ambler A, et al. A longitudinal study of epigenetic variation in twins. Epigenetics 2010; 5:516 - 526
- Terry MB, Ferris JS, Pilsner R, Flom JD, Tehranifar P, Santella RM, et al. Genomic DNA methylation among women in a multiethnic New York City birth cohort. Cancer Epidemiol Biomarkers Prev 2008; 17:2306 - 2310
- Axume J, Smith SS, Pogribny IP, Moriarty DJ, Caudill MA. Global leukocyte DNA methylation is similar in African American and Caucasian women under conditions of controlled folate intake. Epigenetics 2007; 2:66 - 68
- Bollati V, Baccarelli A, Hou L, Bonzini M, Fustinoni S, Cavallo D, et al. Changes in DNA methylation patterns in subjects exposed to low-dose Benzene. Cancer Res 2007; 67:876 - 880
- Xing C, Wang Qf, Li B, Tian H, Ni Y, Yin S, et al. Methylation and expression analysis of tumor suppressor genes p15 and p16 in benzene poisoning. Chem Biol Interact 2010; 184:306 - 309
- Rusiecki JA, Baccarelli A, Bollati V, Tarantini L, Moore LE, Bonefeld-Jorgensen EC. Global DNA hypomethylation is associated with high serum-persistent organic pollutants in Greenlandic Inuit. Environ Health Perspect 2008; 116
- Pilsner JR, Hu H, Ettinger A, Sánchez BN, Wright RO, Cantonwine D, et al. Influence of prenatal lead exposure on genomic methylation of cord blood DNA. Environ Health Perspect 2009; 117
- Wright RO, Schwartz J, Wright RJ, Bollati V, Tarantini L, Park SK, et al. Biomarkers of lead exposure and DNA methylation within retrotransposons. Environ Health Perspect 2010; 118
- Chanda S, Dasgupta UB, GuhaMazumder D, Gupta M, Chaudhuri U, Lahiri S, et al. DNA hypermethylation of promoter of gene p53 and p16 in arsenic-exposed people with and without malignancy. Toxicol Sci 2006; 89:431 - 437
- Zhang AH, Bin HH, Pan XL, Xi XG. Analysis of p16 gene mutation, deletion and methylation in patients with arseniasis produced by indoor unventilated-stove coal usage in Guizhou, China. J Toxicol Environ Health A 2007; 70:970 - 975
- Tarantini L, Bonzini M, Apostoli P, Pegoraro V, Bollati V, Marinelli B, et al. Effects of particulate matter on genomic DNA methylation content and iNOS promoter methylation. Environ Health Perspect 2009; 117
- Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua AA, Suh HH, et al. Rapid DNA methylation changes after exposure to traffic particles. Am J Respir Crit Care Med 2009; 179:572 - 578
- Madrigano J, Baccarelli A, Mittleman MA, Wright RO, Sparrow D, Vokonas PS, et al. Prolonged exposure to particulate pollution, genes associated with glutathione pathways and DNA methylation in a cohort of older men. Environ Health Perspect 2011;
- DeMarini DM. Genotoxicity of tobacco smoke and tobacco smoke condensate: a review. Mutat Res 2004; 567:447 - 474
- Piyathilake CJ, Macaluso M, Hine RJ, Richards EW, Krumdieck CL. Local and systemic effects of cigarette smoking on folate and vitamin B-12. Am J Clin Nutr 1994; 60:559 - 566
- Mortusewicz O, Schermelleh L, Walter J, Cardoso MC, Leonhardt H. Recruitment of DNA methyltransferase I to DNA repair sites. Proc Nat Acad Sci USA 2005; 102:8905 - 8909
- Hillemacher T, Frieling H, Moskau S, Muschler MAN, Semmler A, Kornhuber J, et al. Global DNA methylation is influenced by smoking behaviour. Eur Neuropsychopharmacol 2008; 18:295 - 298
- Figueiredo JC, Grau MV, Wallace K, Levine AJ, Shen L, Hamdan R, et al. Global DNA hypomethylation (LINE-1) in the normal colon and lifestyle characteristics and dietary and genetic factors. Cancer Epidemiol Biomarkers Prev 2009; 18:1041 - 1049
- Leger AJ, Covic L, Kuliopulos A. Protease-activated receptors in cardiovascular diseases. Circulation 2006; 114:1070 - 1077
- Philibert RA, Beach SRH, Gunter TD, Brody GH, Madan A, Gerrard M. The effect of smoking on MAOA promoter methylation in DNA prepared from lymphoblasts and whole blood. Am J Med Genet B Neuropsychiatr Genet 2009; 153:619 - 628
- Xu Q, Ma JZ, Payne TJ, Li MD. Determination of methylated CpG sites in the promoter region of catechol-o-methyltransferase (COMT) and their involvement in the etiology of tobacco smoking. Front Psychiatry 2010; 1:16
- Flom J, Ferris J, Gonzalez K, Santella R, Terry MB. Prenatal tobacco smoke exposure and genomewide methylation in adulthood. Cancer Epidemiol Biomarkers Prev 2011; 20:720
- Toledo-Rodriguez M, Lotfipour S, Leonard G, Perron M, Richer L, Veillette S, et al. Maternal smoking during pregnancy is associated with epigenetic modifications of the brain-derived neurotrophic factor-6 exon in adolescent offspring. Am J Med Genet B Neuropsychiatr Genet 2010; 153:1350 - 1354
- Tobi EW, Heijmans BT, Kremer D, Putter H, Delemarre-van d, Waal HA, Finken MJ, et al. DNA methylation of IGF2, GNASAS, INSIGF and LEP and being born small for gestational age. Epigenetics 2011; 6:171 - 176
- Hamid A, Wani NA, Kaur J. New perspectives on folate transport in relation to alcoholism-induced folate malabsorption—association with epigenome stability and cancer development. FEBS J 2009; 276:2175 - 2191
- Bönsch D, Lenz B, Reulbach U, Kornhuber J, Bleich S. Homocysteine associated genomic DNA hypermethylation in patients with chronic alcoholism. J Neural Transm 2004; 111:1611 - 1616
- Bleich S, Lenz B, Ziegenbein M, Beutler S, Frieling H, Kornhuber J, et al. Epigenetic DNA hypermethylation of the HERP gene promoter induces downregulation of its mRNA expression in patients with alcohol dependence. Alcohol Clin Exp Res 2006; 30:587 - 591
- Zhang FF, Cardarelli R, Carroll J, Zhang S, Fulda KG, Gonzalez K, et al. Physical activity and global genomic DNA methylation in a cancer-free population. Epigenetics 2011; 6:293 - 299
- Piyathilake C, Badiga S, Johanning G, Alvarez R, Partridge E. Predictors and health consequences of epigenetic changes associated with excess body weight in women of child-bearing age. Cancer Epidemiol Biomarkers Prev 2011; 20
- Kok RM, Smith DE, Barto R, Spijkerman AM, Teerlink T, Gellekink HJ, et al. Global DNA methylation measured by liquid chromatography-tandem mass spectrometry: analytical technique, reference values and determinants in healthy subjects. Clin Chem Lab Med 2007; 45:903 - 911
- Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA 2008; 105:17046 - 17049
- Simonetta F, Stefania LF, Hyeran J, J SE, Roberto C, Sang-Woon C. Oestrogen replacement therapy reduces total plasma homocysteine and enhances genomic DNA methylation in postmenopausal women. Br J Nutr 2007; 97:617 - 621
- Jacob RA, Gretz DM, Taylor PC, James SJ, Pogribny IP, Miller BJ, et al. Moderate folate depletion increases plasma homocysteine and decreases lymphocyte DNA methylation in postmenopausal women. J Nutr 1998; 128:1204 - 1212
- Fenech M, Aitken C, Rinaldi J. Folate, vitamin B12, homocysteine status and DNA damage in young Australian adults. Carcinogenesis 1998; 19:1163 - 1171
- Rampersaud GC, Kauwell GP, Hutson AD, Cerda JJ, Bailey LB. Genomic DNA methylation decreases in response to moderate folate depletion in elderly women. Am J Clin Nutr 2000; 72:998 - 1003
- Shelnutt KP, Kauwell GPA, Gregory JF, Maneval DR, Quinlivan EP, Theriaque DW, et al. Methylenetetrahydrofolate reductase 677Câ†'T polymorphism affects DNA methylation in response to controlled folate intake in young women. J Nutr Biochem 2004; 15:554 - 560
- Basten GP, Duthie SJ, Pirie L, Vaughan N, Hill MH, Powers HJ. Sensitivity of markers of DNA stability and DNA repair activity to folate supplementation in healthy volunteers. Br J Cancer 2006; 94:1942 - 1947
- Axume J, Smith SS, Pogribny IP, Moriarty DJ, Caudill MA. The MTHFR 677TT genotype and folate intake interact to lower global leukocyte DNA methylation in young Mexican American women. Nutr Res 2007; 27:1365 - 1317
- Wu HC, Delgado-Cruzata L, Flom JD, Kappil M, Ferris JS, Liao Y, et al. Global methylation profiles in DNA from different blood cell types. Epigenetics 2011; 6:76 - 85
- Quindry JC, Stone WL, King J, Broeder CE. The effects of acute exercise on neutrophils and plasma oxidative stress. Med Sci Sports Exerc 2003; 35:1139 - 1145
- Zorrilla EP, Luborsky L, McKay JR, Rosenthal R, Houldin A, Tax A, et al. The relationship of depression and stressors to immunological assays: a meta-analytic review. Brain Behav Immun 2001; 15:199 - 226
- Tavares-Murta BM, Mendonça MA, Duarte NL, da Silva JA, Mutão TS, Garcia CB, et al. Systemic leukocyte alterations are associated with invasive uterine cervical cancer. Int J Gynecol Cancer 2010; 20:1154 - 1159
- Martin GM. Epigenetic drift in aging identical twins. Proc Nat Acad Sci USA 2005; 102:10413 - 10414