Abstract
Cytosine DNA methylation is an epigenetic mark frequently associated with silencing of genes and transposons. In Arabidopsis, the establishment of cytosine DNA methylation is performed by DOMAINS REARRANGED METHYLTRANSFERASE 2 (DRM2). DRM2 is guided to target sequences by small interfering RNAs (siRNAs) in a pathway termed RNA-directed DNA methylation (RdDM). We performed a screen for mutants that affect the establishment of DNA methylation by investigating genes that contain predicted RNA-interacting domains. After transforming FWA into 429 T-DNA insertion lines, we assayed for mutants that exhibited a late-flowering phenotype due to hypomethylated, thus ectopically expressed, copies of FWA. A T-DNA insertion line within the coding region of the spliceosome gene SR45 (sr45-1) flowered late after FWA transformation. Additionally, sr45-1 mutants display defects in the maintenance of DNA methylation. DNA methylation establishment and maintenance defects present in sr45-1 mutants are enhanced in dcl3-1 mutant background, suggesting a synergistic cooperation between SR45 and DICER-LIKE3 (DCL3) in the RdDM pathway.
T-DNA insertion line within the coding region of the spliceosome gene SR45 (sr45–1) flowered late after FWA transformation. Additionally, sr45–1 mutants display defects in the maintenance of DNA methylation. DNA methylation establishment and maintenance defects present in sr45–1 mutants are enhanced in dcl3–1 mutant background, suggesting a synergistic cooperation between SR45 and DICER-LIKE3 (DCL3) in the RdDM pathway.
Results and Discussion
DRM2 carries out all known DNA methylation establishment—or de novo methylation—in Arabidopsis thaliana.Citation1 Given that DRM2 is guided by both siRNAs and long non-coding RNAs,Citation2,Citation3 we screened a collection of homozygous lines carrying T-DNA insertion in genes containing known or predicted RNA-interacting domains.Citation4 For this screen we used FWA transgene silencing as a reporter system. FWA is a homeodomain transcription factor that has two tandem repeats in its promoter. In wild type plants, the endogenous FWA repeats are stably methylated and FWA is silenced. However, hypomethylation in its promoter region leads to an ectopic expression and a late-flowering phenotype.Citation5 After FWA transformation, plants with an intact de novo methylation machinery are able to methylate and silence the transgenic FWA, while mutants affecting de novo methylation express the transgene and flower late.Citation6,Citation7
Following this strategy, we isolated a line containing a T-DNA insertion in ARGININE/SERINE-RICH 45 (SR45) that flowered slightly late before FWA transformation, but showed a major late flowering phenotype after transformation (, Fig. S1). The sr45–1 mutant has been reported to show a late flowering phenotype due to an increased expression of FLOWERING LOCUS C (FLC).Citation8 To assess whether the observed effect was mediated by FLC alone, we analyzed the methylation status of FWA in sr45–1 mutants after transformation. Bisulfite sequencing analyses revealed that the endogenous FWA methylation was not affected in the CG-dinucleotide context, but did display a defect in non-CG methylation, which is consistent with other mutants in the DRM2 pathway.Citation9 However, the transgenic copy of FWA exhibited reduced methylation levels in every sequence context (CG, CHG, and CHH; H being A, T, or G)(). This reduction in methylation is correlated with the late flowering phenotype and ectopic FWA expression in the transgenic plants (Figs. S2A, B and C). Moreover, the introduction of an SR45 copy into sr45–1, partially restores the ability to methylate FWA to wild type levels (Figs. S2D and E), ruling out that the sole increment in FLC levels accounted for the observed late flowering phenotype. It is worth noting that some individual mutant plants are able to establish methylation at or near wild-type levels, indicating a degree of stochasticity. However, on average, we can safely say that sr45 mutant plants have impaired DNA methylation establishment capacity.
Figure 1.sr45–1 de novo DNA methylation phenotype. (A) Flowering time of Columbia, sr45–1, dcl3–1 and sr45–1, dcl3–1 double before and after FWA transformation. Flowering time is measured as the total number of leaves at the time of flowering. (B) Methylation levels at endogenous and transgenic FWA after FWA transformation. The 594 base-pair repeated region in the 5′ UTR was analyzed. The methylation state of the FWA endogene should remain unaltered by the presence of the FWA transgene. All samples were analyzed in the T1 generation.
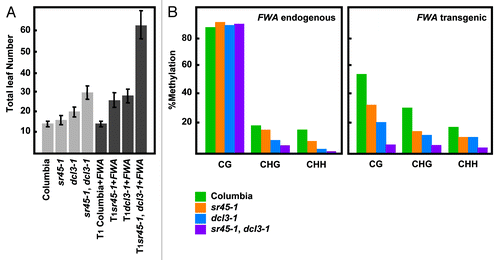
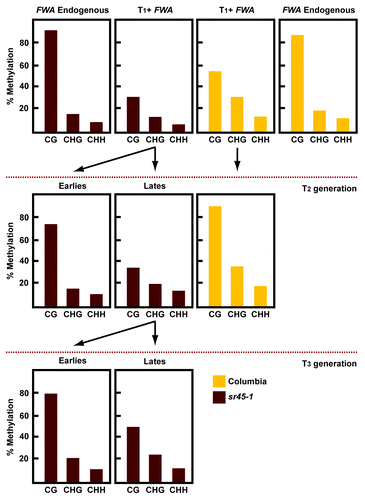
We further analyzed the progression of FWA de novo methylation across subsequent generations in sr45–1. In wild type, the FWA transgene did not reach full levels of methylation until the T2 generation (). In sr45–1 mutants, the levels of methylation at FWA transgene are reduced in the T1 generation, but in some cases complete methylation does occur until the T3 generation or later (). Thus, the sr45–1 mutant only partially impairs the de novo methylation machinery, and wild type methylation levels are regained in later generations. These observations are consistent with the slow intergenerational silencing that has been reported in other transgene systems.Citation10
Figure 2. Analysis of the FWA transgene methylation generationally. Methylation levels at endogenous and transgenic FWA after FWA transformation. Endogenous FWA was only analyzed in the T1 generation. Transgenic FWA was analyzed during three generations after transformation.
To date, every mutant that has been shown to be defective in de novo methylation is also defective in the maintenance of non-CG methylation. We, therefore, examined the methylation status at known RdDM targets such as FWA, MEA-ISR and AtSN1. For this purpose we digested genomic DNA with methylation sensitive enzymes and performed either DNA gel blots or PCR, or we examined individual loci by sequencing following bisulfite treatment. Analysis revealed that the sr45–1 mutant exhibits reduced non-CG methylation for all of the aforementioned loci (). Due to the incompleteness of the phenotype, we generated double mutants with dcl3–1, which been reported to display a weak DNA methylation phenotype.Citation11 We found that sr45–1, dcl3–1 double mutants exhibited an additive effect in methylation phenotype. This enhancement was found at both RdDM target loci tested (, and ). Concordantly, sr45–1 dcl3–1 double mutants also show an enhanced de novo methylation phenotype at FWA transgene (). Together this data indicate that SR45 and DCL3 could cooperate within the RdDM pathway.
Figure 3.sr45–1 maintenance DNA methylation phenotype. (A) Sodium bisulfite analysis of an 180 base-pair region of the MEA-ISR locus. (B) AtSN1 Chop-qPCR assay. Genomic DNA was digested with the methylation sensitive enzyme HaeIII, which recognizes three sites in AtSN1. Amplification of AtSN1 was quantified by Real Time PCR, and signal was normalized to undigested DNA. HaeIII, is blocked by C methylation in GGCC context. (C) MEA-ISR DNA gel blot. MspI digested genomic DNA was probed with MEA-ISR. (D) REP12 and Ta3 DNA gel blot. MspI digested genomic DNA was probed with REP12 or Ta3. MspI is blocked by methylation of the external C in CCGG context. (E) RT-PCR showing expression levels of REP12 and Ta3. UBQ10 expression is showed as a loading control.
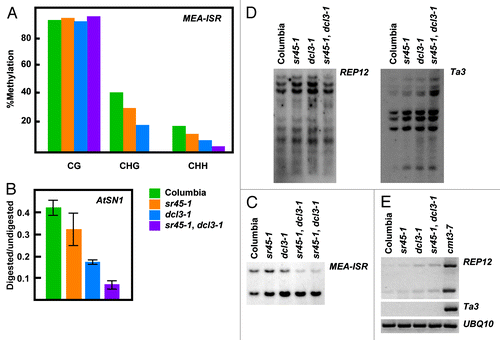
To assess whether the methylation defect is specific to RdDM targets, we analyzed methylation and expression levels at Ta3 and REPEAT12 (REP12) loci by DNA gel blot and RT-PCR–two loci that are known to be methylated in a DRM2-independent manner.Citation12 We observed that sr45–1 as well as sr45–1, dcl3–1 double mutants showed no difference in methylation when compared with wild type, indicating that SR45 function is most likely confined to the DRM2 pathway ( and ).
In order to place SR45 within the context of the RdDM pathway, we analyzed sr45–1 siRNA production by RNA gel blot. The 24-nucleotide siRNAs associated with RdDM are broadly grouped into two types: type I (dependent on both plant specifc RNA polymerases: Pol IV and Pol V) and type II (only dependent on Pol IV).Citation13 Regardless of type, the siRNAs abundance was reduced in sr45–1 mutant plants (), suggesting that SR45 acts in the pathway at steps prior to the production of small RNAs.
Figure 4. Placement of SR45 in RdDM pathway. (A) RNA gel blots showing siRNAs abundance at both type I and II loci. Hybridization with miR163 is shown as a loading control for 5S siRNAs and hybridization with miR159 is shown as a loading control for AtSN1 and siR02. (B) RNA gel blot showing AGO4 expression in leaves, seedlings, and flowers. Hybridization with UBQ10 is shown as a loading control. (C) RT-PCR and protein gel blot showing the expression and abundance of AGO4/AGO4. UBQ10 expression is showed as a loading control for RT-PCR and amido black staining of the RUBISCO large subunit is shown as a loading control for the protein gel blot. (D) Immunofluorescent microscopy showing AGO4 localization in 4xmyc::AGO4 (Columbia) and 4xmyc::AGO4 (sr45–1) backgrounds. White arrows indicate the position of AGO4 in the sr45–1 panel.
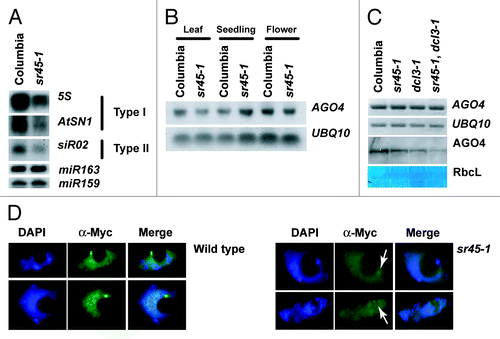
Previous studies have shown that ARGONAUTE 4 (AGO4) protein is destabilized in mutants upstream of siRNA biogenesis.Citation14 To test whether this holds true for sr45–1, we examined AGO4 transcript levels by RT-PCR and RNA gel blots in three different tissues. We observed neither significant alteration in AGO4 expression pattern, nor major splicing variants in sr45–1 relative to wild type ( and ). However, protein gel blot analyses revealed a slight, but reproducible decrease of AGO4 protein in sr45–1 mutants. In addition, the effect of sr45–1 on AGO4 levels was increased in dcl3–1 background (), reinforcing the hypothesis of those two genes cooperating in the regulation of the RdDM pathway. To further confirm the observed reduction of AGO4 levels, we analyzed the nuclear localization pattern of a complementing epitope-tagged version of AGO4 in the sr45–1 background by immunoflourescence. Consistent with protein gel blot data, we observed a decrease in AGO4 abundance in sr45–1; nonetheless the localization pattern of AGO4 was similar to wild type ().
The FLC locus, which is silenced by a DNA methylation-independent mechanismCitation15 is also partially de-repressed in the sr45–1 mutant background (Fig. S4).Citation8 It is interesting to note that DCL3 has been previously reported to be required for FLC silencing despite the lack of transcriptional control by DNA methylation.Citation16 This DCL3 regulation of FLC is probably through small RNAs matching its 3′ region.Citation17 Furthermore, the de-repression of FLC is enhanced in the dcl3–1 sr45–1 double mutant (Fig. S4).
In sum, we have discovered a known spliceosome gene that is required for RdDM. It cannot be ruled out that SR45 may be involved in the splicing of an RdDM factor, thus the methylation phenotype is a secondary effect. Alternatively, given its small RNA phenotype, it potentially has a novel function in siRNA processing. It is worth noting that the nuclear cap-binding complex, which is involved in pre-mRNA splicing, has a role in a distinct DICER-LIKE1-dependent micro RNA pathway.Citation18 This suggests SR45, and perhaps other spliceosome factors, may indeed play a direct role in siRNA accumulation as well. We screened a number of known or putative spliceosome factors as part of our screen; however, sr45 was the only one with an FWA-dependent flowering-time defect (Table S1) Interestingly, sr45 shares a very similar phenotype as dcl3, even at the FLC locus which is not an RdDM target. This suggests that these two proteins likely work in concert to control RNA-mediated silencing.
Materials and Methods
Plant materials. We used the following Arabidopsis strains: The wild type Columbia; the recessive sr45–1 (SALK_004132) and dcl3–1 (SALK_005512); the Myc-tagged complementing AGO4 line used for immunofluorescence and protein gel blots is described in Li.Citation14
FWA transformation and flowering-time analysis: We performed FWA transformation using an AGL0 Agrobacterium tumefaciens strain carrying a pCAMBIA3300 vector with an engineered version of FWA in which an EcoRI site was converted into a BglII site. For selection, we sprayed the resultant T1 population with a 1:1000 dilution of FinaleTM. We measured flowering time of resistant plants as the total number of leaves (rosette and cauline leaves) developed by a plant.
Bisulfite analysis. We performed sodium bisulfite sequencing using EZ DNA Methylation Gold (Zymo Research) reagents for conversion of plant genomic DNA extracted from floral tissue using a standard CTAB protocol. Following amplification of bisulfite treated DNA by PCR, we cloned the resulting PCR fragments into pCR2.1-TOPO (Invitrogen) and analyzed 15 to 22 clones per sample. The FWA transgene was distinguished from the endogene by BglII digestion prior to bisulfite treatment (see FWA Transformation methods) and elimination of any clones containing Col-0 polymorphisms rom the data set after sequencing. All primers are listed in Table S2.
DNA gel blotting. DNA from young flowers was extracted using a standard CTAB protocol. One μg of genomic DNA was digested overnight with MspI. The digestion was run on a 1% agarose gel, transferred to Hybond N+ membranes, blocked and washed according to manufacturer instructions (GE Healthcare). Membranes were probed with a PCR product radiolabeled with α 32P-dCTP using the Megaprime DNA Labeling System. MEA-ISR, Ta3 and REP12 PCR products for probing were generated with primers listed in Table S2.
Small RNA northern blotting. Detection of small RNAs was performed exactly as described in Law.Citation19 Oligonucleotide sequences used for probing can be found in Table S2.
Immunofluorescent microscopy. Detection of Myc-tagged AGO4 protein was performed exactly as described in Li.Citation14 Primary mouse monoclonal anti-Myc (Covance 9E10) was used at a 1:200 dilution. Secondary anti-mouse FITC (Abcam) was used at a 1:200 dilution. DNA was stained using Vectashield mounting medium containing DAPI (Vector Laboratories).
RNA gel blotting. RNA was extracted from the indicated tissue using Trizol reagent (Invitrogen). RNA gel blots were performed as described in Henderson.Citation20AGO4 and UBQ10 PCR products used for probing were generated with primers listed in Table S2.
Bisulfite cutting assay. DNA was extracted and bisulfite treated as described above. The cutting assay was performed exactly as described in Chan.Citation21
| Abbreviations: | ||
| AGO4 | = | ARGONAUTE 4 |
| DCL3 | = | DICER-LIKE 3 |
| DRM2 | = | DOMAINS REARRANGED METHYLTRANSFERASE 2 |
| FLC | = | FLOWERING LOCUS C |
| FWA | = | FLOWERING WAGENINGEN |
| MEA-ISR | = | MEDEA-INTERGENIC SUBTELOMERIC REPEATS |
| REP12 | = | REPEAT 12 |
| RdDM | = | RNA-directed DNA methylation |
| siRNA | = | small interfering RNA |
| SR45 | = | ARGININE/SERINE-RICH 45 |
| UBQ10 | = | UBIQUITIN 10 |
Additional material
Download Zip (1.3 MB)Acknowledgments
We would like to thank Craig Pikaard for kindly donating anti-AGO4 immunosera and the expression vector for the peptide used for purification. We would like to thank members of the S. Jacobsen laboratory for supportive discussions. Jacobsen lab research was supported by US National Institutes of Health grant GM60398. I.A. was supported by a postdoctoral fellowship from the Ministerio de Educacion y Ciencia. M.V.C.G. was supported by USPHS National Research Service Award GM07104. C.F.L. was supported by the Ruth L. Kirschstein National Research Service Award GM07185.
Related Research Data
References
- Cao X, Jacobsen SE. Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr Biol 2002; 12:1138 - 44; http://dx.doi.org/10.1016/S0960-9822(02)00925-9; PMID: 12121623
- Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 2010; 11:204 - 20; http://dx.doi.org/10.1038/nrg2719; PMID: 20142834
- Wierzbicki AT, Haag JR, Pikaard CS. Noncoding Transcription by RNA Polymerase Pol IVb/Pol V Mediates Transcriptional Silencing of Overlapping and Adjacent Genes. Cell 2008; 135:635 - 48; http://dx.doi.org/10.1016/j.cell.2008.09.035; PMID: 19013275
- Ausin I, Mockler TC, Chory J, Jacobsen SE. IDN1 and IDN2 are required for de novo DNA methylation in Arabidopsis thaliana. Nat Struct Mol Biol 2009; 16:1325 - 7; http://dx.doi.org/10.1038/nsmb.1690; PMID: 19915591
- Soppe WJ, Jacobsen SE, Alonso-Blanco C, Jackson JP, Kakutani T, Koornneef M, et al. The late flowering phenotype of fwa mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. Mol Cell 2000; 6:791 - 802; http://dx.doi.org/10.1016/S1097-2765(05)00090-0; PMID: 11090618
- Cao X, Jacobsen SE. Role of the arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr Biol 2002; 12:1138 - 44; http://dx.doi.org/10.1016/S0960-9822(02)00925-9; PMID: 12121623
- Chan SW, Zilberman D, Xie Z, Johansen LK, Carrington JC, Jacobsen SE. RNA silencing genes control de novo DNA methylation. Science 2004; 303:1336; http://dx.doi.org/10.1126/science.1095989; PMID: 14988555
- Ali GS, Palusa SG, Golovkin M, Prasad J, Manley JL, Reddy AS. Regulation of plant developmental processes by a novel splicing factor. PLoS ONE 2007; 2:e471; http://dx.doi.org/10.1371/journal.pone.0000471; PMID: 17534421
- Greenberg MV, Ausin I, Chan SW, Cokus SJ, Cuperus JT, Feng S, et al. Identification of genes required for de novo DNA methylation in Arabidopsis. Epigenetics 2011; 6:344 - 54; http://dx.doi.org/10.4161/epi.6.3.14242; PMID: 21150311
- Cao X, Aufsatz W, Zilberman D, Mette MF, Huang MS, Matzke M, et al. Role of the DRM and CMT3 methyltransferases in RNA-directed DNA methylation. Curr Biol 2003; 13:2212 - 7; http://dx.doi.org/10.1016/j.cub.2003.11.052; PMID: 14680640
- Greenberg MV, Ausin I, Chan SW, Cokus SJ, Cuperus JT, Feng S, et al. Identification of genes required for de novo DNA methylation in Arabidopsis. Epigenetics 2011; 6:344 - 54; http://dx.doi.org/10.4161/epi.6.3.14242; PMID: 21150311
- Cao X, Jacobsen SE. Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc Natl Acad Sci USA 2002; 99:Suppl 4 16491 - 8; http://dx.doi.org/10.1073/pnas.162371599; PMID: 12151602
- Zheng B, Wang Z, Li S, Yu B, Liu JY, Chen X. Intergenic transcription by RNA polymerase II coordinates Pol IV and Pol V in siRNA-directed transcriptional gene silencing in Arabidopsis. Genes Dev 2009; 23:2850 - 60; http://dx.doi.org/10.1101/gad.1868009; PMID: 19948763
- Li CF, Pontes O, El-Shami M, Henderson IR, Bernatavichute YV, Chan SW, et al. An ARGONAUTE4-containing nuclear processing center colocalized with Cajal bodies in Arabidopsis thaliana. Cell 2006; 126:93 - 106; http://dx.doi.org/10.1016/j.cell.2006.05.032; PMID: 16839879
- Jean Finnegan E, Kovac KA, Jaligot E, Sheldon CC, James Peacock W, Dennis ES. The downregulation of FLOWERING LOCUS C (FLC) expression in plants with low levels of DNA methylation and by vernalization occurs by distinct mechanisms. Plant J 2005; 44:420 - 32; http://dx.doi.org/10.1111/j.1365-313X.2005.02541.x; PMID: 16236152
- Schmitz RJ, Amasino RM. Vernalization: a model for investigating epigenetics and eukaryotic gene regulation in plants. Biochim Biophys Acta 2007; 1769:269 - 75; PMID: 17383745
- Swiezewski S, Crevillen P, Liu F, Ecker JR, Jerzmanowski A, Dean C. Small RNA-mediated chromatin silencing directed to the 3′ region of the Arabidopsis gene encoding the developmental regulator, FLC. Proc Natl Acad Sci USA 2007; 104:3633 - 8; http://dx.doi.org/10.1073/pnas.0611459104; PMID: 17360694
- Laubinger S, Sachsenberg T, Zeller G, Busch W, Lohmann JU, Ratsch G, et al. Dual roles of the nuclear cap-binding complex and SERRATE in pre-mRNA splicing and microRNA processing in Arabidopsis thaliana. Proc Natl Acad Sci USA 2008; 105:8795 - 800; http://dx.doi.org/10.1073/pnas.0802493105; PMID: 18550839
- Law JA, Vashisht AA, Wohlschlegel JA, Jacobsen SE. SHH1, a Homeodomain Protein Required for DNA Methylation, As Well As RDR2, RDM4, and Chromatin Remodeling Factors, Associate with RNA Polymerase IV. PLoS Genet 2011; 7:e1002195; http://dx.doi.org/10.1371/journal.pgen.1002195; PMID: 21811420
- Henderson IR, Zhang X, Lu C, Johnson L, Meyers BC, Green PJ, et al. Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nat Genet 2006; 38:721 - 5; http://dx.doi.org/10.1038/ng1804; PMID: 16699516
- Chan SW, Zhang X, Bernatavichute YV, Jacobsen SE. Two-step recruitment of RNA-directed DNA methylation to tandem repeats. PLoS Biol 2006; 4:e363; http://dx.doi.org/10.1371/journal.pbio.0040363; PMID: 17105345