Abstract
The microenvironment plays a significant role in human cancer progression. However, the role of the tumor microenvironment in the epigenetic control of genes critical to cancer progression remains unclear. As transient E-cadherin expression is central to many stages of neoplasia and is sensitive to regulation by the microenvironment, we have studied if microenvironmental control of E-cadherin expression is linked to transient epigenetic regulation of its promoter, contributing to the unstable and reversible expression of E-cadherin seen during tumor progression. We used 3D, bioengineered human tissue constructs that mimic the complexity of their in vivo counterparts, to show that the tumor microenvironment can direct the re-expression of E-cadherin through the reversal of methylation-mediated silencing of its promoter. This loss of DNA methylation results from the induction of homotypic cell-cell interactions as cells undergo tissue organization. E-cadherin re-expression is associated with multiple epigenetic changes including altered methylation of a small number of CpGs, specific histone modifications, and control of miR-148a expression. These epigenetic changes may drive the plasticity of E-cadherin-mediated adhesion in different tissue microenvironments during tumor cell invasion and metastasis. Thus, we suggest that epigenetic regulation is a mechanism through which tumor cell colonization of metastatic sites occurs as E-cadherin-expressing cells arise from E-cadherin-deficient cells.
Introduction
It is well established that the tissue microenvironment has a significant impact on the phenotype and behavior of a broad spectrum of human cancers. Microenvironmental factors may modulate both the transition from pre-cancer to cancer,Citation1,Citation2 and later stages in which tumor cells acquire invasive and metastatic properties.Citation3-Citation5 Tumor cells at these developmental stages encounter distinct microenvironments through which their fate and behavior are influenced by cell-cell and cell-matrix interactions.Citation6,Citation7 Additionally, tumor cells manifest epigenetic plasticity that is known to modulate their fate and phenotype.Citation8,Citation9 However, it remains unclear if epigenetic control of tumor suppressor genes, whose expression is critical to the invasive and metastatic stages of cancer progression, can be programmed through interactions between tumor cells and their immediate microenvironment.
E-cadherin is a gene whose expression may be controlled by both microenvironmental and epigenetic factors during cancer progression. Loss of E-cadherin expression is permissive for the transition from pre-cancer to invasion in squamous cell carcinoma (SCC)Citation10-Citation13 and breast cancerCitation14,Citation15 and correlates with metastasis and patient prognosis.Citation16 Loss of E-cadherin expression due to hypermethylation of its promoter has been reported in numerous tumor types including oral SCC,Citation17-Citation19 breast,Citation20-Citation23 and colon.Citation24 However, loss of E-cadherin appears to be reversible, as both breast cancer cells and OSCC cells that demonstrate loss of E-cadherin at their primary site of invasion can restore expression of E-cadherin at sites of metastasis.Citation25-Citation29 In addition, when breast cancer cells that lack E-cadherin expression due to promoter methylation are grown as tumor spheroids, the E-cadherin promoter is demethylated and E-cadherin is re-expressed.Citation30 Thus, it is possible that epigenetic gene silencing of E-cadherin is reversible and linked to heterogeneous patterns of E-cadherin expression seen during cancer progression. This supports studies demonstrating that the metastatic phenotype is unstable and that alterations in intercellular adhesion are transient and reversible.Citation28,Citation31
It is well established that the acquisition of invasive properties in tumor cells at primary sites of cancer development is due, in part, to epithelial-mesenchymal transition (EMT) and loss of epithelial phenotype. The process of EMT occurs over time and is marked by abrogation of homotypic cell-cell adhesion that occurs in the absence of E-cadherin expression, reorganization of the cytoskeleton, and loss of apical-basal polarity that are linked to increased cellular motility. The invasive phenotype of cells undergoing EMT is further aided by the secretion of enzymes that degrade the surrounding extracellular matrix, thus allowing cancer cells access to vascular and lymphatic networks that disseminate these cells to metastatic sites. In contrast, it is possible that reestablishment of cell-cell adhesion upon re-expression of E-cadherin may result in a mesenchymal-epithelial transition (MET) that can provide an advantage for colonization, survival, and proliferative growth of metastases.Citation32,Citation33 However, mechanisms through which MET may occur at metastatic sites are poorly understood and may be related to the plasticity of E-cadherin gene methylation.
Previous studies examining the reversible nature of E-cadherin promoter methylation and how the microenvironment may modulate this process have focused on breast cancer cell lines and lacked the complexity of a tissue microenvironment that contains basement membrane, stromal fibroblasts and extracellular matrix (ECM) proteins. Therefore, the role of these other tissue components has not been well linked to the reversal of methylation-mediated suppression leading to E-cadherin expression. To extend these previous studies and further investigate how the epigenetic control of E-cadherin expression may be linked to the tumor microenvironment, we explored the interplay between the establishment of homotypic cell-cell interactions and epigenetic control of E-cadherin expression during SCC progression by adapting engineered human 3D tissues that mimic the in vivo tumor microenvironment.Citation34-Citation36 We demonstrate that epigenetic regulation of E-cadherin expression is dynamic and sensitive to the induction of complex homotypic cell-cell interactions in a 3D tissue. Furthermore, we show that microenvironmental control of E-cadherin methylation may be related to the pattern of histone modifications associated with the E-cadherin promoter and the expression of a particular miRNA. These findings suggest an epigenetic mechanism through which E-cadherin expression undergoes differential regulation in primary vs. metastatic sites that can provide a selective advantage for both the invasion of epigenetically-silenced, E-cadherin-deficient cells and the metastatic colonization of E-cadherin-competent cells in which epigenetic silencing has been reversed.
Results
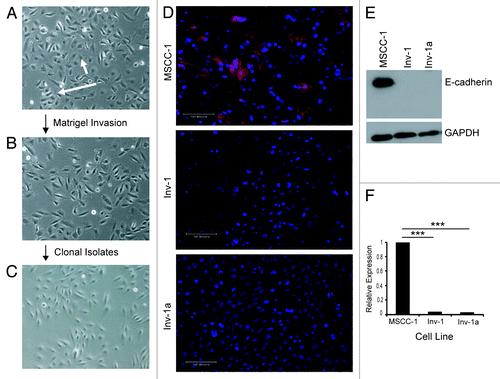
Generation of clonal cell populations that lack E-cadherin expression. To study the effects of the tumor microenvironment on the epigenetic regulation of E-cadherin expression, we derived clonal cell lines that manifested complete loss of E-cadherin expression (). The parental line (MSCC-1) () was derived from a lymph node metastasis that originated in the gingiva,Citation37 and a highly migratory subpopulation of cells (Inv-1) () was subsequently isolated from MSCC-1 using a Matrigel invasion assay and found to lack E-cadherin expression in most cells due to promoter hypermethylation.Citation38 E-cadherin expression was found in many of the parental MSCC-1 cells but not the Inv-1 cells upon immunofluorescence staining (), and protein gel blot analysis (). However, since the Inv-1 cell line was not isolated clonally, the presence of E-cadherin-positive cells could not be ruled out. Therefore, we clonally isolated 10 E-cadherin-deficient cell lines. Here we show the data generated from a representative line (Inv-1a) (). Interestingly, the clonal cell populations could only be isolated from Type I Collagen coated tissue culture plates as the cells were unable to grow out as viable clones on tissue culture treated plastic plates (data not shown). This same phenotype was not observed with the MSCC-1 cells. The complete absence of E-cadherin expression in the Inv-1a cells was confirmed by immunofluorescence, protein gel blot, and qPCR ().
Figure 1. Isolation and characterization of E-cadherin expression in OSCC cell lines. (A-C) Cell lineage map of Inv-1a isolation. (A) MSCC-1harbor cells that form adhesive clusters (long arrow) and individual, spindle shaped cells (short arrow). (B) The majority of Inv-1 cells appear as individual, spindle shaped cells. (C) All Inv-1a cells are spindle shaped and appear as single cells. (D) Immunofluorescent staining of cells for E-cadherin (red) and dapi (blue). Only MSCC-1 cells show positive E-cadherin expression localized to the cell-cell junctions. Scale bars = 100 µm. (E) protein gel blot analysis of E-cadherin and GAPDH in protein extracts from MSCC-1, Inv-1, and Inv-1a. (F) qRT-PCR for E-cadherin mRNA. Data are compared with MSCC-1 and normalized to GAPDH. ***p < 0.00001.
Silencing of E-cadherin in Inv-1a cells is linked to limited DNA methylation of 4 CpG sites within the E-cadherin promoter. To confirm DNA methylation as the cause of E-cadherin deficiency in the Inv-1a cells, we used methylation-specific PCR (MSP) and treatment with the DNA methyltransferase inhibitor, 5-aza-2’-deoxycytidine (5-aza-CdR). MSP revealed that the E-cadherin promoter in Inv-1a cells was methylated while Inv-1 cells showed a low level of methylation and DNA methylation was almost undetectable in MSCC-1 (). To confirm that promoter methylation was linked to the suppression of E-cadherin, all cell lines were exposed to 5-aza-CdR. 5-aza-CdR induced demethylation of the E-cadherin promoter as seen by both renewed transcription of E-cadherin mRNA () and re-expression of E-cadherin protein in Inv-1a (), that were linked to the loss of methylation of the E-cadherin promoter () in Inv-1a cells.
Figure 2. Characterization of the method of E-cadherin suppression in the Inv-1a cell line. (A) MSP for E-cadherin promoter methylation in MSCC-1, Inv-1, and Inv-1a cells. MDA-MB435s cells were used for the positive control as they have been shown to harbor a highly methylated E-cadherin promoter while MCF7 cells were used as the negative control as they have been shown to express E-cadherin. (B) qRT-PCR for E-cadherin transcription following treatment of cells with 5-aza-CdR. (C) Immunofluorescent staining for E-cadherin (red) and dapi (blue) in Inv-1a cells following treatment with 5-aza-CdR. E-cadherin is expressed and localized to the cell-cell junctions. Scale bar = 100 µm. (D) MSP for E-cadherin promoter methylation in Inv-1a cells following treatment with 5-aza-CdR. (E) protein gel blot analysis reveals high DNMT1 protein expression in both the Inv-1 and Inv-1a cell lines compared with the MSCC-1 cell line. (F) qRT-PCR for DNMT1. Data are relative to MSCC-1 and is normalized to GAPDH. (G) qRT-PCR for DNMT1 and E-cadherin in Inv-1a cells transfected with siRNA against DNMT1. Data are normalized to GAPDH. (H) qRT-PCR for DNMT1 and E-cadherin in Inv-1a cells infected with shRNA against DNMT1. Data are normalized to GAPDH.
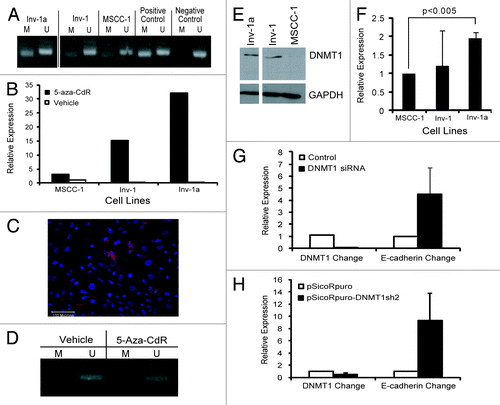
Since the effect of 5-aza-CdR on our cells could be due to a side effect rather than a direct effect upon DNA methylation, we examined the role of DNMT1 in the microenvironment-mediated, epigenetic regulation of E-cadherin expression in Inv-1a cells. Protein gel blot analysis showed DNMT1 protein expression in both the Inv-1 and Inv-1a cell lines, while very little DNMT1 was present in the MSCC-1 cells (). qPCR () showed elevated expression of DNMT1 mRNA in both the Inv-1a and Inv-1 cells compared with the MSCC-1 cells. To further study the relationship between DNMT1 and E-cadherin and confirm DNA methylation as the mechanism of E-cadherin regulation in our cells, we explored how suppression of DNMT1 could alter expression of E-cadherin in Inv-1a cells. When Inv-1a cells were transfected with siRNA directed against DNMT1, knockdown of DNMT1 transcript led to an elevation of E-cadherin transcription (). In addition, shRNA infection, utilizing a different RNAi sequence, resulted in a significant decrease in DNMT1 mRNA that was also linked to an increase in E-cadherin transcript levels in Inv-1a cells (). These findings indicate that the mechanism of E-cadherin suppression in the Inv-1a cell line is DNA methylation.
To further understand the DNA methylation-mediated regulation of E-cadherin, we used bisulfite sequencing (BSP) to examine the location and number of methylated CpGs within the E-cadherin promoter of Inv-1a cells grown in monolayer culture (). Inv-1a cells had more methylated CpG sites than Inv-1 cells, while parental MSCC-1 cells showed an appreciably smaller number of methylated CpGs. The pattern of methylation was sparse in the Inv-1a cells, with consistent methylation at only 4 CpG sites, but always a degree of methylation between 27% and 31%. The degree of methylation at these 4 sites in the Inv-1a cells was consistent among all alleles and found to be significantly different from the same sites in both the Inv-1 and MSCC-1 cell lines with a p-value < 0.01 (). Previous data examining CpG methylation within the E-cadherin promoter has shown that above a threshold of 20% to 30% methylation, E-cadherin gene expression is silenced.Citation39 Significantly, this same study showed consistent methylation at one specific CpG, which correlates to CpG #4 in our BSP study. This low percentage of CpG methylation suggests that the methylation-mediated silencing of E-cadherin within the Inv-1a cells may be weak and easily reversed. Importantly, the 4 consistent sites of methylation were not found to be sites of transcription factor binding, indicating that the mechanism of silencing in the Inv-1a cells via CpG methylation was not linked to interference of transcriptional machinery at these sites.
Figure 3. The methylation-mediated regulation of E-cadherin expression within the Inv-1a cells is related to specific CpG methylation. (A) Schematic illustration of the CpG island within the E-cadherin promoter. Vertical lines indicate CpGs. The bent arrow indicates the transcriptional start site. The small horizontal arrows indicate the primer set used for MSP while the line indicates the region amplified for BSP analysis. (B) BSP data for Inv-1a, Inv-1, and MSCC-1 cells lines. The white circles indicate unmethylated CpGs while the black circles indicate those CpGs that are methylated. The transcriptional start site is indicated by the bent black arrow. The large rectangles indicate the 4 CpGs found to be significantly differentially methylated between Inv-1a and MSCC-1 and Inv-1 **p < 0.01 (C) Statistical analysis of the methylation of different CpGs in the Inv-1a E-cadherin promoter compared with either the Inv-1 or MSCC-1. *p < 0.1, **p < 0.01

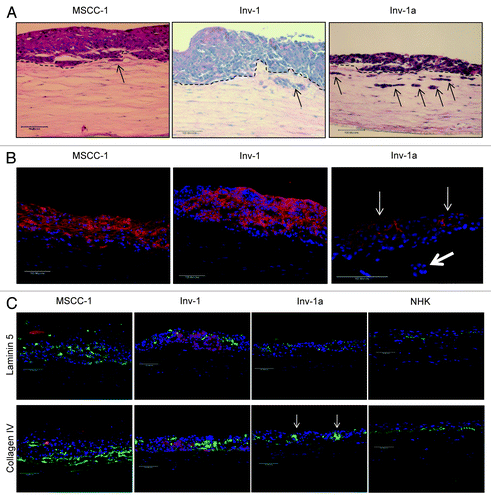
Induction of homotypic cell-cell interactions in a 3D tissue context leads to re-expression of E-cadherin due to loss of E-cadherin promoter methylation. We next determined if methylation patterns in the E-cadherin promoter of Inv-1a cells were reversible and susceptible to microenvironmental control when grown in a 3D network of cell-cell interactions. To accomplish this, we fabricated bioengineered, 3D tissues that mimic the microenvironment in which cells would establish complex cell-cell and cell-matrix interactions upon colonization of metastatic sites. Cells were seeded onto the surface of contracted Type I Collagen gels containing human fibroblasts and grown at an air liquid interface for 21 d. All cell lines generated multilayer tissues that showed varying degrees of tissue organization (). Tissues harboring either MSCC-1 or Inv-1 were thicker and showed invasion of a small number of cells just below the basement membrane interface while tissues constructed with Inv-1a cells showed larger numbers of invading cells into the underlying stroma (, black arrows). This suggests that E-cadherin expression by these cells does not completely inhibit cancer cell invasion or that the MSCC-1 and Inv-1 cell lines both harbor very small numbers of E-cadherin-deficient cells in these parental lines. Immunofluorescent staining revealed E-cadherin expression localized to cell junctions in all tissues fabricated, but in highest concentrations in both the MSCC-1 and Inv-1 tissues. Significantly, 3D tissues composed of Inv-1a cells, that had demonstrated silencing of E-cadherin expression in 2D cultures, expressed this protein in the suprabasal layers of the tissue, where cells had organized into adhesive clusters (, thin arrow). In contrast, individual Inv-1a cells that had invaded into the stromal compartment maintained loss of E-cadherin expression (, thick arrow). Additionally, while all fabricated tissues showed improper deposition of the basement membrane proteins Collagen IV and Laminin, most likely due to their cancer background, the tissues formed with Inv-1a cells only expressed these proteins in the presence of the cells that remained E-cadherin negative (, arrow). Thus, E-cadherin-deficient cells were able to re-express E-cadherin when grown in a tissue microenvironment that enabled them to form complex homotypic cell-cell interactions in a 3D context of neighboring epithelial cells. E-cadherin re-expression was dependent upon the loss of ECM contact as cells formed clusters in the epithelium and maintained loss of E-cadherin expression when in contact with underlying stromal proteins. Importantly, this same pattern of E-cadherin re-expression was seen in other Inv-1 clonal isolates that also lacked E-cadherin expression due to promoter hypermethylation (data not shown).
Figure 4. E-cadherin is re-expressed in Inv-1a cells when grown in the context of a 3D tissue. (A) Tissues generated from MSCC-1, Inv-1, and Inv-1a cell lines were stained with hematoxylin and eosin to examine the tissue architecture. The dashed black lines indicate the interface of the epithelium and the underlying stroma. Black arrows indicate the invading cells. (B) Immunohistochemical staining of 3D tissues for E-cadherin (red) and dapi (blue) reveal E-cadherin staining in all three tissue types. The E-cadherin positive Inv-1a cells are indicated by thin white arrows, while the thick white arrow indicates Inv-1a cells that have invaded into the stroma but do not express E-cadherin. (C) Immunohistochemical staining of 3D tissues for the basement membrane proteins (green) Laminin 5 and Collagen IV in relation to E-cadherin (red) and dapi (blue) in all three tissue types. Tissue made of normal human keratinocytes served as a control. E-cadherin negative Inv-1a cells in conjunction with abnormal Collagen IV staining are indicated by the white arrows. Scale bars = 100 µm.
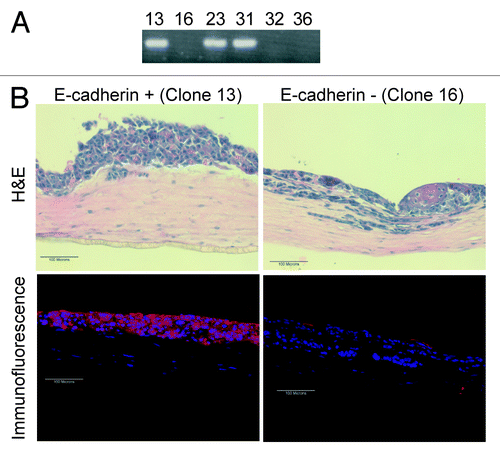
To confirm the role of E-cadherin in the invasion of cells into the underlying stroma, we re-expressed E-cadherin in Inv-1a cells using a retrovirus and isolated 3 E-cadherin-expressing clones and 3 E-cadherin-deficient clones, grew these in 2D monolayer culture, and checked their E-cadherin expression by RT-PCR (). When these E-cadherin-expressing Inv-1a cells were incorporated into 3D tissues, thick epithelial tissues were generated that contained small numbers of invasive cells, similar to those seen in tissues harboring MSCC-1 cells (), and many E-cadherin-expressing cells were identified throughout the tissues (). Thus, re-expression of E-cadherin within the Inv-1a cells was able to shift the 3D tissues to a phenotype that was more similar to the parental, MSCC-1 tissues than to the highly invasive tissues harboring only E-cadherin-deficient cells.
Figure 5. Re-expression of E-cadherin within the Inv-1a cells results in a thickened tissue formation and a reduction in the number of invasive cells. (A) RT-PCR for E-cadherin in Inv-1a clones that were infected with an E-cadherin-expressing retroviral vector showing E-cadherin expression. (B) H&E and immunofluorescence (E-cadherin – red; dapi – blue) staining of 3D tissues composed of either E-cadherin positive or E-cadherin negative Inv-1a cells. Scale bars = 100 µm.
DNA methylation and DNMT1 expression in 3D tissues. We next determined if re-expression of E-cadherin in 3D tissues was linked to a change in the methylation of the E-cadherin promoter. We compared the DNA methylation status of Inv-1a cells that re-expressed E-cadherin in 3D tissues to cells in the same tissue that remained E-cadherin deficient. Laser capture microdissection (LCM) was used to isolate either E-cadherin-expressing cells or E-cadherin-deficient cells from serial sections of tissues constructed with Inv-1a cells (). MSP analysis of the E-cadherin promoter revealed loss of methylation in the Inv-1a cells that stained positively for E-cadherin (, black arrow), while cells dissected from regions of the tissue that maintained loss of E-cadherin expression showed continued methylation of the promoter ().
Figure 6. Gain of E-cadherin expression in the Inv-1a tissues is due to loss of DNA methylation, increased stromal invasion and elevated DNMT1 expression. (A) E-cadherin staining in a paraffin section of Inv-1a tissue used for LCM. The black arrow indicates the E-cadherin positive cells that were removed for DNA isolation. (B) MSP analysis of the DNA isolated from microdissected E-cadherin negative and positive cells in the Inv-1a tissue. Inv-1a cells grown in 2D culture were used as a control. (C) Immunohistochemical staining of E-cadherin (red) and DNMT1 (green) in 3D tissues composed of Inv-1a cell. The white boxes indicate regions of the tissue that stain for E-cadherin but have low levels of DNMT1 expression. Scale bars = 100 µm.
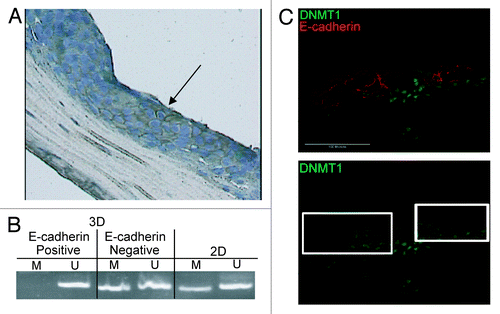
When tissues composed of Inv-1a cells were stained for both DNMT1 and E-cadherin, E-cadherin was present only in the cells that had reduced DNMT1 when compared with adjacent E-cadherin-deficient cells (, white boxes). Additionally, when we tried to grow DNMT1 deficient Inv-1a cells as 3D human tissues, stratified tissues did not form. This is consistent with data revealing that DNMT1 expression is required in the basal layer of normal epithelium to promote cell stratification and differentiation.Citation40 These findings establish that the local tissue microenvironment can directly regulate methylation of the E-cadherin promoter to reverse silencing of E-cadherin expression. This data suggests that individual E-cadherin-deficient cells may undergo metastasis and reform as E-cadherin-competent cell clusters at distant sites.
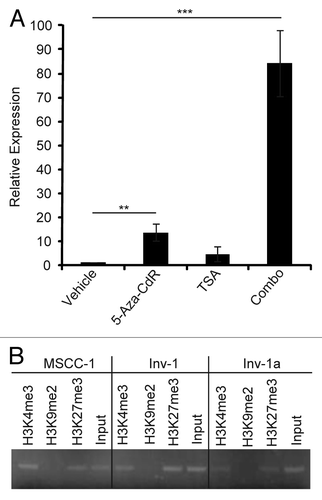
Histone modification patterns indicate the potentially reversible nature of E-cadherin epigenetic silencing. It has been proposed that a particular chromatin modification pattern may place a gene in a state of high plasticity in regards to its transcription. This has mainly been considered in terms of lineage specific genes in embryonic stem cells and has been termed the “bivalent” chromatin domain, H3K4me and H3K27me3 methylation without H3K9 methylation.Citation41,Citation42 Since the E-cadherin promoter has been shown to demonstrate a “bivalent domain,” with both histone H3K4me and H3K27me3 methylationCitation43,Citation44 that may silence expression while leaving it poised for activation, we investigated whether this pattern of histone methylation may also play a role in modulation of expression in E-cadherin-deficient, Inv-1a cells. To assess histone involvement we first treated E-cadherin-deficient Inv-1a cells with Trichostatin A (TSA), an HDAC inhibitor, either alone or in combination with 5-aza-CdR (). Treatment of Inv-1a cells with TSA resulted in renewed E-cadherin transcription but to a lesser extent than upon exposure to 5-aza-CdR. Combined treatment of the Inv-1a cells with both 5-aza-CdR and TSA resulted in an approximately 80-fold increase in E-cadherin transcription that was significantly higher than either treatment alone, indicating that DNA methylation and histone modifications are likely to play a synergistic role in the silencing of E-cadherin. To determine if histone modifications silencing E-cadherin in the Inv-1a cells were indicative of the “bivalent domain,” we performed chromatin immunoprecipitation (ChIP) to identify methylation of histones H3K4me3, H3K9me2, and H3K27me3 (). Inv-1a, Inv-1, and MSCC-1 all showed histone H3K4me3 and H3K27me3 methylation that was related to the E-cadherin promoter. Histone H3K9me2 methylation was not evident in any cell line. This suggests that the reversible silencing of E-cadherin may be due to the combination of a low level of overall DNA methylation and the bivalent chromatin domain signature.
Figure 7. Specific histone modifications provide evidence for the plasticity of E-cadherin epigenetic regulation. (A) qRT-PCR for E-cadherin in Inv-1a cells after treatment with 5-aza-CdR, TSA, or the combination of the two (Combo). Data are normalized to GAPDH. **p < 0.03, ***p < 0.009 (B) ChIP analysis of methylated histones H3K4, H3K9, and H3K27 in association with the E-cadherin promoter in MSCC-1, Inv-1, and Inv-1a cells. No association of the E-cadherin promoter with methylated H3K9 was detected in any cell line.
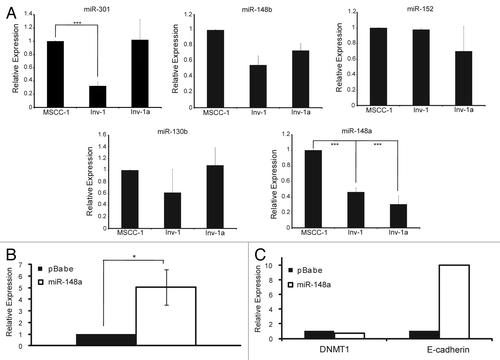
DNMT1 expression and subsequent E-cadherin expression is linked to miR-148a expression. We next explored what factors may be involved in the transient methylation of the E-cadherin promoter. Since we observed that DNMT1 inhibition resulted in renewed E-cadherin expression, we examined the role of putative DNMT1 regulating microRNAs (miRNA). We began by examining the expression of 5 miRNAs shown to have consensus binding sequences for DNMT1, miR-148a, miR-148b, miR-152, miR-130b, and miR-301 (). Only miR-148a showed a significant decrease in expression in both the Inv-1 and Inv-1a cell lines. From this information, we hypothesized that the low amount of miR-148a in the Inv-1 and Inv-1a cell lines in comparison to the MSCC-1 cell line may allow for the significant overexpression of DNMT1 seen in these two cell lines in comparison to MSCC-1. To test this hypothesis, we overexpressed miR-148a in the Inv-1a cell line () and examined the expression levels of both DNMT1 and E-cadherin mRNA (). Overexpression of miR-148a led to a decrease in DNMT1 expression that was linked to an increase in E-cadherin expression within the Inv-1a cell line. The role of miR-148 is significant as it has been shown to be capable of regulating DNMT3bCitation45 while miR-148a specifically has been shown to directly regulate DNMT1 in CD4+T cellsCitation46 and cholangiocarcinoma cells.Citation47 This data suggests that regulation of miR-148a may serve as a method of regulation for DNMT1, resulting in reversible silencing of E-cadherin.
Figure 8. Putative DNMT1 binding miR-148a affects DNMT1 and E-cadherin expression levels. (A) qPCR for miRNAs likely to bind to DNMT1. All data are normalized to U6 expression. ***p < 0.0001. (B) qPCR analysis of miR-148a expression after Inv-1a infection with either empty vector (pBabe) or the retroviral construct coding for miR-148a. (C) qPCR analysis of the mRNA levels of DNMT1 and E-cadherin in Inv-1a cells infected with either the empty pBabe vector or the vector encoding miR-148a. ***p < 0.02.
Discussion
Widespread changes in DNA methylation during tumorigenesis may be quite variable within tumors,Citation48-Citation50 suggesting that epigenetic plasticity may be linked to the phenotypic heterogeneity of cell populations present in primary tumors and at metastatic sites.Citation51,Citation52 We studied if this epigenetic plasticity, specifically the epigenetic control of E-cadherin expression, was modulated by the tissue microenvironment. To address this, we studied if cells derived from oral SCC that demonstrate loss of E-cadherin due to promoter hypermethylation, can be epigenetically reprogrammed and serve as a model system to help explain organizational growth of cancer cells at metastatic sites. Our findings demonstrate that the epigenetic plasticity of these cells is linked to their cellular microenvironment, as methylation-mediated gene silencing of the E-cadherin promoter is dynamically influenced by induction of complex cell-cell and cell-matrix interactions in the context of a 3D tissue. Additionally, we have shown that the state of epigenetic silencing within the E-cadherin promoter may make it more susceptible to such microenvironmental influences.
We studied the phenotype and behavior of a clonal cell population derived from an oral SCC cell line that lacked E-cadherin expression due to promoter hypermethylation when grown in monolayer culture. Tumor cells lost methylation-mediated silencing and re-expressed E-cadherin upon establishment of cell-cell interactions in a 3D tissue context. In contrast, promoter methylation was maintained and E-cadherin was not expressed in tumor cells that invaded into the underlying stroma of these tissues and formed cell-matrix interactions. This heterogeneous pattern of E-cadherin expression was similar to patterns of E-cadherin localization commonly seen in vivo, where cells in contact with surrounding stroma at the tumor periphery more frequently lose E-cadherin expression, while cells in the center of tumor islands maintain expression.Citation53 Our findings link heterogeneous patterns of E-cadherin expression within primary tumors to changes in epigenetic gene regulation that are dependent on whether tumor cells encounter a stromal or intercellular microenvironment. Furthermore, our findings support in vivo studies that have shown that tumors lacking E-cadherin expression at primary tumor sites due to promoter hypermethylation can re-express E-cadherin at metastatic sitesCitation28,Citation54,Citation55. Our observations in 3D tissues provide direct evidence of a link between the microenvironment and variable patterns of E-cadherin expression that occur during the invasive and metastatic stages of SCC progression.
Initial steps in cancer cell dissemination are dependent upon EMT that enable cancer cells to develop migratory and invasive properties.Citation56,Citation57 In contrast, metastatic foci can only develop in a target organ when cells are able to reestablish organizational growth as a solid, secondary tumor foci.Citation58 Tse and Kalluri have proposed that this return to an “epithelial identity,” which mimics their primary tumor phenotype, occurs through a MET. It is thought that reestablishment of E-cadherin-mediated adhesion is critical to MET in order to reverse the migratory phenotype that enabled cells to reach their metastatic site. Thus, while epigenetic suppression of E-cadherin expression aids in cancer cell dissemination, we have found that re-expression of E-cadherin through dynamic epigenetic remodeling is likely to enable metastatic cells to colonize distant tissue microenvironments. Those tumor cells that are capable of epigenetic remodeling may account for the extremely low number of tumor cells that are actually capable of forming metastases. Thus, our data, in conjunction with previous reports showing 5-aza-CdR can upregulate pro-metastatic genes,Citation59-Citation62 suggests that the delivery of agents directed against epigenetic targets must be timed correctly as demethylation may not only promote the invasion of metastatic cells but also the colonization of metastatic sites by tumor cells that are already present at these sites but unable to organize due to epigenetic gene silencing.
Epigenetic reprogramming has been suggested during embryonic stem cell differentiation as being linked to the chromatin “bivalent domain” found in genes that are in a “transcriptionally-ready” state.Citation63,Citation64 This domain is defined as a combination of H3K4me and H3K27me3 methylation and a lack of H3K9me2 methylation. We have shown the presence of these histone modifications in the E-cadherin promoter of our tumor cells. However, this mark is not sufficient to cause the silencing of E-cadherin as the E-cadherin-competent cells, MSCC-1, have the same histone modifications but express E-cadherin. We found that the addition of 4 specific methylated CpGs in the context of a range of 27% to 31% overall promoter methylation results in the silencing of E-cadherin in the Inv-1a cells. While some E-cadherin promoter methylation data has shown much higher percentages of DNA methylation, or data are consistent with previous findings showing that a threshold of 20% to 30% is sufficient for E-cadherin gene silencing.Citation65 This supports evidence that while histone modifications may prime a gene for silencing and cause low expression, the addition of DNA methylation is necessary to induce complete silencing.Citation66-Citation68 Therefore, a gradient of epigenetic gene silencing may exist during the progression of cancer that dictates which genes are irreversibly silenced and which remain in a transient state of gene expression through the acquisition of different histone modifications and degrees of DNA methylation. Different tumor microenvironments may work in distinct ways to regulate these divergent epigenetic modifications. As has recently been pointed out,Citation69 insight into the complex and dynamic nature of epigenetic regulation during cancer progression will be essential to establishing a better understanding of patient responses to epigenetic therapies for human cancers.
Our work builds upon previous studies that have suggested a potential role for cell-cell interactions in the modulation of DNA methylation. For example, re-expression of E-cadherin mediated by promoter demethylation has been shown when MDA-MB-231 breast cancer cells were cultured as tumor spheroids in forced suspension.Citation70 However, such spheroids do not fully recapitulate the complexity of the in vivo tumor microenvironment, as they lack complex interactions with ECM and other cell types that exist in tissues. Beyond this, a role for the ECM microenvironment in epigenetic control of E-cadherin expression has recently been supported by the finding that Laminin-1 induces E-cadherin expression in 3D cultures of breast cancer cells by inhibiting DNMT1 and reversing promoter methylation.Citation71 Our findings extend these results by showing similar microenvironment mediated epigenetic changes in oral SCC and revealing that homotypic cell-cell interactions may impart a similar response as that induced by the ECM.
Finally, our miRNA data suggests the involvement of miR-148a in the dynamic epigenetic regulation of E-cadherin through its ability to silence DNMT1 expression. This is significant, as miR-148a has been shown to be associated with proteins involved in cancer metastasis-related functions. Additionally, its expression was found to be significantly reduced in gastric cancer tissues compared with normal controls,Citation72,Citation73 correlated with increased gastric tumor size,Citation74 and correlated well with low levels of patient survival compared with patients expressing high levels of miR-148a.Citation72 It will be interesting to examine the expression of this miRNA in OSCC tumors. Additionally, since miR-148a does contain a CpG islandCitation75 and has been shown to be hypermethylated in pancreatic tumors,Citation76 it is possible that DNA methylation in tumors is highly regulated through both the hypermethylation of a miRNA controlling DNMT1 and DNMT1 resulting activities. Closer inspection of the regulation of miR-148a will need to be accomplished to understand if ECM components play a role in its regulation.
Overall, our findings illustrate that defining microenvironmental factors that play key roles as regulators of epigenetic plasticity are necessary and will be dependent on the development of 3D tissue models that mimic the complexity seen in human tumors, as patterns of DNA methylation and gene expression differ greatly between 2D, monolayer culture systems and 3D reconstructed tissues. We extend the use of engineered tissues to directly study how epigenetic regulation of gene expression is directed by the microenvironment. These studies may make it possible to adapt such tissue models to screen agents directed against epigenetic targets that may significantly advance the development of potential therapeutics.
In summary, we show a dynamic mechanism of E-cadherin promoter methylation that helps explain the heterogeneous pattern of E-cadherin expression observed during tumor progression and implicates the tumor microenvironment in the regulation of methylation-mediated control of E-cadherin expression. Our finding of specific CpG methylation sites combined with a specific ES cell linked histone methylation signature within the E-cadherin promoter provides additional evidence of a dynamic epigenetic plasticity in SCC cells. Thus, methylation of specific CpG sites and histones within the E-cadherin promoter may regulate a “metastasis signature” that reflects the reacquisition of E-cadherin function leading to MET that enables the organizational growth of metastatic deposits. Future studies are necessary to understand more precise mechanisms of microenvironmental regulation of E-cadherin methylation by examining pathways essential to methylation machinery. These results support accumulating evidence that implicate the critical role of the tumor microenvironment in progression of many cancers and extend this microenvironmental impact to epigenetic control of gene expression that may be crucial for initiation of metastasis.
Methods
Cell culture. MSCC-1, Inv-1, and Inv-1a cells were grown in Keratinocyte-SFM containing Bovine Pituitary Extract and Epidermal Growth Factor (Invitrogen, #17005–042) at 37°C in 5% CO2. Human dermal fibroblasts used for organotypic cultures were grown in DMEM containing 10% fetal bovine serum at 37°C in 7.5% CO2. E-cadherin-deficient clones were isolated from Inv-1 cells by seeding 50 cells onto a p100 tissue culture plate coated with Type I Collagen and isolating colonies with cloning rings.
Phase contrast, fluorescence microscopy, and laser capture microdissection analysis. Phase contrast images of cells were captured with PixeLINK software 4.5 (Ottawa, ON, Canada), using an inverted Axiovert 40C Zeiss microscope (Gottingen, Germany) equipped with a PixeLINK Camera PL-A600 series (Ottawa, ON, Canada). For immunocytochemistry, cultures were grown on coverslips and fixed with 4% paraformaldehyde in PBS for 5 min. Cells were immunostained with mouse anti-E-cadherin (Invitrogen, #13–1700), rabbit anti-DNMT1 (Abcam Inc., #ab19905), mouse anti-Laminin (Abcam Inc., #ab49726), or rabbit anti-collagen IV (Abcam Inc., #ab6586) for 1 h followed by Alexa 594-conjugated goat anti-mouse secondary antibody or Alexa 488-conjugated goat anti-rabbit secondary antibody (Invitrogen, #A-11005, #A-11008) for 30 min at room temperature. For Immunohistochemistry, tissues were frozen in OCT in liquid nitrogen vapors and 6µm sections were fixed in 4% paraformaldehyde in PBS and stained in the same manner as for immunocytochemistry. Tissue sections were counterstained with DAPI in Vectashield mounting medium (Vector Labs, #H-1200). For tissue morphology, tissues were fixed in 10% formalin and embedded in paraffin. Hematoxylin and eosin (H&E) staining was performed on 6µm tissue sections. Immunofluorescent and H&E images were captured with Spot Advanced Program 4.5, using a Nikon Eclipse 80i microscope (Nikon Instruments Inc., Melville, NY) equipped with a SPOT RT camera (Diagnostic Instruments, Sterling Heights, MI). For laser capture microdissection, 8 µm sections of paraffin embedded tissues were stained with mouse anti-E-cadherin (Invitrogen, #13–1700) followed by a biotinylated goat anti-mouse secondary antibody. The secondary antibody was visualized with Vectastain ABC kit (Vector labs, #PK-4000) followed by DAB Peroxidase Substrate (Vector Labs, #SK-4100). The cells were counterstained with toluidine blue (Sigma Aldrich, #89640). Microdissection was performed on an Arcturus PixCell IIe system (Molecular Devices, Sunnyvale, CA) using CapSure HS LCM caps (Molecular Devices, #08A16E). DNA was isolated from the caps using QIAamp DNA micro kit (Qiagen Inc., #56304).
protein gel blot analysis. Protein was extracted with RIPA buffer (150 mM NaCl, 50 mM TRIS-HCl (pH 7.5), 10 mM EDTA, 1% Triton X-100, 10 mM NaF, 10 µg mL−1 aprotinin, 10 µg mL−1 leupeptin, 2 µg mL−1 pepstatin, 1 mM PMSF, 200 mM NaVO4). Protein concentrations were measured using BCA Protein Assay Kit (PIERCE, #23225) and 10 µg protein samples were separated on 7.5% SDS-PAGE gels and transferred onto nitrocellulose membrane (Bio-Rad, #162–0115). The blot was probed with mouse anti-E-cadherin (BD Biosciences, #610404) or rabbit anti-DNMT1 (Abcam Inc., #ab19905) followed by horseradish peroxidase-conjugated secondary antibody (Amersham, #NXA931–1ml, #NA934–100UL). Membranes were stripped and re-blotted with mouse anti-GAPDH (Abcam Inc., #ab9484) for loading controls. Proteins were visualized using SuperSignal West Pico Chemiluminescent Substrate (PIERCE, #34077).
RNA isolation and qPCR. RNA was extracted from cell cultures using the QIAshredder to lyse the cells and the RNeasy RNA isolation kit to purify the RNA (Qiagen Inc., #74104). Purified RNA was quantified using a NanoDrop 1000 (Thermo Scientific, Wilmington, DE). One µg of RNA was converted into cDNA using the iScript cDNA synthesis kit (Bio-Rad, #170–8840). Amplification was performed using iQ SYBR Green supermix (Bio-Rad, #170–8880). Primers are listed in . Real-time PCR was performed on the iCycler iQ5 Optical System using the iQ5 Optical System software (Bio-Rad, Hercules, CA). The cDNA was amplified with an annealing temperature of 58°C for 40 cycles. All reactions were normalized to GAPDH and performed in triplicate. Fold change was calculated using the Pfaffl method.
Table 1. A listing of all primers used in qPCR, MSP, BSP, and ChIP
DNA isolation, methylation-specific PCR (MSP) and bisulfite sequencing (BSP). DNA was extracted and purified from cell cultures using the DNA mini kit (Qiagen Inc., #51304) and quantified using a NanoDrop 1000 (Thermo Scientific, Wilmington, DE). Bisulfite conversion was performed with the MethylCode Bisulfite Conversion kit (Invitrogen, #MECOV-50). All PCR reactions were performed with Platinum PCR Supermix (Invitrogen, #11306–016). All primers are listed in . For MSP, 2 µL of bisulfite converted DNA was amplified using a nested PCR technique. The initial round of PCR used the external primers and an annealing temperature of 56°C for 35 cycles. The second round of PCR used 1.5 µL of the external PCR product and utilized specific primers to differentiate between methylated and unmethylated DNA with both sets using an annealing temperature of 60°C and 30 cycles. The product was visualized on a 2% agarose gel containing ethidium bromide. For BSP, 2 µL of bisulfite converted DNA was amplified through 2 rounds of PCR. The first round of PCR was run for 5 cycles with an annealing temperature of 43.8°C for 90 sec and an extension at 72°C for 120 sec followed by 25 cycles with an extension time of 90 sec each cycle. The second round of PCR was run with 2 µL of product from the first round. The same 5 and 25 cycles were used with an annealing temperature of 37°C. The product was visualized on a 2% agarose gel containing ethidium bromide to ensure the formation of a 350 bp product. The amplified DNA was purified with PCR Purification kit (Qiagen Inc., #28104) and quantified with NanoDrop 1000. Twenty ng of purified DNA was inserted into the pGEM-T easy vector (Promega, #A1380) and transformed into JM109 cells (Promega, #L1001). Twenty clones were selected for each condition, grown up overnight in Luria broth (LB), purified with Spin miniprep kit (Qiagen Inc., #27106), quantified with NanoDrop 1000, and sequenced using T7 primers on an ABI 3130XL DNA sequencer. Sequencing data was analyzed for CpG methylation using CpGViewer. Statistical analysis was performed with QUMA.Citation77
5-aza-2’deoxycytidine and Trichostatin A treatment. 5-aza-2’deoxycytidine (5-aza-CdR) and Trichostatin A (TSA) were purchased from (Sigma Aldrich, #A3656 and #T1952). Cells were seeded 24 h prior to treatment at either 100,000 cells per well of a 6-well plate or 500,000 cells per 10 cm2 plate. 5-aza-CdR treatment occurred every 24 h for 3 d and consisted of 1 µM of 5-aza-CdR along with fresh media with each treatment. RNA and DNA were isolated as previously described. Cells treated with TSA were given 250 nM 24 h after seeding and allowed to grow for 24 h at which time RNA was isolated as previously described. For treatment with both drugs, cells were seeded and allowed to grow for 24 h. They were then treated with 5-aza-CdR as described. After the third treatment, they were given fresh media and 250 nM TSA for 24 h. RNA was then extracted as described.
3D organotypic constructs. 3D organotypic cultures were prepared as previously described.Citation78 Briefly, human dermal fibroblasts were added to neutralized Type I Collagen (Organogenesis, Canton, MA) and incubated for 7 d in media containing DMEM and 10% fetal calf serum. At this time, a total of 6x105 cells were seeded on top of the contracted collagen gel. Cultures were maintained submerged in low calcium media for 2 d, submerged for 2 d in normal calcium media, and raised to the air-liquid interface in normal calcium cornification medium for 21 d. All organotypic cultures were performed in triplicate.
Chromatin immunoprecipitation (ChIP). Cells were grown to approximately 75% confluency on p150 cell culture plates and chromatin was sheared and isolated using ChIP-IT enzymatic shearing kit (Active Motif, #53035). Immunoprecipitation was performed using Protein G magnetic beads (Active Motif, #53014). All immunoprecipitations were allowed to run overnight at 4°C. Antibodies used were, rabbit anti-human histone H3 trimethyl Lys4 (Active Motif, #39915), anti-human trimethyl-histone H3 (Lys27) (Millipore, #17–622) and anti-human histone H3 dimethyl K9 (Abcam, #ab1220). Reactions were cleaned, and 5 µL was used for PCR analysis using Platinum PCR supermix (Invitrogen, #11306–016). The PCR was run for 35 cycles with an annealing temperature of 54°C. Primers are listed in . PCR products were visualized on a 2% agarose gel containing ethidium bromide.
siRNA, shRNA, and E-cadherin re-expression. siRNA against DNMT1 was purchased from Dharmacon. Cells were transfected with siRNA with Lipofectamine 2000 (Invitrogen, #11668–019). RNA was isolated 72 h after transfection. pSicoR vectors containing shRNA against DNMT1 were a generous gift of Dr. Laurie Jackson-Grusby. After transfection into Phoenix 293FT cells, media containing virus was added to Inv-1a cells along with polybrene for 4 h. Cells were grown for 48 h in fresh media before selection with puromycin. Following 48 h of selection, RNA was isolated as previously described. LZRS retroviral vector for E-cadherin expression were a generous gift or Dr. Panos Anastasiadis and Dr. Albert Reynolds. After transfection into Phoenix 293FT cells, media containing virus was added to Inv-1a cells along with polybrene for 4 h. Cells were grown for 48 h in fresh media before selection of clones using cloning rings. Selected clones were expanded in 2D monolayer culture and RNA was isolated as previously described.
miRNA isolation and Real-time RT-PCR. miRNA was isolated from cultured cells using the mirVana miRNA isolation kit (Ambion, #AM1560). Purified miRNA was quantified using a NanoDrop 1000 (Thermo Scientific, Wilmington, DE). Fifteen ng of miRNA was converted to cDNA and used for real-time RT-PCR using the mirVana qRT-PCR miRNA detection kit and mirVana qRT-PCR primer sets (Ambion, #AM1558, U6 #AM30303). Real-time PCR was performed on the iCycler iQ5 Optical System using the iQ5 Optical System software (Bio-Rad, Hercules, CA). The kit directions were followed for cDNA amplification and all reactions were normalized to U6 and performed in triplicate.
miRNA retrovirus synthesis and infection. Primers were designed to amplify a 300 bp region of chromosome 7 that encodes miR-148a. PCR was run using DNA isolated from normal human keratinocytes (NHK) and Platinum PCR Supermix (Invitrogen, #11306–016). The product was run on a 2% agarose gel containing ethidium bromide to ensure the correct size of the amplicon. The PCR product was purified using QIAquick PCR Purification kit (Qiagen Inc., #28104) and quantified using a NanoDrop 1000 (Thermo Scientific, Wilmington, DE). Both the retroviral vector, pBabe, and the DNA were cut with the restriction enzymes EcoR1 and Sal1 (New England Biolabs, #R0101L and #R0138L) overnight at 37°C. The resulting linearized vector and sticky end DNA were cleaned up with QIAquick PCR Purification kit (Qiagen Inc., #28104) and quantified using a NanoDrop 1000 (Thermo Scientific, Wilmington, DE). The DNA was ligated into the linear pBabe vector for 5 min at room temperature, and transformed into JM109 cells (Promega, #L1001). Clones were isolated and grown overnight at 37°C in 4 mL of Luria Broth containing ampicillin. 150 µL of the overnight culture was transferred to 100 mL of Luria Broth containing ampicillin and again grown overnight at 37°C. These cultures were purified using HiSpeed Plasmid Purification kit (Qiagen, Inc., #12643). Both the pBabe vector containing the miR-148a coding region and the empty vector were transfected into Phoenix 293 cells using Lipofectamine 2000 (Invitrogen, #11668019). The media was changed 24 h after transfection, and harvested 48 h later. The viral containing supernatant was filtered through a 0.45 µm filter and 1.5 mL was added to a p100 containing the Inv-1a cells along with 3.2 µL polybrene (10mg/mL). The media was changed 4 h later and the cells were allowed to grow for 48 h before selection with puromycin.
| Abbreviations: | ||
| SCC | = | Squamous Cell Carcinoma |
| EMT | = | epithelial-mesenchymal transition |
| MET | = | mesenchymal-epithelial transition |
| ECM | = | extracellular matrix |
| MSP | = | methylation-specific PCR |
| 5-aza-CdR | = | 5-aza-2’ deoxycytidine |
| BSP | = | Bisulfite sequencing |
| LCM | = | laser capture microdissection |
| TSA | = | Trichostatin A |
| miRNA | = | microRNA |
| ChIP | = | Chromatin immunoprecipitation |
Acknowledgments
We thank Judith Edwards, Dr. Mark W. Carlson, Dr. Shumin Dong, Adam Sowalsky, Avi Smith, and Marie Rougié for their assistance. We would like to thank Dr. Panos Anastasiadis and Dr. Albert Reynolds for the E-cadherin expression vector. This research was supported in part by grant #DE017143 from the National Institute of Dental and Craniofacial Research.
References
- Andriani F, Garfield J, Fusenig NE, Garlick JA. Basement membrane proteins promote progression of intraepithelial neoplasia in 3-dimensional models of human stratified epithelium. Int J Cancer 2004; 108:348 - 57; http://dx.doi.org/10.1002/ijc.11525; PMID: 14648700
- Margulis A, Zhang W, Alt-Holland A, Pawagi S, Prabhu P, Cao J, et al. Loss of intercellular adhesion activates a transition from low- to high-grade human squamous cell carcinoma. Int J Cancer 2006; 118:821 - 31; http://dx.doi.org/10.1002/ijc.21409; PMID: 16152579
- Andriani F, Garfield J, Fusenig NE, Garlick JA. Basement membrane proteins promote progression of intraepithelial neoplasia in 3-dimensional models of human stratified epithelium. Int J Cancer 2004; 108:348 - 57; http://dx.doi.org/10.1002/ijc.11525; PMID: 14648700
- Itano N, Zhuo L, Kimata K. Impact of the hyaluronan-rich tumor microenvironment on cancer initiation and progression. Cancer Sci 2008; 99:1720 - 5; http://dx.doi.org/10.1111/j.1349-7006.2008.00885.x; PMID: 18564137
- Li H, Fan X, Houghton J. Tumor microenvironment: the role of the tumor stroma in cancer. J Cell Biochem 2007; 101:805 - 15; http://dx.doi.org/10.1002/jcb.21159; PMID: 17226777
- Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer 2001; 1:46 - 54; http://dx.doi.org/10.1038/35094059; PMID: 11900251
- Ziober AF, Falls EM, Ziober BL. The extracellular matrix in oral squamous cell carcinoma: friend or foe?. Head Neck 2006; 28:740 - 9; http://dx.doi.org/10.1002/hed.20382; PMID: 16649214
- Gal-Yam EN, Egger G, Iniguez L, Holster H, Einarsson S, Zhang X, et al. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc Natl Acad Sci USA 2008; 105:12979 - 84; http://dx.doi.org/10.1073/pnas.0806437105; PMID: 18753622
- Graff JR, Gabrielson E, Fujii H, Baylin SB, Herman JG. Methylation patterns of the E-cadherin 5′ CpG island are unstable and reflect the dynamic, heterogeneous loss of E-cadherin expression during metastatic progression. J Biol Chem 2000; 275:2727 - 32; http://dx.doi.org/10.1074/jbc.275.4.2727; PMID: 10644736
- Alt-Holland A, Shamis Y, Riley KN, DesRochers TM, Fusenig NE, Herman IM, et al. E-cadherin suppression directs cytoskeletal rearrangement and intraepithelial tumor cell migration in 3D human skin equivalents. J Invest Dermatol 2008; 128:2498 - 507; http://dx.doi.org/10.1038/jid.2008.102; PMID: 18528437
- Behrens J. The role of cell adhesion molecules in cancer invasion and metastasis. Breast Cancer Res Treat 1993; 24:175 - 84; http://dx.doi.org/10.1007/BF01833258; PMID: 8435473
- Birchmeier W, Hulsken J, Behrens J. E-cadherin as an invasion suppressor. Ciba Found Symp 1995; 189:124 - 36; PMID: 7587628
- Margulis A, Zhang W, Alt-Holland A, Crawford HC, Fusenig NE, Garlick JA. E-cadherin suppression accelerates squamous cell carcinoma progression in three-dimensional, human tissue constructs. Cancer Res 2005; 65:1783 - 91; http://dx.doi.org/10.1158/0008-5472.CAN-04-3399; PMID: 15753375
- Zschiesche W, Schonborn I, Behrens J, Herrenknecht K, Hartveit F, Lilleng P, et al. Expression of E-cadherin and catenins in invasive mammary carcinomas. Anticancer Res 1997; 17:561 - 7; PMID: 9066580
- Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol 2010; 12:247 - 56; PMID: 20173740
- Diniz-Freitas M, García-Caballero T, Antúnez-López J, Gándara-Rey JM, García-García A. Reduced E-cadherin expression is an indicator of unfavourable prognosis in oral squamous cell carcinoma. Oral Oncol 2006; 42:190 - 200; http://dx.doi.org/10.1016/j.oraloncology.2005.07.010; PMID: 16249116
- Supić G, Kozomara R, Brankovic-Magic M, Jovic N, Magic Z. Gene hypermethylation in tumor tissue of advanced oral squamous cell carcinoma patients. Oral Oncol 2009; 45:1051 - 7; http://dx.doi.org/10.1016/j.oraloncology.2009.07.007; PMID: 19665921
- Ha PK, Califano JA. Promoter methylation and inactivation of tumour-suppressor genes in oral squamous-cell carcinoma. Lancet Oncol 2006; 7:77 - 82; http://dx.doi.org/10.1016/S1470-2045(05)70540-4; PMID: 16389187
- Hung KF, Chang CS, Liu CJ, Lui MT, Cheng CY, Kao SY. Differential expression of E-cadherin in metastatic lesions comparing to primary oral squamous cell carcinoma. J Oral Pathol Med 2006; 35:589 - 94; http://dx.doi.org/10.1111/j.1600-0714.2006.00474.x; PMID: 17032390
- Caldeira JR, Prando EC, Quevedo FC, Neto FA, Rainho CA, Rogatto SR. CDH1 promoter hypermethylation and E-cadherin protein expression in infiltrating breast cancer. BMC Cancer 2006; 6:48; http://dx.doi.org/10.1186/1471-2407-6-48; PMID: 16512896
- Graff JR, Herman JG, Lapidus RG, Chopra H, Xu R, Jarrard DF, et al. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res 1995; 55:5195 - 9; PMID: 7585573
- Hiraguri S, Godfrey T, Nakamura H, Graff J, Collins C, Shayesteh L, et al. Mechanisms of inactivation of E-cadherin in breast cancer cell lines. Cancer Res 1998; 58:1972 - 7; PMID: 9581841
- Lombaerts M, van Wezel T, Philippo K, Dierssen JW, Zimmerman RM, Oosting J, et al. E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-to-mesenchymal transition in breast cancer cell lines. Br J Cancer 2006; 94:661 - 71; PMID: 16495925
- Lind GE, Thorstensen L, Lovig T, Meling GI, Hamelin R, Rognum TO, et al. A CpG island hypermethylation profile of primary colorectal carcinomas and colon cancer cell lines. Mol Cancer 2004; 3:28; http://dx.doi.org/10.1186/1476-4598-3-28; PMID: 15476557
- Hung KF, Chang CS, Liu CJ, Lui MT, Cheng CY, Kao SY. Differential expression of E-cadherin in metastatic lesions comparing to primary oral squamous cell carcinoma. J Oral Pathol Med 2006; 35:589 - 94; http://dx.doi.org/10.1111/j.1600-0714.2006.00474.x; PMID: 17032390
- Zhong LP, Li J, Zhang CP, Zhu HG, Sun J, Zhang ZY. Expression of E-cadherin in cervical lymph nodes from primary oral squamous cell carcinoma patients. Arch Oral Biol 2007; 52:740 - 7; http://dx.doi.org/10.1016/j.archoralbio.2007.01.013; PMID: 17331461
- Bukholm IK, Nesland JM, Borresen-Dale AL. Re-expression of E-cadherin, alpha-catenin and beta-catenin, but not of gamma-catenin, in metastatic tissue from breast cancer patients [seecomments]. J Pathol 2000; 190:15 - 9; http://dx.doi.org/10.1002/(SICI)1096-9896(200001)190:1<15::AID-PATH489>3.0.CO;2-L; PMID: 10640987
- Kowalski PJ, Rubin MA, Kleer CG. E-cadherin expression in primary carcinomas of the breast and its distant metastases. Breast Cancer Res 2003; 5:R217 - 22; http://dx.doi.org/10.1186/bcr651; PMID: 14580257
- Chao YL, Shepard CR, Wells A. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol Cancer 2010; 9:179; http://dx.doi.org/10.1186/1476-4598-9-179; PMID: 20609236
- Graff JR, Gabrielson E, Fujii H, Baylin SB, Herman JG. Methylation patterns of the E-cadherin 5′ CpG island are unstable and reflect the dynamic, heterogeneous loss of E-cadherin expression during metastatic progression. J Biol Chem 2000; 275:2727 - 32; http://dx.doi.org/10.1074/jbc.275.4.2727; PMID: 10644736
- Schipper JH, Frixen UH, Behrens J, Unger A, Jahnke K, Birchmeier W. E-cadherin expression in squamous cell carcinomas of head and neck: inverse correlation with tumor dedifferentiation and lymph node metastasis. Cancer Res 1991; 51:6328 - 37; PMID: 1933895
- Tse JC, Kalluri R. Mechanisms of metastasis: epithelial-to-mesenchymal transition and contribution of tumor microenvironment. J Cell Biochem 2007; 101:816 - 29; http://dx.doi.org/10.1002/jcb.21215; PMID: 17243120
- Wells A, Yates C, Shepard CR. E-cadherin as an indicator of mesenchymal to epithelial reverting transitions during the metastatic seeding of disseminated carcinomas. Clin Exp Metastasis 2008; 25:621 - 8; http://dx.doi.org/10.1007/s10585-008-9167-1; PMID: 18600305
- Alt-Holland A, Shamis Y, Riley KN, DesRochers TM, Fusenig NE, Herman IM, et al. E-cadherin suppression directs cytoskeletal rearrangement and intraepithelial tumor cell migration in 3D human skin equivalents. J Invest Dermatol 2008; 128:2498 - 507; http://dx.doi.org/10.1038/jid.2008.102; PMID: 18528437
- Margulis A, Zhang W, Alt-Holland A, Crawford HC, Fusenig NE, Garlick JA. E-cadherin suppression accelerates squamous cell carcinoma progression in three-dimensional, human tissue constructs. Cancer Res 2005; 65:1783 - 91; http://dx.doi.org/10.1158/0008-5472.CAN-04-3399; PMID: 15753375
- Alt-Holland A, Zhang W, Margulis A, Garlick JA. Microenvironmental control of premalignant disease: the role of intercellular adhesion in the progression of squamous cell carcinoma. Semin Cancer Biol 2005; 15:84 - 96; http://dx.doi.org/10.1016/j.semcancer.2004.08.007; PMID: 15652453
- Kudo Y, Kitajjma S, Sato S, Miyauchi M, Ogawa I, Takata T. Establishment of an oral squamous cell carcinoma cell line with high invasive and p27 degradation activities from a lymph node metastasis. Oral Oncol 2003; 39:515 - 20; http://dx.doi.org/10.1016/S1368-8375(03)00015-0; PMID: 12747977
- Kudo Y, Kitajima S, Ogawa I, Hiraoka M, Sargolzaei S, Keikhaee MR, et al. Invasion and metastasis of oral cancer cells require methylation of E-cadherin and/or degradation of membranous beta-catenin. Clin Cancer Res 2004; 10:5455 - 63; http://dx.doi.org/10.1158/1078-0432.CCR-04-0372; PMID: 15328184
- Reinhold WC, Reimers MA, Maunakea AK, Kim S, Lababidi S, Scherf U, et al. Detailed DNA methylation profiles of the E-cadherin promoter in the NCI-60 cancer cells. Mol Cancer Ther 2007; 6:391 - 403; http://dx.doi.org/10.1158/1535-7163.MCT-06-0609; PMID: 17272646
- Sen GL, Webster DE, Barragan DI, Chang HY, Khavari PA. Control of differentiation in a self-renewing mammalian tissue by the histone demethylase JMJD3. Genes Dev 2008; 22:1865 - 70; http://dx.doi.org/10.1101/gad.1673508; PMID: 18628393
- Kondo Y, Shen L, Cheng AS, Ahmed S, Boumber Y, Charo C, et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet 2008; 40:741 - 50; http://dx.doi.org/10.1038/ng.159; PMID: 18488029
- Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet 2007; 39:232 - 6; http://dx.doi.org/10.1038/ng1950; PMID: 17200670
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006; 125:315 - 26; http://dx.doi.org/10.1016/j.cell.2006.02.041; PMID: 16630819
- Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 2007; 39:237 - 42; http://dx.doi.org/10.1038/ng1972; PMID: 17211412
- Duursma AM, Kedde M, Schrier M, le Sage C, Agami R. miR-148 targets human DNMT3b protein coding region. RNA 2008; 14:872 - 7; http://dx.doi.org/10.1261/rna.972008; PMID: 18367714
- Pan W, Zhu S, Yuan M, Cui H, Wang L, Luo X, et al. MicroRNA-21 and microRNA-148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J Immunol 2010; 184:6773 - 81; http://dx.doi.org/10.4049/jimmunol.0904060; PMID: 20483747
- Braconi C, Huang N, Patel T. MicroRNA-dependent regulation of DNA methyltransferase-1 and tumor suppressor gene expression by interleukin-6 in human malignant cholangiocytes. Hepatology 2010; 51:881 - 90; PMID: 20146264
- Graff JR, Gabrielson E, Fujii H, Baylin SB, Herman JG. Methylation patterns of the E-cadherin 5′ CpG island are unstable and reflect the dynamic, heterogeneous loss of E-cadherin expression during metastatic progression. J Biol Chem 2000; 275:2727 - 32; http://dx.doi.org/10.1074/jbc.275.4.2727; PMID: 10644736
- Bukholm IK, Nesland JM, Borresen-Dale AL. Re-expression of E-cadherin, alpha-catenin and beta-catenin, but not of gamma-catenin, in metastatic tissue from breast cancer patients. [seecomments] J Pathol 2000; 190:15 - 9; http://dx.doi.org/10.1002/(SICI)1096-9896(200001)190:1<15::AID-PATH489>3.0.CO;2-L; PMID: 10640987
- Dumont N, Wilson MB, Crawford YG, Reynolds PA, Sigaroudinia M, Tlsty TD. Sustained induction of epithelial to mesenchymal transition activates DNA methylation of genes silenced in basal-like breast cancers. Proc Natl Acad Sci USA 2008; 105:14867 - 72; http://dx.doi.org/10.1073/pnas.0807146105; PMID: 18806226
- Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer 2001; 1:46 - 54; http://dx.doi.org/10.1038/35094059; PMID: 11900251
- Alt-Holland A, Zhang W, Margulis A, Garlick JA. Microenvironmental control of premalignant disease: the role of intercellular adhesion in the progression of squamous cell carcinoma. Semin Cancer Biol 2005; 15:84 - 96; http://dx.doi.org/10.1016/j.semcancer.2004.08.007; PMID: 15652453
- Eriksen JG, Steiniche T, Sogaard H, Overgaard J. Expression of integrins and E-cadherin in squamous cell carcinomas of the head and neck. APMIS 2004; 112:560 - 8; http://dx.doi.org/10.1111/j.1600-0463.2004.apm1120902.x; PMID: 15601304
- Hung KF, Chang CS, Liu CJ, Lui MT, Cheng CY, Kao SY. Differential expression of E-cadherin in metastatic lesions comparing to primary oral squamous cell carcinoma. J Oral Pathol Med 2006; 35:589 - 94; http://dx.doi.org/10.1111/j.1600-0714.2006.00474.x; PMID: 17032390
- Bukholm IK, Nesland JM, Borresen-Dale AL. Re-expression of E-cadherin, alpha-catenin and beta-catenin, but not of gamma-catenin, in metastatic tissue from breast cancer patients [seecomments]. J Pathol 2000; 190:15 - 9; http://dx.doi.org/10.1002/(SICI)1096-9896(200001)190:1<15::AID-PATH489>3.0.CO;2-L; PMID: 10640987
- Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 2008; 14:79 - 89; http://dx.doi.org/10.1016/j.ccr.2008.06.005; PMID: 18598946
- Weinberg RA. Twisted epithelial-mesenchymal transition blocks senescence. Nat Cell Biol 2008; 10:1021 - 3; http://dx.doi.org/10.1038/ncb0908-1021; PMID: 18758491
- Tse JC, Kalluri R. Mechanisms of metastasis: epithelial-to-mesenchymal transition and contribution of tumor microenvironment. J Cell Biochem 2007; 101:816 - 29; http://dx.doi.org/10.1002/jcb.21215; PMID: 17243120
- Ateeq B, Unterberger A, Szyf M, Rabbani SA. Pharmacological inhibition of DNA methylation induces proinvasive and prometastatic genes in vitro and in vivo. Neoplasia 2008; 10:266 - 78; PMID: 18320071
- Chik F, Szyf M. Effects of specific DNMT gene depletion on cancer cell transformation and breast cancer cell invasion; toward selective DNMT inhibitors. Carcinogenesis 2011; 32:224 - 32; http://dx.doi.org/10.1093/carcin/bgq221; PMID: 20980350
- Frost P, Kerbel RS, Hunt B, Man S, Pathak S. Selection of metastatic variants with identifiable karyotypic changes from a nonmetastatic murine tumor after treatment with 2'-deoxy-5-azacytidine or hydroxyurea: implications for the mechanisms of tumor progression. Cancer Res 1987; 47:2690 - 5; PMID: 2436755
- Yu Y, Zeng P, Xiong J, Liu Z, Berger SL, Merlino G. Epigenetic drugs can stimulate metastasis through enhanced expression of the pro-metastatic Ezrin gene. PLoS One 2010; 5:e12710; http://dx.doi.org/10.1371/journal.pone.0012710; PMID: 20856924
- Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 2007; 39:237 - 42; http://dx.doi.org/10.1038/ng1972; PMID: 17211412
- Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, et al. Epigenetic stem cell signature in cancer. Nat Genet 2007; 39:157 - 8; http://dx.doi.org/10.1038/ng1941; PMID: 17200673
- Reinhold WC, Reimers MA, Maunakea AK, Kim S, Lababidi S, Scherf U, et al. Detailed DNA methylation profiles of the E-cadherin promoter in the NCI-60 cancer cells. Mol Cancer Ther 2007; 6:391 - 403; http://dx.doi.org/10.1158/1535-7163.MCT-06-0609; PMID: 17272646
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006; 125:315 - 26; http://dx.doi.org/10.1016/j.cell.2006.02.041; PMID: 16630819
- Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 2007; 39:237 - 42; http://dx.doi.org/10.1038/ng1972; PMID: 17211412
- Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev 2006; 20:1123 - 36; http://dx.doi.org/10.1101/gad.381706; PMID: 16618801
- Issa JP, Kantarjian HM. Targeting DNA methylation. Clin Cancer Res 2009; 15:3938 - 46; http://dx.doi.org/10.1158/1078-0432.CCR-08-2783; PMID: 19509174
- Graff JR, Gabrielson E, Fujii H, Baylin SB, Herman JG. Methylation patterns of the E-cadherin 5′ CpG island are unstable and reflect the dynamic, heterogeneous loss of E-cadherin expression during metastatic progression. J Biol Chem 2000; 275:2727 - 32; http://dx.doi.org/10.1074/jbc.275.4.2727; PMID: 10644736
- Benton G, Crooke E, George J. Laminin-1 induces E-cadherin expression in 3-dimensional cultured breast cancer cells by inhibiting DNA methyltransferase 1 and reversing promoter methylation status. FASEB J 2009; 23:3884 - 95; http://dx.doi.org/10.1096/fj.08-128702; PMID: 19635753
- Tseng CW, Lin CC, Chen CN, Huang HC, Juan HF. Integrative network analysis reveals active microRNAs and their functions in gastric cancer. BMC Syst Biol 2011; 5:99; http://dx.doi.org/10.1186/1752-0509-5-99; PMID: 21703006
- Chen Y, Song Y, Wang Z, Yue Z, Xu H, Xing C, et al. Altered expression of MiR-148a and MiR-152 in gastrointestinal cancers and its clinical significance. J Gastrointest Surg 2010; 14:1170 - 9; http://dx.doi.org/10.1007/s11605-010-1202-2; PMID: 20422307
- Chen Y, Song Y, Wang Z, Yue Z, Xu H, Xing C, et al. Altered expression of MiR-148a and MiR-152 in gastrointestinal cancers and its clinical significance. J Gastrointest Surg 2010; 14:1170 - 9; http://dx.doi.org/10.1007/s11605-010-1202-2; PMID: 20422307
- Deng S, Calin GA, Croce CM, Coukos G, Zhang L. Mechanisms of microRNA deregulation in human cancer. Cell Cycle 2008; 7:2643 - 6; http://dx.doi.org/10.4161/cc.7.17.6597; PMID: 18719391
- Hanoun N, Delpu Y, Suriawinata AA, Bournet B, Bureau C, Selves J, et al. The silencing of microRNA 148a production by DNA hypermethylation is an early event in pancreatic carcinogenesis. Clin Chem 2010; 56:1107 - 18; http://dx.doi.org/10.1373/clinchem.2010.144709; PMID: 20431052
- Kumaki Y, Oda M, Okano M. QUMA: quantification tool for methylation analysis. Nucleic Acids Res 2008; 36:W170 - 5; http://dx.doi.org/10.1093/nar/gkn294; PMID: 18487274
- Carlson MW, Alt-Holland A, Egles C, Garlick JA. Three-dimensional tissue models of normal and diseased skin. Curr Protoc Cell Biol 2008; Chapter 19:Unit 19.9; PMID: 19085986