Abstract
5-Aza-2'-deoxycytidine (decitabine) is a drug targeting the epigenetic abnormalities of tumors. The basis for its limited efficacy in solid tumors is unresolved, but may relate to their indolent growth, their p53 genotype or both. We report that the primary molecular mechanism of decitabine—depletion of DNA methyltransferase-1 following its “suicide” inactivation—is not absolutely associated with cell cycle progression in HCT 116 colon cancer cells, but is associated with their p53 genotype. Control experiments affirmed that the secondary molecular effects of decitabine on global and promoter-specific CpG methylation and MAGE-A1 mRNA expression were S-phase dependent, as expected. Secondary changes in CpG methylation occurred only in growing cells ~24–48 h after decitabine treatment; these epigenetic changes coincided with p53 accumulation, an index of DNA damage. Conversely, primary depletion of DNA methyltransferase-1 began immediately after a single exposure to 300 nM decitabine and it progressed to completion within ~8 h, even in confluent cells arrested in G1 and G2/M. Our results suggest that DNA repair and remodeling activity in arrested, confluent cells may be sufficient to support the primary molecular action of decitabine, while its secondary, epigenetic effects require cell cycle progression through S-phase.
Introduction
5-Aza-2′-deoxycytidine (decitabine) targets the epigenetic instability of tumors.Citation1,Citation2 When administered at low doses, which favor its inhibition of DNA methyltransferase enzymes (DNMT) over its clastogenic effects, decitabine can slow progression of myelodysplastic syndrome (MDS) and chronic or acute myeloid leukemias (CML, AML).Citation2–Citation4 In contrast, decitabine has poor efficacy against solid tumors.Citation2 The reasons for this are unclear but current hypotheses implicate slower growth rates,Citation2,Citation5,Citation6 more frequent dysregulation of the p53 tumor suppressorCitation7–Citation9 and pleiotropic resistance in solid tumors.Citation10
Decitabine relies on incorporation of its metabolite, 5-aza-2′-deoxycytidine triphosphate (ZdC), into DNA for its efficacy. ZdC is a “suicide” substrate which covalently inactivates DNMT.Citation11–Citation13 Fusion of DNMT1 with ZdC-DNA has two consequences. First, it depletes the DNMT1 enzyme from cells, which leads secondarily to replacement of 5′MeCpG with CpG.Citation11,Citation13 It is axiomatic that replacement of genomic 5′MeCpG with CpG eventually requires S-phase duplication of hemimethylated DNA.Citation14–Citation16 However, we hypothesized that the prerequisite depletion of DNMT1 enzyme might not be replication dependent. Reports that anti-mitotic agents have inconsistent effects on ZdC-mediated disposal of DNMT lend support to this hypothesis. For example, hydroxyurea prevents loss of DNMT in cells exposed to decitabine,Citation11 whereas aphidicolin has no effect,Citation17 or only a partial effect.Citation18 Furthermore, DNMT1 associates with chromatin in G2/M, as well as S-phase of the cell cycle,Citation19 and DNMT1 is recruited to DNA repair sites in arrested cells, where it may play a role in restoring epigenetic information.Citation20 The influence of cell cycle progression on decitabine-mediated disposal of DNMT1 enzyme merits further study, because of its potential implications for treatment of indolent, solid tumors.
We compared the kinetics of DNMT1 depletion in growing, confluent and serum-starved HCT 116 isogenic cell lines that differed with respect to their p53 genotype (wild type versus null). Our results show that the cell cycle dependence of decitabine is not absolute. As expected, its secondary, epigenetic effects were replication dependent—changes in global and promoter specific CpG methylation and re-expression of the somatically silenced MAGE-A1 gene occurred at 24–48 h after a single exposure to 300 nM decitabine. In contrast, its primary effect—DNMT1 enzyme depletion—occurred immediately with elimination half-lives that were unrelated to the fraction of cells in S-phase, but inversely related to [3H] thymidine uptake. Thus, the rates of DNMT1 depletion varied as a function of the metabolic and replication state of the cells, but they were not absolutely cell cycle dependent. A p53 null genotype slowed depletion of DNMT1 by ∼5-fold, in both growing and confluent HCT 116 cells, compared to a p53 wild type genotype. Our results suggest that DNA repair and remodeling activity in arrested, confluent cells may be sufficient to support the primary molecular action of decitabine, while its secondary, epigenetic effects require cell cycle progression through S-phase.
Results
Cell cycle progression is unnecessary for depletion of DNMT 1 enzyme in HCT 116 cells exposed to decitabine.
In growing HCT 116 cells, 0.1–5.0 µM Decitabine caused dose-dependent depletion of DNMT1 enzyme. The half-maximal effect (EC50) occurred with ∼300 nM decitabine, a clinically achievable serum level with minimal adverse effects.Citation22
The time course for depletion of DNMT1 varied as a function of the metabolic and replication state of the cells, but it was not absolutely dependent on cell cycle progression (). Half lives (t½), calculated from immunoblots of DNMT1 enzyme in HCT 116 cells exposed once to 300 nM decitabine showed that depletion was fastest in confluent cells, with t½ = 1.4 h (metabolically active/slow replication, 11% S-phase at the start of experiment); intermediate in the growing cells, with t½ = 2.5 h (metabolically active/fast replication, 29% S-phase at the start of experiment); and slowest in serum-starved cells, with t½ = 10.1 h (metabolically quiescent/slow replication, 12% S-phase at the start of experiment) ( and B). These t½ did not correlate with the fraction of cells in S-phase (), e.g., serum-starved and confluent HCT 116 cells treated once with 300 nM decitabine each had ∼10–12% of cells in S-phase prior to the addition of decitabine, but a 7-fold difference in their DNMT1 half lives—10.1 h versus 1.4 h, respectively. If the responses to decitabine were solely or mainly, attributable to the sub-populations of S-phase cells, one would predict uniform elimination kinetics, coupled with variable levels of DNMT1 enzyme expression at the start of the experiment. However, we found the opposite: the t½ of DNMT1 enzyme depletion varied and the baseline expression of DNMT1 enzyme at t = 0 was equivalent among growing, confluent and serum-starved HCT 116 cells (). There was no detectable recovery of DNMT1 enzyme expression in HCT 116 cells up to 72 h after a single exposure to 300 nM decitabine (data not shown). From the same membranes depicted in there was no change in DNMT3a enzyme levels, but DNMT3b levels showed a moderate drop by 6 h (data not shown). As expected, 6-azacytidine used as a procedural control did not cause depletion of DNMT1 enzyme levels () because its metabolite cannot bond covalently with DNMT1 to generate DNA-DNMT1 adducts.
To affirm that HCT 116 cells expressed DNMT1 at different stages of the cell cycle, not just S-phase, we used flow cytometry to sort the cells and then measured the respective content of DNMT1 enzyme via immunoblots. S-phase and non-S phase cells (G/M) expressed appreciable DNMT1 protein, consistent with reports by othersCitation11,Citation18–Citation20 (Sup. Fig. 1).
Cells import and use nucleosides for processes other than chromosomal duplication during S-phase. For example, DNA repair occurs during cell cycle arrest and DNMT1 is recruited to DNA repair sites in arrested cells.Citation20 Therefore, to supplement our flow cytometry experiments we also measured [3H] thymidine uptake by HCT 116 cells treated with decitabine. Confluent, growing and starved cells assimilated 21, 15 and <5%, respectively, of added [3H]thymidine, during their exposure to 300 nM decitabine. This index of nucleoside utilization correlated inversely (r = −0.99) with the corresponding t½ of 1.4, 2.5 and 10.1 h for DNMT1 depletion.
The p53 tumor suppressor genotype influences the rate of depletion of DNMT 1 in HCT 116 cells exposed to decitabine.
Because the p53 tumor suppressor modulates the cytotoxicity of decitabine in HCT 116 cellsCitation7 we examined its influence on the depletion of DNMT1 in HCT 116 cells. The time course for DNMT1 depletion varied as a function of their p53 genotype in both confluent and growing HCT 116 isogenic cell lines (). Half lives (t½), calculated from immunoblots of DNMT1 enzyme in confluent HCT 116 cells exposed once to 300 nM decitabine showed that depletion was faster in p53 wild type (+/+) cells, with t½ = 1.4 h (metabolically active/slow replication, 11% S-phase at the start of the experiment) and slower in the p53 null (−/−) cells, with t½ = 6.4 h (metabolically active/slow replication, 12% S-phase at the start of the experiment) ( and B). These t½ values did not correlate with the fraction of cells in S-phase (), e.g., confluent cells with p53 (+/+) wild type and p53(−/−) null genotype each had ∼10–12% of cells in S-phase prior to exposure to decitabine, but ∼4.5-fold difference in DNMT1 half-lives. Results were comparable in growing HCT 116 isogenic cells, with ∼4-fold difference in DNMT1 half-lives, i.e., faster depletion with t½ = 2.5 h in growing p53 wild-type (+/+) cells and slower depletion, with t½ = 9.5 h in growing p53 null (−/−) cells.
Epigenetic effects of decitabine in HCT 116 cells are cell cycle dependent and coincide with a p53 DNA damage response.
As a control, we measured the secondary, epigenetic effects of decitabine. As expected, detectable replacement of genomic 5′MeCpG with CpG required replication and S-phase duplication of hemi-methylated DNA in HCT 116 cells exposed to decitabine. At 24–48 h after treatment, genomic DNA methylation and promoter-specific demethylation of the MAGE-A1 gene declined only in growing cells, but not confluent cells (). Likewise, the MAGE-A1 gene was released from “silencing” and its mRNA was expressed only in growing cells, but not in confluent cells (). However, if these confluent HCT 116 cells were released from growth arrest; then allowed to replicate, they too showed declines in genomic DNA methylation and MAGE-A1 promoter methylation, coincident with increased expression of MAGE-A1 mRNA, all of which were quantitatively equivalent to that measured in growing cells ( and ). Evidently, confluent HCT 116 cells could import decitabine and incorporate its metabolite, ZdC, into DNA which caused a prompt, cell cycle independent depletion of DNMT1 enzyme ( and ); followed 24–48 h later—only after they were released from arrest—by cell cycle dependent epigenetic effects ( and ).
In wild type p53(+/+) HCT 116 cells the time course of the p53 response, and index of DNA damage, and the epigenetic effects corresponded closely. Accumulation of p53 protein levels increased by 2-, 2.4- and 3-fold, respectively, at 24, 36 and 48 h post-treatment with decitabine in growing HCT 116 (). There was no accumulation of p53 protein from 0–72 h in confluent HCT 116 cells ().
Discussion
Our results show that depletion of DNMT1 enzyme from HCT 116 cells treated once with 300 nM decitabine is not absolutely dependent on mitosis and cell cycle transit. Expression of DNMT1 mRNA is reportedly greatest in S-phase cells;Citation23 nevertheless, some studies contradict this point, and several others show that DNMT1 enzyme expression is not restricted to S-phase. For instance, DNMT1 mRNA and protein levels did not correlate with proliferation markers in normal and malignant uroepithelial cells;Citation24 and DNMT1 enzyme expression in HCT 116 cells was unchanged after radiation treatment, even though such treatment typically causes cell cycle arrest.Citation18 Our results also support numerous studies that consistently show DNMT1 enzyme expression during G2/M and G1 arrest.Citation11,Citation18–Citation20,Citation25,Citation26 Importantly, our results did not originate solely from the sub-populations of S-phase cells present in growing, confluent or serum starved conditions, before their exposure to decitabine. If expression and subsequent depletion of DNMT1 were attributable to the S-phase cells, one would expect uniform elimination kinetics (similar t½) and different starting levels of DNMT1 enzyme at t = 0 in our experiments. This expectation is opposite that shown in our data ( and ).
While the t½ for DNMT1 protein depletion did not correlate with the proportion of cells in S-phase at the start of the experiment, it did correlate well (r = −0.99) with their uptake of [3H] thymidine, an index of nucleoside import and utilization for DNA repair processes, as well as mitosis during their exposure to decitabine. Fastest depletion of DNMT1 and highest uptake of [3H] thymidine occurred in confluent cells; while slowest depletion of DNMT1 and lowest uptake of [3H] thymidine occurred in serum-starved cells. These results align with the reported downregulation of DNA repair genes by serum deprivation;Citation27 upregulation of DNA repair machinery by confluenceCitation28 and regulation of DNA repair by the p53 tumor suppressor.Citation29 While speculative, a connection between DNA repair processes and decitabine-mediated DNMT1 depletion is compatible with the active recruitment of DNMT1 to DNA repair sites in arrested cells, where it may play a role in restoring epigenetic information.Citation19 DNA repair processes might also factor into the contrasting effects of anti-mitotic agents—hydroxyurea and aphidicolin—on DNMT1 depletion. Hydroxyurea, which inhibits DNA excision repair in non-cycling quiescent cells,Citation30 fully antagonized decitabine-mediated depletion of DNMT1.Citation11 Conversely, aphidicolin, which preferentially inhibits S-phase duplication of DNA, failed to block or only partially blocked, decitabine mediated DNMT1 in HeLaCitation17 and HCT 116 cells.Citation18
Generally, our results and conclusions agree with those of Karpf et al.Citation7 linking the p53 tumor suppressor genotype and decitabine pharmacology; however, they diverge from Jutterman et al.Citation13 and results recently reported by Patel et al.Citation18 The kinetics of p53 accumulation reported by Karpf et al.Citation7 were nearly identical to ours—p53 protein levels rose appreciably by 24 h and continued to rise gradually for 48 () or 120 h.Citation7 Mechanistically, a p53 response could reflect DNA damage derived from DNMT1-DNA adducts;Citation7,Citation13 or genomic demethylation;Citation31 or both. In this regard our results align better with the genomic demethylation hypothesisCitation31 since rapid depletion of DNMT1, without demethylation, in confluent HCT 116 cells did not provoke a detectable p53 response, at least at the 300 nM dose of decitabine we used. However, in growing HCT 116 cells the slow, prolonged p53 response coincided with epigenetic demethylation at 24–48 h, which followed the complete elimination of DNMT1. It is worth noting that Jutterman et al.Citation13 used murine embryonic stem cells, which do not rely on normal p53 checkpoints to manage DNA damage;Citation32 this may have influenced their results and conclusions. While the p53 genotype modulated the rate of DNMT1 depletion in our experiments, it did not do so in experiments recently reported by Patel et al.Citation18 Since we and Patel et al.Citation18 each used isogenic HCT 116 cell lines the differences may relate to our emphasis on depletion kinetics of DNMT1 as a function of the cell cycle, at a single concentration of decitabine; versus their emphasis on dose-response at a single time point. Otherwise, it is difficult to explain this discrepancy. We chose to study decitabine at a concentration of 300 nM for two main reasons. Firstly, 300 nM was the concentration causing half-maximal depletion of DNMT1 in our dose-response experiments. Secondly, it approximates the serum steady-state concentration which had the least adverse effects in human clinical trials.Citation22
Our observation of a relationship between p53 genotype and DNMT1 depletion kinetics is interesting. Ordinarily, cells maintain wild type p53 protein at a low basal level by degrading it at the proteasome. Various stresses, e.g., DNA damage, provoke post-translational modifications of p53 protein that slow its degradation, and enhance its nuclear localization as a homotetramer that modulates transcriptional activation of its target genes. Our results ( and related text) show that the rate of DNMT1 depletion is influenced by the p53 genotype—but in a time frame that precedes any significant induction of p53 protein. Notably, p53-regulated proteins often occur at appreciable basal levels and p53 is responsible for maintaining basal transcription of its downstream genes in some cell types, especially epithelial cell lines and fibroblasts. This has important implications for nucleotide excision repair (NER) because NER normally occurs within ∼3 h after DNA damage. This time frame seldom allows for full induction of p53-regulated genes; instead, basal transcription of p53-regulated genes, in a p53-dependent manner facilitates temporal coupling between the p53 pathway and cellular NER.Citation33 Likewise, the presence or absence of p53 affects DNA polymerase β (β-pol), an enzyme involved in base excision repair (BER) synthesis. In fact, β-pol repair synthesis in vitro was affected by p53, even when p53 was added as a recombinant protein.Citation34 Lastly; recent discoveries show that p53 can modulate cellular stress pathways via mechanisms that do not rely solely on its role as a transcription factor, but on its direct interactions with mitochondria and other cellular proteins.Citation35
In summary, most of the efforts to relate decitabine function with gene re-expression have focused on methylation-dependent processes aloneCitation2,Citation5,Citation22,Citation36–Citation39 which do require S-phase transit. Our results reinforce the hypothesis that the genotype of the p53 tumor suppressor, plus the replication and metabolic status of malignant cells are likely to have complex effects on solid tumor responses to decitabine. DNMT1 and p53 coordinately regulate gene expression via methylation-dependent and -independent pathways. DNMT1 can also directly repress transcription of some genes, thus its depletion might translate into re-expression of some genes, without requiring S-phase transit.
Materials and Methods
Materials.
We used cell culture media and antibiotics (Gibco®, Invitrogen, Carlsbad, CA); fetal bovine serum (FBS) (Hyclone Media, Logan, UT); 5-aza-2′-deoxycytidine and 2-deoxycytidine (Sigma-Aldrich, St. Louis, MO, #A3656 and #D3897 respectively); 6-azacytidine (Developmental Therapeutics Program NCI/NIH, Bethesda, MD); 5-methyl-2-deoxycytidine (#PY7635, Berry & Associates Inc., Dexter, MI); [3H]-thymidine (#MT 846W, Moravek Biochemicals and Radiochemicals Inc., Brea, CA); antibodies to detect DNMT1, DNMT3a and DNMT3b (Novus Biologicals, Littleton, CO, #NB100-264, #NB100-265, #NB100-266, respectively); β-tubulin antibody (Invitrogen, Carlsbad, CA #32-2600); goat anti-human p53 FL393 antibody, donkey anti-rabbit HRP and donkey anti-goat HRP (Santa Cruz Biotechnology, Santa Cruz, CA, #SC-6243, #SC-2077, #SC-2020, respectively); DNeasy®, RNeasy® and Qiaquick® DNA clean up kits (Qiagen, Valencia, CA, #69504, #74104, #28106 respectively); nuclease P and alkaline phosphatase (Sigma #N8630 and #P4252 respectively); HpaII restriction endonuclease (New England Biolabs, Ipswich, MA, #R0171S); 2× LightCycler 480 SYBR Green I Master mix (Roche Applied Science, Indianapolis, IN, #04 707 516 001); first strand cDNA synthesis kit (Fermentas Inc., Glen Burnie, MD, #K611); XTT cell growth assay kit (Roche Applied Science, #11 465 015 001); 8% precast tris-glycine gels (Invitrogen #EC60185); enhanced chemiluminescence (ECL) reagents (Amersham Pharmacia, Piscataway, NJ #RPN2132) and Vybrant DyeCycle Violet (Molecular Probes®, Invitrogen #V35003). Nucleoside analogs were freshly prepared in sterile water before each experiment and the concentrations were determined spectrophotometrically, using the extinction coefficient of ε = 8,767 (Merck Manual). Cell lysis buffer consisted of 50 mM of Tris, pH 7.4, 0.1 M NaCl, 2 mM EDTA, 1% SDS, 1% deoxycholate, 1 mM NaF, 1 mM sodium orthovanadate and 1× complete protease inhibitors (Roche Applied Science, #04 693 124 001).
Cell culture.
HCT116 p53+/+ (ATCC, Manassas, VA, # CCL-247) and HCT 116 p53−/− cellsCitation21 were propagated in McCoy's 5A with penicillin/streptomycin (50 U/ml), 1 mM sodium pyruvate, 2 mM glutamine and 10% v/v FBS at 37°C in a humidified incubator with 5% CO2. Cells were seeded at 3 × 105 or 9 × 105 per well and allowed to grow for 24 h before treatments. These are designated as growing and confluent cells, respectively. Cells were seeded at 6 × 105 per well, allowed to grow for 24 h, then FBS was removed 24 h prior to any treatments. These are designated serum starved cells. Lastly, confluent cells treated with decitabine were allowed 24 h to grow, then trypsinized and seeded again at 9 × 105 per well and allowed to re-grow for the indicated times. These cells are designated confluent-released from arrest. All cells were treated once with 300 nM decitabine, 1 µM 6-azacytidine or phosphate buffered saline (PBS) vehicle. Cells were harvested at various time points shown in the results.
Western immunoblotting.
After treatment with 300 nM decitabine, 1 µM 6-azacytidine or vehicle, HCT 116 cells were washed in ice cold PBS and harvested at 0, 45, 90, 180, 360 min and 12, 24, 48 and 72 h in cell lysis buffer with protease inhibitors. Cells were flash frozen, then allowed to thaw on ice and centrifuged at 17,900× g for 10 min at 4°C. Protein concentration was determined spectrophotometrically. Lysates (15 µg) were fractionated using 8% denaturing tris-glycine gels and transferred onto a polyvinyl difluoride (PVDF) membrane. Membranes were blocked with 5% non-fat dry milk powder in TBST, incubated overnight at 4°C with primary antibody, followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibody (1:4,000). Antigen-antibody complexes were detected with enhanced chemiluminescence (ECL) reagents. Protein band intensities were estimated by scanning the blots with the Kodak Image Station.
Cell cycle analysis.
FACS analysis of the cell cycle in growing, confluent and serum-starved cells was done to determine the relative content of cells in S-phase and G2/M before any exposure to decitabine. 106–107 cells were washed in ice cold PBS, trypsinized, centrifuged at 400× g for 5 min, and re-suspended into a single cell suspension in 1 ml ice cold PBS. Two ml of methanol (−20°C) were slowly added while vortexing. Cells were incubated at 4°C for a minimum of 2 h then centrifuged at 400× g for 5 min. The supernatant was removed and cells were gently resuspended in 1 ml of freshly made 50 µg/ml propidium iodide/10 µg RNase A/0.1% triton-X. Cells were then incubated for 30 min at 25°C in the dark before being analyzed using a cell lab quanta cytometer (Beckman Coulter, Brea, California). Live cells were resuspended in PBS, stained with Vybrant DyeCycle Violet and sorted using a FACSVantage SE Turbo. [3H] Thymidine incorporation by confluent, growing and serum-starved cells was also measured by washing cells, extracting their DNA and quantifying [3H]-thymidine via scintillation counting. In contrast to the FACS analysis, the measurement of [3H]-thymidine uptake was done in cells exposed to decitabine. When [3H]thymidine and decitabine were added together the rate of [3H]-thymidine uptake is a proxy for the rate of decitabine uptake, and reflects the rate of uptake in the presence of cell cycle arrest and DNA damage.
Global methylation analysis.
The cellular content of 2-deoxycytidine and 5-methyl-2-deoxycytidine was quantified by high performance liquid chromatography (HPLC).Citation5 Treated and untreated HCT116 cells were harvested and genomic DNA extracted using the DNeasy® kit, following the manufacturer's protocol. Genomic DNA (10 µg) was sheared and treated with nuclease P1 and bacterial alkaline phosphatase. Digested DNA samples (150 µl in duplicate for each experiment) were analyzed using a Beckman System Gold HPLC system equipped with a Phenomenex Synergi Max-RP C12, 250 × 4.6 mm column and a photodiode array detector. Samples were assayed in duplicate and the experiment done in triplicate. Results are presented as mean ± SD.
MAGE-A1 promoter methylation assay by qPCR.
Genomic DNA (2 µg) from the same samples used in the global methylation assay were analyzed for MAGE-A1 promoter methylation as described.Citation5 Samples were digested using HpaII, a methylation-sensitive restriction enzyme. Digested samples were purified using a DNA clean up kit. Two primer sets are used; CDS20/EPD4 serves as the methylation sensitive primer set, while CDS21/EPD4 serves as a control set. PCR parameters were 94°C for 10 min, followed by 35 cycles of 94°C for 15 seconds, 61°C for 15 seconds, 72°C for 20 seconds and plate read. Copy numbers were calculated using the software provided with the instrument (Opticon Chromo4 thermocycler, Eurogentec, Fremont, California). Samples and standards were in a 2× LightCycler 480 SYBR Green I Master mix. For each sample, the copy number obtained from the test reaction (using CDS20/EPD4 primers) was divided by the copy number derived from the control reaction (CDS21/EPD4 primers) to generate a ratio. This ratio directly reflects the average methylation state of the MAGE-A1 promoter HpaII sites. Samples were assayed in triplicate and the experiment done in triplicate. Results are presented as mean ± SD.
MAGE-A1 re-expression assay.
RNA was extracted from HCT 116 cells using the RNeasy® kit following the manufacturer's protocol, with on column DNA digestion performed. Total RNA concentration was measured spectrophotometrically. Using a first strand cDNA synthesis kit with the oligo(dT) primer, 2.5 µg of RNA was converted to cDNA. PCR was then performed on 2 µl of the resulting cDNA using the following primers for MAGE-A1. Forward AGA GGC AAC CCA GTG AG, reverse CAG CCA CCT TCT TAG TGA T. PCR parameters were 95°C for 2 min, followed by 35 cycles of 95°C for 30 seconds, 60°C for 30 seconds, 72°C for 1 min. Actin or GAPDH primers were used as controls.
Statistics.
Statistical significance was assessed by analysis of variance (ANOVA) with Bonferroni's post-hoc test for comparisons among groups.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Abbreviations
| AML | = | acute myeloid leukemia |
| BER | = | base excision repair |
| CML | = | chronic myeloid leukemia |
| DNMT | = | DNA methyltransferase |
| ECL | = | enhanced chemiluminescence |
| FBS | = | fetal bovine serum |
| HRP | = | horseradish peroxidase |
| MDS | = | myelodysplastic syndrome |
| NER | = | nucleotide excision repair |
| TBST | = | tris buffered saline with tween |
| ZdC | = | 5-aza-2′-deoxycytidinetriphosphate |
Figures and Tables
Figure 1 Cell cycle progression is unnecessary for depletion of DNMT1 enzyme in HCT 116 cells exposed to decitabine. (A) Western immunoblots of DNMT1 enzyme and β-tubulin. Serum-starved, growing or confluent HCT116 p53+/+ cells were treated once with 300 nM decitabine or 1 µM 6-azacytidine. DNMT1 was detected in the cell lysates by western immunoblotting. β-tubulin served as a loading control. The blots are representative of five experiments for the growing and confluent cells and two experiments for the starved cells. (B) Time course of DNMT1 depletion by decitabine. Plots of the relative depletion of DNMT1 versus time were based on the densitometry of DNMT1 immunoblots normalized to β-tubulin. *Denotes statistical significance (p < 0.05), error bars are 1 SD (n = 5 confluent and growing, n = 2 starved). The Table shows the t½ ± SD of DNMT1 depletion for each experimental condition and the corresponding percentage of the cell population in s-phase.
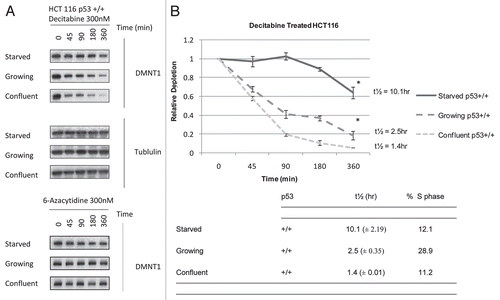
Figure 2 The p53 tumor suppressor modulates the rate of depletion of DNMT1 in HCT 116 cells exposed to decitabine. (A) Immunoblots of DNMT1 enzyme and tubulin Confluent HCT 116 cells that were wild type p53+/+ or null p53−/− were treated once with 300 nM decitabine. DNMT1 enzyme levels and β-tubulin loading control were detected by western immunoblotting. The blots are representative of five experiments for the wild type p53+/+ and two experiments for null p53−/− HCT 116 cells. (B) Time course of DNMT1 depletion by decitabine. Plots of the relative depletion of DNMT1 versus time were based on the densitometry of the DNMT1 immunoblots normalized to β-tubulin. *Denotes statistical significance, error bars are 1 SD. The table shows the t½ ± SD of DNMT1 depletion for each experimental condition and the corresponding percentage of the cell population in s-phase.
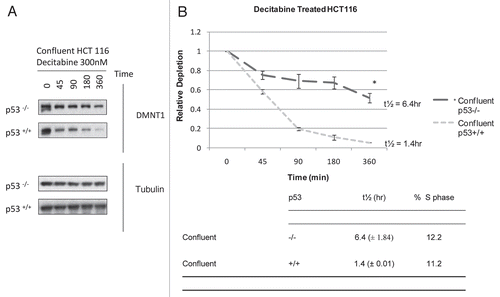
Figure 3 Epigenetic effects of decitabine in HCT 116 cells are cell cycle dependent: MAGE-A1 re-expression is s-phase dependent. Reverse transcription PCR and ethidium bromide detection of MAGE-A1 mRNA expression at 24 and 48 h after a single exposure to 300 nM decitabine in growing, confluent or confluent HCT 116 cells released from arrest. GAPDH and actin were used as procedural controls. Agarose gels shown are representative of three experiments.
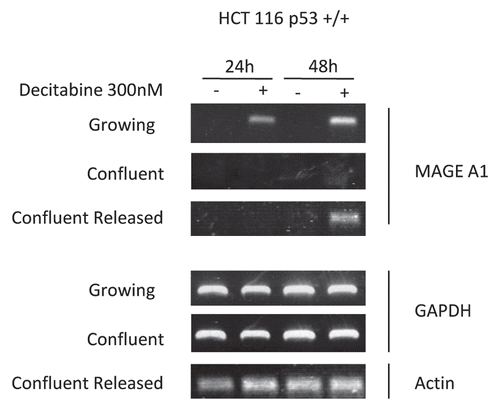
Figure 4 A p53 response to DNA damage coincides with the epigenetic effects of decitabine. (A) Immunoblot of p53 tumor suppressor protein and tubulin in growing cells. Growing wild-type p53+/+ HCT116 cells were treated once with 300 nM decitabine. p53 Protein and α-tubulin loading control in cell lysates were measured at 12 h intervals by western immunoblotting. (B) Immunoblot of p53 tumor suppressor protein and tubulin in confluent cells. Confluent wild-type p53+/+ HCT116 cells were treated once with 300 nM 5decitabine. p53 Protein and β-tubulin loading control in cell lysates were measured at 12 h intervals by western immunoblotting.
Table 1 Epigenetic effects of eecitabine are replication dependent
Additional material
Download Zip (113 KB)Acknowledgments
We thank Dr. Burt Vogelstein for the isogenic HCT 116 cells and Dr. David A. Jones, Huntsman Cancer Institute, for helpful discussion. 6-Azacytidine was provided by the Developmental Therapeutics Program of the National Cancer Institute. NCI 5R21CA122270 (W.S.), Huntsman Cancer Foundation (M.A.S. and F.A.F.) and an ASCO young investigator award (M.Y.) funded this investigation.
References
- Esteller M. Epigenetics in cancer. N Engl J Med 2008; 358:1148 - 1159; PMID: 18337604
- Issa JP, Kantarjian HM. Targeting DNA Methylation. Clin Cancer Res 2009; 15:3938 - 3946; PMID: 19509174
- Wijermans P, Lubbert M, Verhoef G, Bosly A, Ravoet C, Andre M, et al. Low-dose 5-aza-2′-deoxycytidine, a DNA hypomethylating agent, for the treatment of high-risk myelodysplastic syndrome: a multicenter phase II study in elderly patients. J Clin Oncol 2000; 18:956 - 962; PMID: 10694544
- Issa JP, Garcia-Manero G, Giles FJ, Mannari R, Thomas D, Faderl S, et al. Phase I study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (Decitabine) in hematopoietic malignancies. Blood 2004; 103:1635 - 1640; PMID: 14604977
- Samlowski WE, Leachman SA, Wade M, Cassidy P, Porter-Gill P, Busby L, et al. Evaluation of a 7-day continuous intravenous infusion of decitabine: inhibition of promoter-specific and global genomic DNA methylation. J Clin Oncol 2006; 23:3897 - 3905; PMID: 15753459
- Aparicio A, Eads CA, Leong LA, Laird PW, Newman EM, Synold TW, et al. Phase I trial of continuous infusion 5-aza-2′-deoxycytidine Cancer Chemother. Pharmacol 2003; 51:231 - 239; PMID: 12655442
- Karpf AR, Moore BC, Ririe TO, Jones DA. Activation of the p53 DNA damage response pathway after inhibition of DNA methyltransferase by 5-aza-2′-deoxycytidine. Mol Pharmacol 2001; 59:751 - 757; PMID: 11259619
- Link PA, Baer MR, James SR, Jones DA, Karpf AR. p53-inducible ribonucleotide reductase (p53R2/RRM2B) is a DNA hypomethylation-independent decitabine gene target that correlates with clinical response in myelodysplastic syndrome/acute myelogenous leukemia. Cancer Res 2008; 68:9358 - 9366; PMID: 19010910
- Peller S. Clinical implications of p53: effect on prognosis, tumor progression and chemotherapy response. Semin Cancer Biol 1998; 8:379 - 387; PMID: 10101803
- Stewart DJ, Issa JP, Kurzrock R, Nunez MI, Jelinek J, Hong D, et al. Decitabine effect on tumor global DNA methylation and other parameters in a phase I trial in refractory solid tumors and lymphomas. Clin Cancer Res 2009; 15:3881 - 3888; PMID: 19470736
- Creusot F, Acs G, Christman JK. Inhibition of DNA methyltransferase and induction of Friend erythroleukemia cell differentiation by 5-azacytidine and 5-aza-2′-deoxycytidine. J Biol Chem 1982; 257:2041 - 2048; PMID: 6173384
- Santi DV, Norment A, Garrett CE. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc Natl Acad Sci USA 1984; 81:6993 - 6997; PMID: 6209710
- Juttermann R, Li E, Jaenisch R. Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc Natl Acad Sci USA 1994; 91:11797 - 11801; PMID: 7527544
- Bender CM, Gonzalgo ML, Gonzales FA, Nguyen CT, Robertson KD, Jones PA. Roles of cell division and gene transcription in the methylation of CpG islands. Mol Cell Biol 1999; 19:6690 - 6698; PMID: 10490608
- Hermann A, Goyal R, Jeltsch A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J Biol Chem 2004; 279:48350 - 48359; PMID: 15339928
- Velicescu M, Weisenberger DJ, Gonzales FA, Tsai YC, Nguyen CT, Jones PA. Cell division is required for de novo methylation of CpG islands in bladder cancer cells. Cancer Res 2002; 62:2378 - 2384; PMID: 11956100
- Ghoshal K, Datta J, Majumder S, Bai S, Kutay H, Motiwala T, et al. 5-Aza-Deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain and nuclear localization signal. Mol Cell Biol 2005; 25:4727 - 4741; PMID: 15899874
- Patel K, Dickson J, Din S, Macleod K, Jodrell D, Ramsahoye B. Targeting of 5-aza-2′-deoxycytidine residues by chromatin-associated DNMT1 induces proteasomal degradation of the free enzyme. Nucleic Acids Res 2010; 38:4313 - 4324; PMID: 20348135
- Easwaran HP, Schermelleh L, Leonhardt H, Cardoso MC. Replication-independent chromatin loading of Dnmt1 during G2 and M phases. EMBO Rep 2004; 5:1181 - 1186; PMID: 15550930
- Mortusewicz O, Schermelleh L, Walter J, Cardoso MC, Leonhardt H. Recruitment of DNA methyltransferase I to DNA repair sites. Proc Natl Acad Sci USA 2005; 102:8905 - 8909; PMID: 15956212
- Bunz F, Hwang PM, Torrance C, Waldman T, Zhang Y, Dillehay L, et al. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest 1999; 104:263 - 269; PMID: 10430607
- Appleton K, Mackay HJ, Judson I, Plumb JA, McCormick C, Strathdee G, et al. Phase I and pharmacodynamic trial of the DNA methyltransferase inhibitor decitabine and carboplatin in solid tumors. J Clin Oncol 2007; 25:4603 - 4609; PMID: 17925555
- Robertson KD, Keyomarsi K, Gonzales FA, Velicescu M, Jones PA. Differential mRNA expression of the human DNA methyltransferases (DNMTs) 1, 3a and 3b during the G(0)/G(1) to S phase transition in normal and tumor cells. Nucleic Acids Res 2000; 28:2108 - 2113; PMID: 10773079
- Kimura F, Seifert HH, Florl AR, Santourlidis S, Steinhoff C, Swiatkowski S, et al. Decrease of DNA methyltransferase 1 expression relative to cell proliferation in transitional cell carcinoma. Int J Cancer 2003; 104:568 - 578; PMID: 12594811
- Schermelleh L, Haemmer A, Spada F, Rösing N, Meilinger D, Rothbauer U, et al. Dynamics of Dnmt1 interaction with the replication machinery and its role in postreplicative maintenance of DNA methylation. Nucleic Acids Res 2007; 35:4301 - 4312; PMID: 17576694
- Tatematsu KI, Yamazaki T, Ishikawa F. MBD2-MBD3 complex binds to hemi-methylated DNA and forms a complex containing DNMT1 at the replication foci in late S phase. Genes Cells 2000; 5:677 - 688; PMID: 10947852
- Iwanaga R, Komori H, Ohtani K. Differential regulation of expression of the mammalian DNA repair genes by growth stimulation. Oncogene 2004; 23:8581 - 8590; PMID: 15467751
- Rancourt RC, Hayes DD, Chess PR, Keng PC, O'Reilly MA. Growth arrest in G1 protects against oxygen-induced DNA damage and cell death. J Cell Physiol 2002; 193:26 - 36; PMID: 12209877
- Ljungman M. Activation of DNA damage signaling. Mutat Res 2005; 577:203 - 216; PMID: 15922368
- Snyder RD. Inhibitors of ribonucleotide reductase alter DNA repair in human fibroblasts through specific depletion of purine deoxynucleotide triphosphates. Cell Biol Toxicol 1984; 1:81 - 94; PMID: 6401127
- Jackson-Grusby L, Beard C, Possemato R, Tudor M, Fambrough D, Csankovszki G, et al. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat Genet 2001; 27:31 - 39; PMID: 11137995
- Aladjem MI, Spike BT, Rodewald LW, Hope TJ, Klemm M, Jaenisch R, et al. ES cells do not activate p53-dependent stress responses and undergo p53-independent apoptosis in response to DNA damage. Curr Biol 1998; 8:145 - 155; PMID: 9443911
- Smith ML, Seo YR. p53 regulation of DNA excision repair. Mutagenesis 2002; 17:149 - 156; PMID: 11880544
- Zhou J, Ahn J, Wilson SH, Prives C. A role for p53 in base excision repair. EMBO J 2001; 20:914 - 923; PMID: 11179235
- Moll UM, Wolff S, Speidel D, Deppert W. Transcription-independent pro-apoptotic functions of p53. Curr Opin Cell Biol 2005; 17:631 - 636; PMID: 16226451
- Lin RK, Wu CY, Chang JW, Juan LJ, Hsu HS, Chen CY, et al. Dysregulation of p53/Sp1 control leads to DNA methyltransferase-1 overexpression in lung cancer. Cancer Res 2010; 70:5807 - 5817; PMID: 20570896
- Estève PO, Chin HG, Pradhan S. Human maintenance DNA (cytosine-5)-methyltransferase and p53 modulate expression of p53-repressed promoters. Proc Natl Acad Sci USA 2005; 102:1000 - 1005; PMID: 15657147
- Robertson KD. DNA methylation, methyltransferases and cancer. Oncogene 2001; 20:3139 - 3155
- Karpf AR, Jones DA. Reactivating the expression of methylation silenced genes in human cancer. Oncogene 2002; 21:5496 - 5503; PMID: 12154399