Abstract
Chromatin dynamics across cellular differentiation states is an emerging perspective from which the mechanism of global gene expression regulation may be better understood. While the roles of some histone marks have been partially interpreted in terms of their association with gene transcription, the dynamics of histone marks from a loci-specific perspective during cellular differentiation is not well studied. We established a method to systematically assess the histone modification variations of genes across various cellular differentiation states. We calculated the histone modification variation scores of H3K4me3, H3K27me3 and H3K36me3 for over 1300 curated transcription factors (TFs) during human blood cell differentiation. Hematopoietic-specific TFs (identified by literature mining) were significantly overrepresented by TFs with higher histone modification variation scores. Hierarchical clustering of all TFs based on the histone modification variation scores defined a group of TFs where known or potential hematopoietic-specific TFs were remarkably enriched. Our results suggest that local chromatin state dynamics of transcription factors across cellular differentiation states could imply cell lineage-specific functions. More importantly, our method can be applied to broader systems, holding the promise to discover de novo, lineage-specific TFs by interrogating their histone modification dynamics across cell lineages.
Introduction
Hematopoiesis, which is the formation and development of blood cells, represents one of the best-studied models of cellular differentiation. While dozens of blood cell lineages have been identified, all can be traced back to hematopoietic stem cells (HSCs) as the common progenitor. HSCs continually renew themselves to maintain a pool of blood stem cells; however, upon stimulation by cytokines, HSCs give rise to multipotent progenitor cells (hematopoietic multipotent cells: HPCs) with restricted differentiation potential, which can further develop into various terminally differentiated blood cells.Citation1-Citation3 The diversity of cell types and clear tractability of cell differentiation pathways makes hematopoiesis a very attractive model for the investigation of cellular differentiation.
Cells of different identities have unique gene expression profiles, which are hierarchically regulated by interactions of transcription factors (TFs).Citation4 Cellular differentiation, the transformation of a cell from one identity (i.e., progenitor cells) to another (i.e., progeny cells), can be attributed to the alternation of varying TF complexes. Over last two decades, the involvement of key TFs in establishing specific hematopoietic lineages has been revealed.Citation1,Citation5 A recent genome-wide characterization of the binding sites of 10 hematopoietic-specific TFs in a blood progenitor cell line highlighted the multi-player nature of the TF complexes involved in hematopoiesis.Citation6
The histone octamer, on which the DNA of eukaryotic organisms is wound, plays a pivotal role in the regulation of gene expression primarily through conformational changes, which control the accessibility of the transcriptional machinery to a DNA segment. The N-terminal tails of histones are subjected to various covalent chemical modifications, such as methylation, acetylation and ubiquitination. Recent histone modification ChIP‑seq assays (chromatin immunoprecipitation coupled with high-throughput sequencing) have provided an extraordinarily detailed atlas of histone modifications at a genome-wide scale.Citation7-Citation10 The availability of these resources prompted a series of follow-up studies that expanded the arsenal of computational tools in bioinformatics and shed light on the roles that histone modifications may play in gene transcription.Citation11-Citation14
It has been established that histone modifications vary in their distribution over a certain genomic region and the direction in which they are linked to gene expression levels. Indeed, H3K4me3 is enriched at the TSS (transcription start site) and is often associated with gene activation.Citation7 H3K36me3, an active mark highly correlated with transcriptional elongation, spreads over the entirety of the gene, peaking at the TTS (transcription termination site).Citation7 H3K27me3 is a repressive mark that can stretch from the promoter region of a gene to regions downstream of the TTS.Citation7 Intuitively, one may postulate that histone modifications are connected to gene expression levels. Through the use of computational measures, it has been shown that histone modifications are indeed predictive of gene expression levels.Citation15
While disputes may remain over whether the role of histone modification in gene expression is causal or concomitant, it has been established that certain combinatorial histone modification patterns have significant implications on gene expression during cellular differentiation. Bivalent genes, concurrently marked with both H3K4me3 and H3K27me3, were first identified as being abundant in embryonic stem cells (ES cells).Citation16 Over time, the significance of bivalency in stem cell differentiation has been better understood. In fact, bivalency represents a parsimonious and fixable mechanism that enables either activation, by removing H3K27me3, or repression, by removing H3K4me3 during cellular differentiation. Previous studies focused on the global histone modification dynamics during cellular differentiation.Citation17,Citation18 For instance, by comparing the epigenomic landscapes of human ES cells, Hawkins et al. found that the repressive marks, H3K9me3 and H3K27me3, expand significantly in fibroblasts.Citation18
Characterization of the histone modification dynamics from a loci-specific perspective during cellular differentiation holds promise to determine key players for the cellular differentiation process. To verify our hypothesis, we investigated the histone modification variations of human transcription factors (TFs) using a publicly available, histone modification ChIP-seq data set containing 4 blood cell lineages, including CD133+ cells (HSCs/ HPCs),Citation19 CD36+ cells (erythrocyte precursors),Citation19 GM12878 cells (B lymphoblastoid) and CD4+ T cells. We sought to establish a method to assess the variations of histone modifications across cellular differentiation states. Our results indicated that, in most cases, hematopoietic-specific TFs are characterized by higher histone modification variations. Hierarchical clustering analysis of all curated human TFs, based on three histone modification variation scores, defined a group of TFs where known and potential hematopoietic-specific TFs were remarkably enriched. Our method can be applied to other cellular systems and promises to be a useful tool for the de novo discovery of lineage-specific TF functions.
Results and Discussion
Collection of a comprehensive gene list of hematopoietic-specific transcription factors
Two different approaches were used in parallel to generate a relatively comprehensive gene list of hematopoietic-specific transcription factors (HPTFs). First, keywords encompassing the names of a wide range of hematopoietic cells (such as blood stem cells, lymphocytes, T cells, B cells, and erythroids) were used to retrieve articles from the PubMed database. By manually checking over 2000 preliminarily matched titles and corresponding abstracts, we identified approximately 50 transcription factors described in the literature to be unambiguously involved in hematopoiesis. The gene names, which encode the candidate hematopoietic-specific transcription factors, were queried against HGNC database (www.genenames.org) to obtain the corresponding Ensembl and RefSeq gene IDs. Second, as an alternative approach, the list of 1391 curated, human TFsCitation4 was uploaded to DAVID (david.abcc.ncifcrf.gov) to identify annotations for each TF. Each annotation was manually checked to determine if a given TF was specific to hematopoiesis. Using this method, approximately 60 hematopoietic-specific TF genes were identified. Finally, the two TF lists were combined to obtain the final version consisting of 63 hematopoietic-specific TFs, which were of good representation and comprehensive coverage. The final list of HPTFs containing the gene symbols and Ensembl and RefSeq IDs is available in additional file 1.
TFs with higher histone modification variation scores were significantly enriched in the HPTF list
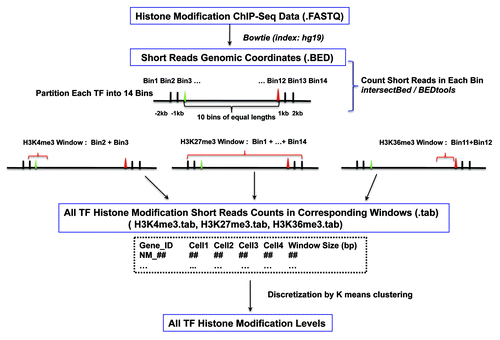
Following the pipeline illustrated in , histone modification variation scores for all 1329 TFs in 4 blood cell lineages were calculated for each histone mark (see Materials and Methods for details). For a given TF, when calculating the variation score for a histone mark, the maximum Euclidean distance between two cells was used as the histone variation score for the TF. Based on our method, histone modification variation scores of TFs fall into 4 categories [i.e., “high,” “medium,” “low” or “none” (no observable variation)].
Figure 1. Schematic representation of the analysis pipeline.
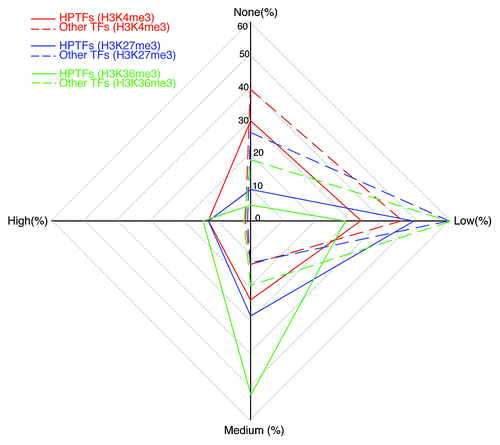
Using the list of HPTFs obtained through literature mining, comparisons of the histone modification variation scores between HPTFs and non-HPTFs were performed. Significant differences between these two groups of TFs could be appreciated in terms of histone modification variations (). In general, the majority of non-HPTFs were characterized by low-to-no variation for all histone marks (e.g., 85.4% of non‑HPTFs show low-to-no variation in H3K4me3 levels). This trend also holds, to some extent, for HPTFs (e.g., 63.5% of the HPTFs show low-to-no variation in H3K4me3 levels). However, in the HPTF list, TFs with medium or high variation scores for all the three histone marks were significantly enriched. The enrichment for TFs with high variation scores was even more dramatic. Indeed, 12.7% of the HPTFs showed high variation in terms of H3K4me3 levels, whereas only 1.5% of the non-HPTFs were characterized by high variation in H3K4me3 levels (Fisher exact test p value = 1.775 × 10−5). Taken together, during blood cell differentiation, HPTFs, on average, were associated with more dramatic chromatin dynamics compared with non‑HPTFs.
Figure 2. HPTFs are characterized by increased histone modification variations during blood cell differentiation. The radar plot has 4 poles, each corresponding to a level of histone modification variation. Level of variation is designated as “none,” “low,” “medium” and “high.” On each of the 4 axes, the percentage of TFs with a corresponding histone modification variation level is noted. For a given histone mark, points representing the variation levels on the 4 axes are connected sequentially. Different histone marks are illustrated by different colors. Dashed lines depict the variations of other TFs (apart from HPTFs), and solid lines depict the variations of HPTFs.
Hierarchical clustering analysis defined a group of TFs where HPTFs were extraordinarily enriched
Our results indicate that HPTFs, on average, had higher variation scores for any histone mark during blood cell differentiation. However, because chromatin states are often dictated by combinatorial histone marks and each histone also has its specific implications, we explored the combinatorial histone modification variations across all TFs. In an attempt to find some combinatorial histone modification variation patterns that may be common in HPTFs, a hierarchical clustering analysis was performed. Each TF was represented by 3 values corresponding to variation scores for H3K4me3, H3K27me3 and H3K36me3. We cut the tree generated by hierarchical clustering into 5 clusters, and the overlap of TFs of each cluster with the HPTF list was investigated (, ). As shown in , 9 out of 21 (43%) TFs in Group E are known to be HPTFs. Because the HPTFs represent less than 5% of the total TFs (63/1329), there is an extraordinary enrichment of HPTFs in Group E (p value = 5.6 × 10−9).
Figure 3. Hierarchical clustering of all TFs based on the variation scores of three histone marks. The tree was cut into 5 groups, each encompassing a variable number of TFs. The order of the groups (A to E) was based on the group size.
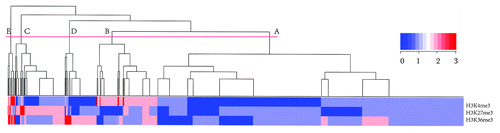
Table 1. Features of the Five Subclasses of TFs and Overlap with HPTFs
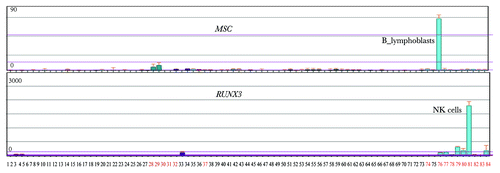
Curious about the functions of the remaining 12 TFs in Group E that did not match HPTFs list, we checked the annotations for these 12 TFs in Biogps (biogps.org). Surprisingly, 6 TFs displayed either blood cell-specific expression patterns or are known to interact with HPTFs (). As illustrated in , RUNX3 is exclusively expressed in blood cells, and there is in vivo evidence indicating that RUNX3 interacts with AML, a known HPTF (). Therefore, it is highly likely that RUNX3 is a potential HPTF.
Table 2. Potential HPTFs in Group E
Figure 4. Two potential HPTFs within group E show blood cell-specific expression patterns. A total of 84 human cell lines or primary tissues were characterized by expression profiling. Tissues or cell lines, denoted by numbers with black colors, represent a broad range of non-blood tissues (or cell lines), such as kidney, thymus, liver, lung, prostate, heart and others. Red numbers 28–37 denote MOLT-4, K562, lymphoma, HL‑60, Raji and early erythroid cells. Red numbers 74 to 84 denote CD34+ cells, B lymphoblasts, CD19+ B cells, dendritic cells, CD8+ T cells, CD4+ T cells, CD56+ NK cells, CD33+ myeloid cells, CD14+ monocytes and whole blood. The expression profiles for MSC and RUNX3 were obtained from Biogps (biogps.org).
The remarkable enrichment of HPTFs in Group E could lead to the conclusion that some TFs of related or similar functions (e.g., involvement in hematopoiesis) in a set of cells share similar combinatorial histone modification variation patterns during cellular differentiation. A closer look into the 5 groups of TFs (Groups A-E) revealed marked differences in terms of histone modification variation patterns (). Group A comprised the largest number of TFs (893 in total), and none of the TFs in Group A showed medium/high variation in H3K4me3 levels or H3K27me3 levels. Only 10.9% of the TFs in Group A showed medium/high variations in H3K36me3 levels. Therefore, in general, TFs in Group A were characterized by minor variations for almost all three histone mark levels. Although TFs in Groups B-D showed major variations in H3K4me3, H3K27me3 and H3K36me3 respectively, there was no dramatic enrichment of HPTFs in any of the three groups. Noticeably, the TFs in Group E were characterized by major variations for both H3K4me3 and H3K36me3, while 66.7% of the TFs in Group E showed high/medium variations in H3K27me3 levels.
Histone modification dynamics of TFs correlated with their lineage-specific functions
Our method enabled a systematic assessment of histone modification dynamics of all TFs during blood cell differentiation. These results show that HPTFs have dramatic histone modification variations during these processes. It would be highly desirable to investigate whether these dramatic histone modification dynamics of HPTFs have functional implications. To this end, we focused on the HPTFs belonging to Group E. Loci-specific wiggle (WIG) files were generated from the ChIP‑seq BED files and uploaded to the UCSC genome browser as custom tracks. Combinatorial signals of the three histone marks could define the chromatin state of the corresponding gene. As shown in , GATA1 was characterized by low signals of both H3K4me3 and H3K36me3 and high signals for H3K27me3, a typical repressed state. In erythrocyte precursor cells, the signal for H3K4me3 was sharply increased, while significant signals for H3K36me3 were observed, and the signal for H3K27me3 was decreased. In contrast, GATA1 in B lymphoblastoid cells remained in a repressed state. GATA1 is a known TF that functions in erythrocyte differentiation but does not play a role in lymphocyte development; therefore, the chromatin dynamics of GATA1 during blood cell differentiation perfectly matches its function.
Figure 5. Visualization of the histone modification dynamics of some HPTFs. From left to right, the histone modification profiles are displayed for the genomic loci of HPTFs GATA1, Aiolos and PAX5 in CD133+ cells (HSCs/ HPCs), CD36+ cells (erythrocyte precursors), GM12878 cells (B lymphoblastoids) and CD4+ T cells. The histone modification ChIP‑seq tag wiggle files were uploaded to the UCSC genome browser and visualized as custom tracks. The panels in red, black and blue denote the profiles of H3K4me3, H3K27me3 and H3K36me3, respectively. The structures of genes are shown at the top panels.
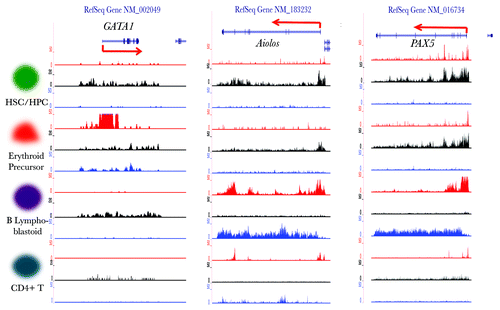
Two more examples are shown in , namely Aiolos and PAX5, both of which have been reported to function specifically during B lymphocyte development. The chromatin states of these two genes become selectively activated in B lymphoblastoid cells ().
Comparison of the predictive performance of our method with that of microarray method
By systematic assessment of histone modification dynamics of TFs during blood cell differentiation, we have shown that, HPTFs tend to have higher histone modification variations. In addition, clustering analysis has revealed that, TFs with simultaneous high variations for H3K4me3, H3K27me3 and H3K36me3 are more likely to be TFs with lineage-specific functions. These observations point to the possibility of predicting TFs with lineage-specific functions by assessing histone modification dynamics (hereafter refered to as the epigenetical approach).
We made predictions of HPTFs using the method as detailed in the materials and method section. As it is shown in , single histone marks all showed similar specificities as that of microarray. While H3K36me3 displayed a significantly higher sensitivity than that of microarray (p < 0.001), the rest two histone marks (H3K4me3 and H3K27me3) were both inferior to microarray due to significantly lower sensitivities (p < 0.001). When it comes to double or triple histone marks, again their sensitivities were largely compromised. However, in any case of the combinations of histone marks, the specificity always outperformed that of microarray. As it was assessed by MCC (Matthews correlation coefficient), a balanced measure that takes into account true and false positives and negatives, microarray only showed a moderate performance, e.g., H3K36me3 coupled with H3K4me3 was superior to microarray.
Table 3. Prediction of lineage-specific TFs: a comparison of the epigenetical approach with microarray analysis
In summary, when it comes to the comparison of predictive performance of our epigenetical approach with microarray method, we have shown that H3K36me3 alone is at least as good as microarray in both sensitivity and specificity. This result stands to reason as H3K36me3 density as assigned to a gene is explicitly linked to the gene expression level.Citation7 However, the other two histone marks, namely H3K4me3 and H3K27me3, are linked to gene expression level in a much more subtle way. H3K4me3 marks the promoter regions and corresponds to RNAP II binding sites, however, RNAP II binding does not necessarily mean gene transcription actually occursCitation20. Besides, H3K27me3, an earlier defined repressive mark, has been revealed to play a more complex role in differentiation than one has appreciated over the last few years.Citation16 It has been very recently reported that, in some way, H3K27me3 densities could be positively associated with gene expression levels.Citation21 Our results suggest that, when there are ChIP‑seq data available for multiple histone marks in different cell lineages sharing the same progenitor cells, it holds promise to really capture some crucial TFs by picking those TFs of simultaneous high variations for multiple histone marks.
Materials and Methods
Histone modification ChIP-seq raw data collection and preprocessing
The H3K4me3, H3K27me3 and H3K36me3 ChIP‑seq raw data of CD133+ cells (HSCs/ HPCs), CD36+ cells (erythrocyte precursors), and GM12878 cells (B lymphoblastoid) were downloaded as FASTQ format files. The Solexa short reads were mapped to the human genome using Bowtie (index file: hg19.fa) with default parameters.Citation22 Each of the three histone modification ChIP‑seq data sets of CD4+ T cells were downloaded as BED format files. Because the BED files were generated based on human genome assembly hg18, LiftOver (UCSC genome browser) was used to transform the hg18-based BED files into hg19-based BED files. The detailed information concerning the sources of the histone modification data and cell identities is described in additional file 2. PCR redundancies were removed from all BED files by counting the multiple reads that mapped to the same genomic position (i.e., chromosome names, start and end positions and strand types that were identical) before further analysis was performed. This was performed to reduce noises attributable to genomic regional biases during the PCR amplification step in the ChIP-seq assays.
Establishment of bin-based histone modification read count vectors of all curated human TFs
The 1391 curated human TF gene IDs from the Ensembl Genome Browser (Ensembl) were downloaded from the supplementary files of Vaquerizas et al. (2009).Citation4 All gene IDs were uploaded to DAVID (david.abcc.ncifcrf.gov), and 1366 out of the 1391 total Ensembl gene IDs could be unambiguously converted to RefSeq IDs. The genomic coordinates of the RefSeq IDs were extracted from the hg19 assembly based on the gene annotation sheet downloaded from the UCSC table browser. Approximately 18 RefSeq IDs had multiple genomic coordinates, likely representing genome duplication. Because the role of histone modification in genome duplication is not within the scope of this study and no evidence has shown that there is a bias in the probabilities of certain classes of TFs being duplicated, these 18 RefSeq IDs were removed. There were also several cases where a given RefSeq ID was matched to multiple Ensembl IDs. A closer look into these cases revealed that redundant Ensembl IDs represented either pseudo-genes or alias IDs. In such cases, pseudo-genes was removed and only the most recently updated Ensembl IDs were kept for each given RefSeq ID. In the end, the genomic coordinates of 2113 RefSeq IDs (representing 1329 ensemble gene IDs) were identified.
Each TF out of the total 2113 RefSeq IDs was partitioned into 14 bins. The first two bins were 2000 basepairs (bp) upstream, 1000 bp upstream, and 1000 bp upstream of the TSS. The last two bins were from the TTS to 1000 bp downstream, 1000 bp downstream and 2000 bp downstream. The open reading frame of each gene, from TSS to TTS, was partitioned into 10 equal parts based on gene length.
To make comparisons of all TFs in terms of histone modification variations, genes in the list of 1329 TFs that had more than one corresponding RefSeq ID were represented by the RefSeq genes with longest transcripts.
Calculation of histone modification variation scores
For each cell type, histone modification tag densities, as calculated by dividing the read counts by window sizes (), were split into 4 nominal variables representing high, moderate, low and extremely low levels (dubbed H, M, L, and LL, respectively). The boundaries between groups were determined by k-means clustering. One hundred repetitions were performed for each histone modification density vector for a given cell type, and the medians were used to define the boundaries. The histone modification tag densities (H, M, L and LL) corresponded to the numeric values 4, 3, 2 and 1. For a given histone modification type, each TF was represented by a vector of 4 numerical values representing the histone modification levels of the 4 blood cell lineages. For a given histone modification type, the histone modification variation score (HMV) for a given TF in each of the 4 blood cell lineages was calculated using the formula:
HMV = max {Dist (Ci, Cj)} where 1 < = i < j < = 4
Ci represents a specific blood cell lineage and is characterized by discrete values for the three histone marks. “Dist” represents the Euclidian distance of Ci and Cj. Because each histone modification was categorized into 4 levels, the HMV was also represented by 4 levels (i.e., 0, 1, 2 and 3).
Microarray data analysis and prediction of HPTFs by microarray data and histone modification dynamics
The microarray raw data for the 4 types of blood cells were downloaded from GEO database (GEO IDs are GSE12646, GSE26312). As these microarray raw data were from different platforms, it was not applicable to do expression indexing for them together (e.g., using RMA algorithm). Therefore, we performed present/absent calls using MAS5.0 algorithm from Bioconductor package (www.bioconductor.org). The rationale for making prediction of HPTFs by microarray was that, if a TF encoding gene showed ON/OFF variations across the 4 blood cells, this TF was predicted as a HPTF.
In order to make prediction of HPTFs based on histone modification dynamics, we made binary partition of all TFs’ histone modification variation scores (i.e., combining histone modification variation scores “0” and “1 ” into “Low_V,” “2” and “3” into “High_V”). For each single histone mark, a TF with a “High_V” was predicted as a HPTF. When prediction was made based on more than one histone mark, a TF with simultaneous “High_Vs” was predicted. The list of 63 HPTFs as detailed in additional file 1 was used as the benchmark to compare the performances of predictions made by microarray method, single histone marks and combinations of histone marks.
Conclusion
As epigenetics takes center stage, interest in unraveling the role of histone modifications during cellular differentiation is increasing. Here, we presented a pipeline for assessing the histone modification variations of genes involved in hematopoietic differentiation. Our results showed that, in most of cases, hematopoietic-specific TFs had higher variation scores for all three histone marks (H3K4me3, H3K27me3 or H3K36me3). These higher variations of histone modifications corresponding to hematopoietic-specific TFs represent dramatic chromatin state changes leading to the activation or repression of certain genes. Interestingly, clustering of TFs based on histone modification variation scores defined a group of TFs where known or potential hematopoietic-specific TFs were remarkably enriched. Our results strongly suggest that investigation of loci-specific chromatin dynamics during cellular differentiation holds promise to identify TFs of lineage-specific function.
| Abbreviations: | ||
| TF | = | transcription factor |
| HPTF | = | hematopoietic-specific transcription factor |
Additional material
Download Zip (16.2 KB)Acknowledgments
The authors would like to thank Xiaole Shirley Liu, Tonghua Li, Jiangming Sun, Cizhong Jiang and Cheng Li for very helpful suggestions and critical comments. We thank Min Li, Meng Zhou and Kai Fu for help with downloading part of the raw data, and Tao Liu for sharing several python scripts. This study was supported by funding from the National Natural Science Foundation of China (31071114), the National Basic Research Program of China (973 Program; Nos. 2010CB944904 and 2011CB965104) and the Shanghai Rising-Star Program (10QA1407300).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell 2008; 132:631 - 44; http://dx.doi.org/10.1016/j.cell.2008.01.025; PMID: 18295580
- Orkin SH. Diversification of haematopoietic stem cells to specific lineages. Nat Rev Genet 2000; 1:57 - 64; http://dx.doi.org/10.1038/35049577; PMID: 11262875
- Orkin SH. Transcription factors and hematopoietic development. J Biol Chem 1995; 270:4955 - 8; PMID: 7890597
- Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. A census of human transcription factors: function, expression and evolution. Nat Rev Genet 2009; 10:252 - 63; http://dx.doi.org/10.1038/nrg2538; PMID: 19274049
- Zhang P, Zhang X, Iwama A, Yu C, Smith KA, Mueller BU, et al. PU.1 inhibits GATA-1 function and erythroid differentiation by blocking GATA-1 DNA binding. Blood 2000; 96:2641 - 8; PMID: 11023493
- Wilson NK, Foster SD, Wang X, Knezevic K, Schutte J, Kaimakis P, et al. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell 2010; 7:532 - 44; http://dx.doi.org/10.1016/j.stem.2010.07.016; PMID: 20887958
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell 2007; 129:823 - 37; http://dx.doi.org/10.1016/j.cell.2007.05.009; PMID: 17512414
- Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 2008; 40:897 - 903; http://dx.doi.org/10.1038/ng.154; PMID: 18552846
- Koch CM, Andrews RM, Flicek P, Dillon SC, Karaoz U, Clelland GK, et al. The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res 2007; 17:691 - 707; http://dx.doi.org/10.1101/gr.5704207; PMID: 17567990
- Zhang Y, Shin H, Song JS, Lei Y, Liu XS. Identifying positioned nucleosomes with epigenetic marks in human from ChIP-Seq. BMC Genomics 2008; 9:537; http://dx.doi.org/10.1186/1471-2164-9-537; PMID: 19014516
- Yu H, Zhu S, Zhou B, Xue H, Han JD. Inferring causal relationships among different histone modifications and gene expression. Genome Res 2008; 18:1314 - 24; http://dx.doi.org/10.1101/gr.073080.107; PMID: 18562678
- Xu H, Wei CL, Lin F, Sung WK. An HMM approach to genome-wide identification of differential histone modification sites from ChIP-seq data. Bioinformatics 2008; 24:2344 - 9; http://dx.doi.org/10.1093/bioinformatics/btn402; PMID: 18667444
- He HH, Meyer CA, Shin H, Bailey ST, Wei G, Wang Q, et al. Nucleosome dynamics define transcriptional enhancers. Nat Genet 2010; 42:343 - 7; http://dx.doi.org/10.1038/ng.545; PMID: 20208536
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008; 9:R137; http://dx.doi.org/10.1186/gb-2008-9-9-r137; PMID: 18798982
- Karlić R, Chung HR, Lasserre J, Vlahovicek K, Vingron M. Histone modification levels are predictive for gene expression. Proc Natl Acad Sci USA 2010; 107:2926 - 31; http://dx.doi.org/10.1073/pnas.0909344107; PMID: 20133639
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006; 125:315 - 26; http://dx.doi.org/10.1016/j.cell.2006.02.041; PMID: 16630819
- Attema JL, Papathanasiou P, Forsberg EC, Xu J, Smale ST, Weissman IL. Epigenetic characterization of hematopoietic stem cell differentiation using miniChIP and bisulfite sequencing analysis. Proc Natl Acad Sci USA 2007; 104:12371 - 6; http://dx.doi.org/10.1073/pnas.0704468104; PMID: 17640913
- Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 2010; 6:479 - 91; http://dx.doi.org/10.1016/j.stem.2010.03.018; PMID: 20452322
- Cui K, Zang C, Roh TY, Schones DE, Childs RW, Peng W, et al. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell 2009; 4:80 - 93; http://dx.doi.org/10.1016/j.stem.2008.11.011; PMID: 19128795
- Muse GW, Gilchrist DA, Nechaev S, Shah R, Parker JS, Grissom SF, et al. RNA polymerase is poised for activation across the genome. Nat Genet 2007; 39:1507 - 11; http://dx.doi.org/10.1038/ng.2007.21; PMID: 17994021
- Young MD, Willson TA, Wakefield MJ, Trounson E, Hilton DJ, Blewitt ME, et al. ChIP-seq analysis reveals distinct H3K27me3 profiles that correlate with transcriptional activity. Nucleic Acids Res 2011; 39:7415 - 27; http://dx.doi.org/10.1093/nar/gkr416; PMID: 21652639
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25; http://dx.doi.org/10.1186/gb-2009-10-3-r25; PMID: 19261174