Abstract
We present here the first high resolution melt (HRM) assay to quantitatively analyze differences in murine DNA methylation levels utilizing CpG methylation of Long Interspersed Elements-1 (LINE1 or L1). By calculating the integral difference in melt temperature between samples and a methylated control, and biasing PCR primers for unmethylated CpGs, the assay demonstrates enhanced sensitivity to detect changes in methylation in a cell line treated with low doses of 5-aza-2’-deoxycytidine (5-aza). The L1 assay was confirmed to be a good marker of changes in DNA methylation of L1 elements at multiple regions across the genome when compared with total 5-methyl-cytosine content, measured by Liquid Chromatography-Mass Spectrometry (LC-MS). The assay design was also used to detect changes in methylation at other murine repeat elements (B1 and Intracisternal-A-particle Long-terminal Repeat elements). Pyrosequencing analysis revealed that L1 methylation changes were non-uniform across the CpGs within the L1-HRM target region, demonstrating that the L1 assay can detect small changes in CpG methylation among a large pool of heterogeneously methylated DNA templates. Application of the assay to various tissues from Balb/c and CBA mice, including previously unreported peripheral blood (PB), revealed a tissue hierarchy (from hypermethylated to hypomethylated) of PB > kidney > liver > prostate > spleen. CBA mice demonstrated overall greater methylation than Balb/c mice, and male mice demonstrated higher tissue methylation compared with female mice in both strains. Changes in DNA methylation have been reported to be an early and fundamental event in the pathogenesis of many human diseases, including cancer. Mouse studies designed to identify modulators of DNA methylation, the critical doses, relevant time points and the tissues affected are limited by the low throughput nature and exorbitant cost of many DNA methylation assays. The L1 assay provides a high throughput, inexpensive and sensitive screening tool for identifying and characterizing DNA methylation changes to L1 elements at multiple regions across the genome.
Introduction
Approximately 2–10% of the cytosine residues in the mammalian genome are methylated (5-methyl-deoxycytosine; 5-mdC), predominantly when in sequence with a guanine (termed a CpG dinucleotide) and largely within transcriptionally silent regions of the genome. CpG-rich regions within transcriptionally active gene promoters are generally unmethylated, although it has been reported that only key CpGs within the promoter region need to be (un)methylated to exert control on gene expression.Citation1 In cancer, DNA methylation changes can occur at individual genes, or across the genome (global methylation), resulting in transcriptionally silent regions becoming demethylated, leading to transcriptional activation, or regions that are normally transcriptionally active rendered silent by aberrant increases in DNA methylation.
The most heavily methylated regions of the genome are DNA sequences belonging to repetitive DNA elements, which comprise approximately 45% of the mammalian genome,Citation2 of which 20% consist of Long Interspersed Elements-1 (LINE1 or L1).Citation2-Citation4 Short Interspersed Elements (SINE) and Long-terminal Repeat elements (LTR) are also distributed throughout the genome and can be methylated. These repeat elements can contain full length coding regions that, upon demethylation, may result in transposition across the genome, which can result in disruption to gene expression and, in some cases, disease.Citation5,Citation6 Loss of repeat element methylation can be an early event in carcinogenesis or tumor progression. For example, loss of methylation at intracisternal-A particle (IAP) repeat elements in mice results in active transcription and transposition leading to insertion of the IAP element into the genomic locus of Notch-1. This retrotransposition induces expression of an oncogenic transcript of Notch-1 in thymic tumors.Citation7 In several cases, it has been reported that a loss of methylation at repeat elements has resulted in increased chromosomal instability as well as the enhanced development of colorectal cancer, non-small cell lung cancer and bladder tumors.Citation8-Citation15
Increasingly, mouse models are being used to study the acute and transgenerational effects of exogenous modulators of global DNA methylation such as diet,Citation16-Citation18 chemical carcinogens,Citation19 ionizing radiation,Citation20-Citation24 as well as the study of DNA methylation in aging animals.Citation25,Citation26 Accordingly, there is an increasing need for sensitive, robust, inexpensive and high throughput methods for the detection of changes to global DNA methylation levels. While High Performance Liquid Chromatography (HPLC) and Liquid Chromatography-Mass Spectrometry (LC-MS) are considered two of the gold standards for assessing total methylated cytosine content due to their high sensitivity, accuracy and reproducibility, they are low-throughput due to the requirements of sample preparation, lengthy analysis procedures, and cost per sample. More recently, whole genome sequencing and array technologies such as the Illumina, SOLiD (Invitrogen), and 454 platforms (Applied Biosystems) have been used to investigate global CpG methylation. While these platforms are able to determine the methylation status of thousands of genes at the single CpG site-level offering the highest level of resolution, they are not amenable for screening large numbers of experimental samples and the technology is not available to all laboratories. Another common sequencing by synthesis technique used in methylation analysis is pyrosequencing, which offers information about the proportion of methylated cytosines in a target sequence. While this is also a powerful tool, it is limited by the size of the target region and cannot be used to analyze highly polymorphic sequences.
High resolution melt (HRM) PCR is now being used in methylation studies and is capable of detecting single nucleotide differences based on differences in melting temperatures.Citation27 The widespread nature and CpG density of L1 elements has resulted in the utilization of L1 elements to investigate changes in DNA methylation at multiple locations within the genome.Citation28-Citation30 We have developed a HRM-PCR to quantitatively detect methylation changes to heterogeneously methylated murine L1 repeat elements as a surrogate marker of widespread alternations to DNA methylation levels. The L1 assay calculates the integral difference in the melt curves of a sample compared with the methylated control sample (termed the Net Temperature Shift – NTS), providing a sensitive, quantitative measurement of differences in methylation. Using the NTS calculation, the assay was shown to detect dose-dependent demethylation of L1 elements induced by a chemical demethylating agent. Application of the assay to various tissues from two common inbred laboratory mouse strains – Balb/c and CBA, demonstrated that the assay could detect differences in methylation between tissue samples that had not been treated with a demethylating agent. We report, for the first time, the relative methylation levels of peripheral blood samples when compared with other murine tissues, which were highly methylated in both mouse strains. The ability to quantitatively measure methylation of mouse PB samples using a high throughput and sensitive screening assay provides a powerful tool for assessing changes to methylation in response to exogenous and endogenous modulators of methylation in comparison to human studies where repeated sampling of subjects is required.
Results
L1-HRM Assay Design
We aimed to develop a high-throughput, quantitative assay for the detection of DNA methylation changes by analyzing the CpG methylation status of murine LINE1 (L1) repeat elements using PCR high-resolution melt curve analysis (HRM). The forward primer to a murine L1 element (Accession number: D84931.1) from Kato et al.Citation31,Citation32 was used as a query sequence for a BLAT search (http://genome.ucsc.edu/). A target CpG island (containing 13 CpGs) upstream of the Kato primer binding site (located within the open reading frame 2 (ORF2) of the consensus sequence) was identified using MethPrimerCitation33 and candidate primer sequences that did not contain CpGs were then used to perform an in silico PCR (http://genome.ucsc.edu/). Primer pairs were chosen based on the number of genomic locations predicted to amplify with the candidate primer pair indicating widespread coverage of the L1 elements. The primers were found to amplify L1 elements at numerous locations on each mouse chromosome (data not shown). Successful amplification of murine L1 elements was confirmed by DNA sequence analysis.
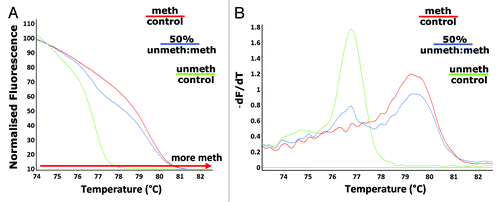
HRM analysis is used to distinguish between single nucleotide differences based on the melting temperatures of amplicons.Citation27,Citation28,Citation34,Citation35 HRM can be used to differentiate between methylated and unmethylated DNA following treatment of genomic DNA with sodium bisulphite. DNA that is heavily methylated will remain cytosine-rich following bisulphite modification, whereas less methylated DNA will contain a lower GC-content due to the presence of uracil in place of the original unmethylated cytosines. These sequence changes result in melting temperature differences (due to differences in the amount of thermostable GC). Methylated mouse genomic control DNA and an unmethylated control were amplified with the F_unbiased_mLINE1 and R_unbiased_mLINE1 primers. A 50% mixture of the two control DNA samples was bisulphite modified and amplified. As expected, the melt curve analysis showed that the methylated control had a higher melt temperature range compared with the unmethylated control DNA. The 50% mixture had a melt temperature range situated between the two control DNA samples (). The graph of the negative first derivative of the melting curve (-dF/dT) () showed that the methylated and unmethylated control samples had only one melt peak each, whereas the 50% mixture of the two control DNA samples had two melt peaks representative of each of the control DNA samples.
Figure 1. Detection of differences in L1 methylation. Melt curve analysis following amplification with the unbiased L1 primers (F_unbiased_mLINE1 and R_unbiased_mLINE1) - (A) the HRM normalized melt curve and (B) the first derivative melt curve (dF/dT), was performed on methylated (red) and unmethylated (green) control DNA as well as a 50% (methylated: unmethylated control) methylated sample (blue).
Detection of heterogeneous DNA demethylation induced by a chemical demethylating agent
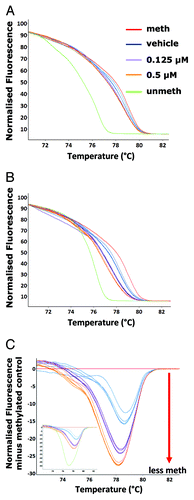
Not all CpG islands will have complete demethylation at every CpG and this will result in a heterogeneous pattern of methylation at any individual CpG dinucleotide.Citation28,Citation36-Citation43 In order to investigate if small changes in methylation patterns across numerous L1 elements distributed throughout the genome could be detected with this assay, a mouse hybridoma cell line was treated with two different low doses (0.125 µM and 0.5 µM) of the chemical demethylating agent 5-aza-2’-deoxycytidine (5-aza). The melt curves of the samples from each treatment group could be distinguished from the melt profile of the methylated control DNA. A significant difference in melting temperature between the 5-aza treated cells and the vehicle (DMSO) treated control cells was not detected (). It has been proposed in several reports, that to increase sensitivity to detect small methylation/demethylation events within a large pool of heterogeneously methylated templates, PCR primers may need to enrich for methylated or unmethylated templates.Citation44-Citation46 Using this approach, a new forward and reverse primer pair was designed to match the bisulphite-modified sequence of an unmethylated CpG dinucleotide at the 3′ end of each oligonucleotide. Thus, the same region could now be amplified with an unbiased-primer pair (not selective for methylation status) or an unmethylated-biased primer pair (preferentially amplifying L1 sequences with both flanking CpG sites unmethylated). Using the unmethylated-biased primers, a distinct, dose-dependent difference in melt curves was observed for the samples treated with 5-aza compared with the vehicle control and between the two 5-aza doses (). All L1-HRM assays were subsequently performed using the unmethylated-biased primers unless otherwise stated.
Figure 2. Detection of 5-aza induced partial demethylation. Vehicle control (DMSO) (blue), 0.125 µM (purple) and 0.5 µM (orange) 5-aza treated A11 cell line genomic DNA was amplified following bisulphite modification with the unbiased L1 primers and subjected along with the methylated (red) and unmethylated (green) controls to melt analysis - (A) the normalized HRM melt curve (n = 3 samples per treatment group is displayed). The samples were then amplified with unmethylated-biased primers. Melt analysis was performed on the PCR products – (B) the normalized HRM curve and (C) the difference graph (one PCR replicate for each of the n = 4–5 samples is displayed); inset is the difference graph with the unmethylated control included.
Statistical analysis of methylation differences between samples using the Net Temperature Shift
In order to statistically quantify the differences in melt curves observed, a means of deriving the methylation index of a sample based on the melting profile of amplicons was devised. Within the Rotor-Gene Q software, normalized melt curves of all samples were generated. Following this, a control sample was selected (in this instance, the methylated control DNA) and a difference plot of the melt curves compared with the methylated control was generated (). The difference plot fluorescence value at each temperature point (0.1°C intervals from 74°C to 84°C for a total of 100 readings) within the melt curve was exported to Microsoft Excel. All values within the melt curve were summed and divided by 100 to give the Net Temperature Shift (NTS) value (Table S1). A greater negative NTS value indicates a greater shift from the methylated control and, thus, a less methylated sample. Using the NTS, a significant dose-dependent demethylation of the L1 elements for samples treated with 5-aza compared with the vehicle control (p < 10−5) and between the two 5-aza doses (p < 10−5) was observed ().
Figure 3. Quantitation of methylation differences using the Net Temperature Shift. The normalized fluorescence at each temperature point on the melt curve were used to calculate the mean NTS of the 5-aza treated samples for (A) L1, (B) murine B1 family Mm (B1_Mm) and (C) Intracisternal-Particle A Long-terminal Repeat (IAP_LTR) repeat elements (n = 4–5 samples per treatment group) (*p < 10−5 compared with the vehicle control; + p < 0.05 compared with the 0.125 µM treated 5-aza samples).
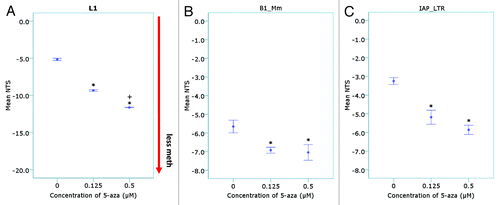
To demonstrate that methylation changes were not restricted to L1 elements and that the assay design could be applied to other repeat elements, the NTS calculation was applied to amplicon melt curves from two other DNA repeat elements: mouse B1 (Short Interspersed Elements; SINE family, Mm; located within the first 92 bp of the ~130 bp monomeric repeat unit) and Intracisternal-A particle Long-terminal Repeat elementsCitation32 (IAP_LTR; located within the 5′LTR of the IAP promoter) (). For both the B1_Mm primers, which were unbiased for methylation, and the unmethylated-biased IAP_LTR element primers, there was a significant difference in the NTS of the 0.125 µM and 0.5 µM 5-aza treated samples compared with the vehicle control (p < 0.05). Both repeat elements showed strong correlation with the L1 elements (p < 0.01); with the IAP_LTR demonstrating greater correlation with the L1 elements than the B1 elements (Table S2).
Validation of the L1-HRM assay
in silico PCR and analysis of L1 repeat element sequences
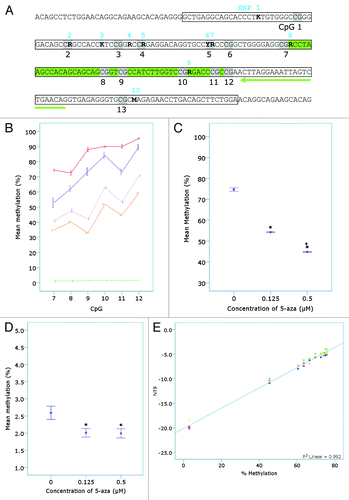
Sequence analysis of L1-PCR products identified six non-CpG nucleotide positions with more than one defined peak on the sequence chromatogram (data not shown), indicating potential sequence variation. Using the original unbiased L1 primer sequences, the mouse genome was searched by in silico PCR (http://genome.ucsc.edu/) to identify the predicted pool of L1 template sequences. Alignment of 20 randomly selected in silico PCR results revealed ten nucleotide positions with sequence variation and that the frequency of the minority variants occurred in 5% to 40% of the analyzed templates (). Sequence variants 2, 5, and 7 occurred at the guanine base in CpG dinucleotides () and as a result, 10 - 15% of the L1 elements did not contain a CpG at these sites. All six sequence variants identified from the sequencing of PCR products were observed in the 20 in silico predicted L1 template sequences.
Table 2. Frequency of detected variants in L1 target sequence
Figure 4. Confirmation of the detection of 5-aza induced demethylation by the L1-assay. (A) The L1 sequence as confirmed by DNA sequencing. The location of sequence variants in the target L1 region (boxed region) in unmodified genomic DNA identified from both sequence analysis of L1-PCR products (amplified with F_unmod_mLINE1 and R_unmod_mLINE1) and in silico PCR analysis (see ) are shown in bold, numbered 1–10 in blue and characterized as an R (A/G), K (T/G) or M (A/C). CpGs are shaded and numbered in black 1–13. The L1-HRM PCR products (n = 4–5 samples per treatment group from n = 1 bisulphite modification and PCR reaction) were then subjected to pyrosequencing. The pyrosequencing primer is highlighted by the green arrow and the region pyrosequenced is shown in green. (B) The percent methylation of each CpG was plotted and separated by 5-aza concentration (vehicle - blue, 0.125 µM - purple and 0.5 µM -orange) or control DNA status (red – methylated control, green – unmethylated control). (C) The mean methylation of all CpGs pyrosequenced (7–12) was plotted vs. 5-aza concentration (*p < 10−5 compared with the vehicle control; + p < 0.05 compared with the 0.125 µM treated 5-aza samples). (D) Genomic DNA samples (n = 4–5 per treatment group) were subjected to Liquid Chromatography – Mass Spectrometry (LC-MS) and mean percent methylation (5-mdC/dG ratio) for each treatment group plotted (*p < 0.05). (E) Linear regression analysis was performed on the mean NTS for samples of a standard curve (R2 = 0.992; p < 0.001). The standard curve was made from mixtures of the vehicle and 0.5 µM 5-aza treated A11 samples (75.6, 75.45, 75.3, 75, 74.09, 72.58, 69.56, 66.54, 63.52, 60.5 and 45.4% methylated) using the mean pyrosequencing values for the two samples. The unmethylated control DNA (~3% methylated) was also included in the L1-HRM PCR reactions. The samples underwent two separate bisulphite modifications and were amplified with the unmethylated-biased primers in two separate PCR reactions per bisulphite modification, prior to melt analysis.
Comparison of the L1-HRM assay with pyrosequencing and LC-MS
It was necessary to determine whether the differences in melting temperatures observed were due to differences in cytosine composition following bisulphite modification and not the result of differences in non-CpG sequence variations (). Analysis of the melt curves for L1 elements amplified from bisulphite unmodified genomic DNA revealed no difference in melt curves between the 5-aza and vehicle control treated samples (data not shown). This indicated that the identified sequence variants were not influencing the differences in melt curves observed, and that the differences were due to cytosine content following bisulphite modification. To confirm this, the L1-HRM PCR products were subjected to pyrosequencing. Pyrosequencing of CpGs 7 - 12 () revealed that each of the six CpG sites in the DNA from the 0.125 µM and 0.5 µM 5-aza treated cells displayed significantly less L1 methylation than the vehicle control (). The mean percent of L1 methylation across all six CpGs revealed a significant dose-dependent reduction in L1 methylation with increasing concentration of 5-aza () and a significant difference between all treatments (p < 10−5). CpG 9 demonstrated a greater reduction in methylation compared with the other CpG sites analyzed in the 5-aza treated samples and in comparison with the vehicle control ().
Liquid Chromatography-Mass Spectrometry (LC-MS) was used to assess total genomic 5-mdC content for comparison with the NTS detection of demethylation. A significant reduction in total genomic 5-mdC content compared with the vehicle control for both 0.125 µM and 0.5 µM treated samples was observed (). Unlike the HRM assay and pyrosequencing, the LC-MS method was unable to distinguish differences in DNA methylation between the 0.125 µM and 0.5 µM 5-aza treated cells. Bivariate correlations with 5-aza dose for the three assays () showed equal and significant negative correlations for NTS vs. 5-aza dose and pyrosequencing mean methylation vs. 5-aza dose, with a weaker but still significant negative correlation between LC-MS 5-mdC content vs. 5-aza dose. The NTS and pyrosequencing mean methylation levels were highly and significantly correlated with each other, while the 5-mdC content by LC-MS, which is not specific for L1 methylation, showed less concordance with the other two methods.
Table 3. Correlation of HRM assay with pyrosequencing and LC-MS
Linearity of the L1-HRM assay
Various ratios of genomic DNA from the 0.5 µM 5-aza treated and vehicle treated samples were made (prior to bisulphite modification) to create heterogeneously methylated samples with predicted mean L1 methylation levels (based on pyrosequencing measurements) between 45.4 and 75.6%. These DNA mixtures were then subjected to the L1-HRM assay to test the sensitivity and linearity of the assay. Linear regression analysis on the standard curve indicated that the NTS was highly linear compared with the predicted L1 methylation levels of the samples (p < 0.001; R2 = 0.992) (). Bonferroni corrected post-hoc analysis revealed that with the number of replicates used here, an absolute difference of 3% mean L1 methylation or greater could be detected between two mixes of heterogeneously methylated DNA (p < 0.05). Comparison of these DNA mixtures by L1-HRM was performed for two separate bisulphite modification reactions and two HRM-PCR runs per modification (shown in ) and revealed that neither bisulphite modification batch nor PCR run made a significant contribution to the overall variance (ANOVA, p < 0.05), with the variance largely explained by the predicted methylation levels of the various mixtures (p < 0.01).
Table 4. L1-HRM inter- and intra-assay variation
Detection of tissue and sex specific methylation levels in murine tissues
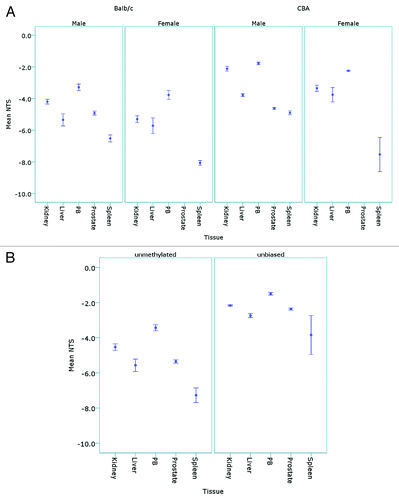
The L1-HRM assay was then used to detect L1 methylation differences between samples that had not undergone 5-aza induced demethylation, but were expected to be of differing (L1) methylation levels, based on previous reports.Citation20,Citation22,Citation47 Mouse strain, gender and the tissues analyzed have been shown to have differences in global DNA methylation levels. Therefore, genomic DNA from male and female Balb/c and CBA mice were analyzed using the unmethylated-biased L1 primers. Analysis of the NTS values revealed that there were both strain and sex based differences in L1 methylation levels; with the peripheral blood samples (PB) displaying the highest L1 methylation and the spleen tissue genomic DNA the lowest. In all mice, the order of L1 methylation from hypermethylated to hypomethylated was as follows: PB > kidney > liver > prostate > spleen (). For Balb/c mice, higher methylation was observed in the male kidney (p = 0.03) and spleen (p = 0.01) tissues compared with the females, and for CBA mice, higher methylation was observed in the male kidney (p = 0.01), PB (p = 0.01) and spleen (p = 0.028) tissues relative to female mice. The CBA mice demonstrated higher L1 methylation of the kidney, liver and PB tissues compared with the Balb/c mice (p < 0.05). A comparison of the unbiased primers with the unmethylated-biased primers on the male Balb/c tissues () showed hierarchy of tissue L1 methylation that correlated with the NTS for the unmethylated-biased primers (p < 0.01, Pearson correlation); however, the unbiased primers did not demonstrate significant differences in L1 methylation between all tissues. For example, the spleen was hypomethylated at L1 elements compared with all other tissues when analyzed with the unmethylated-biased primers (ANOVA, p < 0.05); whereas the unbiased primers only demonstrated significant L1 hypomethylation of the spleen when compared with the PB samples (ANOVA, p = 0.045).
Figure 5. Detection of tissue and sex specific methylation levels in murine tissues. (A) The mean NTS of L1 elements was used to assess DNA methylation levels in the kidney, liver, peripheral blood (PB), prostate and spleen tissues of male (n = 5) and female (n = 5) Balb/c and CBA mice following bisulphite modification and amplification with the unmethylated-biased primers. (B) The mean NTS of the kidney, liver, PB, prostate and spleen of male Balb/c mice when amplified with the unbiased primers for comparison with the unmethylated-biased primers.
Discussion
HRM analysis of bisulphite modified DNA templates is used to investigate DNA methylation, with various quantitative calculations of methylation levels employed. These calculations range from comparing Cq values to a standard curve,Citation28,Citation46 a calculation based on the temperature at which half of the PCR products have melted,Citation48 measuring the height of the differential fluorescence melt peak from a nominated control,Citation49 TaqMan™ probe technologyCitation37 and single dilutions of complex melt curves.Citation36 In these assays, the methylation of a sample is calculated by comparison to a standard curve that is generated using completely methylated and unmethylated control DNA.Citation28,Citation36,Citation37,Citation46,Citation48-Citation50 With the exception of Stanzer et al.,Citation28 all assays were utilized to investigate gross changes in CpG methylation at single genomic loci in cancer samples. Analysis of the methylation levels of L1 elements using HRM presents a unique challenge due to the number of CpGs within the CpG islands and the abundance and sequence diversity of LINE1 elements throughout the genome. Methylation profiles will be heterogeneous and will produce complex melt curves that cannot be quantified by comparison to a standard curve of methylated/unmethylated DNA. Our aim was to utilize HRM technology to detect small changes in DNA methylation to L1 elements throughout the genome that can result from perturbations to the homeostatic environment such as chemical or radiation exposure, dietary changes and aging in mouse studies. As a proof-of-principle, the chemical demethylating agent 5-aza was used to demonstrate the detection of L1 demethylation using HRM. Treatment with 5-aza induces significant changes to total genomic 5-mdC levels.Citation51,Citation52 In this study, lower doses of 5-aza were used to treat the A11 cells, which underwent only one cell division in order to induce low level demethylation, as would be expected following low level exposure to exogenous or endogenous modulators of DNA methylation. We proposed that treatment would cause partial demethylation resulting in increased heterogeneity across the L1 elements. Initial analysis of the resulting melt curves following amplification with primers unbiased for L1 methylation status demonstrated demethylation of the 5-aza treated cells compared with vehicle treated cells but no difference between the two 5-aza doses. To enhance the detection of small differences in methylation, primers were biased for unmethylated CpGs within the primer binding sites to enrich for the small proportion of samples that had undergone partial demethylation, hypothesizing that CpGs within the target region would also have undergone some partial demethylation. When these primers were applied to the same 5-aza treated samples, we saw differences in the melt curves of the 5-aza treated samples indicating that the biased primers were capable of sensitively detecting small changes in GC content of the L1 elements following bisulphite modification. In order to quantify these small differences, we devised a means of measuring the difference in melt temperature between samples following PCR—termed the Net Temperature Shift (NTS). The NTS quantifies changes in methylation (based on the GC content of the amplicons) compared with a control sample, rather than determining mean percent methylation of a target region, and incorporates all products that melt at each 0.1°C interval within the melt range for an amplified sample. By analyzing the data obtained at all points within the melt curve, all products that are melting are included in the final index of methylation. This approach enables non-uniform melt profiles to be more accurately analyzed in comparison to other reported methods for analyzing HRM data, for example, the T50Citation48 or the differential fluorescence peak,Citation49 where only a single point in the melt curve is measured.
It has been reported that 3′ end primer biasing can introduce amplification bias-false-positive results.Citation44,Citation45,Citation53,Citation54 However, analysis of the L1-HRM PCR products by pyrosequencing confirmed that the L1 elements amplified by the biased primers were heterogeneously methylated and that primer biasing did not enrich for L1 elements that were completely unmethylated at all CpGs. The mean methylation of all CpGs within the L1 target region analyzed by pyrosequencing confirmed that there was dose dependent demethylation with 5-aza treatment compared with vehicle treated control cells; however, it also demonstrated that the level of demethylation induced by 5-aza was not uniform across all of the CpGs. The sequence heterogeneity of the L1 elements arising from non-CpG sequence variants restricted the pyrosequencing analysis to only 6 out of the 13 CpG dinucleotides. As HRM is used to detect non-CpG polymorphisms, there is the potential for these non-CpG variants to influence the melting profiles of the amplified L1 elements. However, the HRM data showed correlation with total 5-mdC content as determined by LC-MS, which demonstrated a significant, negative correlation with 5-aza dose, indicating that the assay is detecting sequence differences arising from cytosine content following bisulphite modification rather than differences due to sequence diversity. While we observed good correlation between the L1 assay and the LC-MS data, the assay is specific for L1 methylation, and may not reflect changes in cytosine content occurring elsewhere. The demonstration of methylation differences between the two 5-aza doses using the L1 assay suggests that either there is a difference in GC content at the L1 elements that is not reflected in the total genomic 5-mdC content, indicating that L1 elements are more sensitive to 5-aza induced demethylation, or that the L1 assay has greater sensitivity to detect very small changes in DNA methylation. L1 elements have been shown in other studies to be relatively resistant to 5-aza induced demethylation compared with other sequences in the genome.Citation55 This suggests that the L1 assay is a sensitive assay capable of detecting changes in L1 DNA methylation across the genome.
Irahara et al.Citation56 analyzed the precision and reproducibility of pyrosequencing to detect methylation differences, which is reported to be one of the most sensitive methods for methylation analysis. They demonstrated that variability arising from bisulphite modification ranged from 1.2 – 4.2%, and run to run variation for the pyrosequencing reactions ranged from 1.9 – 3.8% of the detected mean CpG methylation. The L1 assay presented here was shown to be as sensitive as the gold standard methods, with a 3% difference in NTS between samples detected. The sensitive nature of the L1-HRM assay as a result of biasing the primers was further demonstrated by the strong correlation of the assay with the pyrosequencing results. Assessment of the effect of PCR and bisulphite modification reaction variability on detection of methylation differences confirmed that the differences detected between samples was not due to inherent PCR or bisulphite modification variability, but due to differences in cytosine content.
Compared with pyrosequencing, the L1 assay is not limited by the presence of non-CpG sequence variants or the size of the target region. The target region of a pyrosequencing assay is restricted to ~30 nucleotides, whereas the L1 assay incorporates an entire CpG island consisting of 13 CpGs spanning 192 bp (of the ~6 kb L1 element) and, as we have demonstrated, does not appear to be influenced by the presence of the sequence variants. Economically, the cost to pyrosequence (including specially designed biotin labeled primers, PCR, PCR product purification, and the pyrosequencing analysis cost) is far greater than performing an in-house PCR reaction with HRM analysis.
Application of the assay to two other murine repetitive DNA elements demonstrated that the assay design can also be applied to other repeat elements. The number of CpGs available for analysis within the smaller B1 elements precluded the design and use of biased primers to enhance sensitivity of detection, while the IAP_LTR element primers could be biased for unmethylated CpGs to enhance sensitivity. While not significant, the IAP_LTR elements displayed a greater NTS difference between the 0.125 µM and 0.5 µM treated 5-aza samples relative to the B1 elements. While this may provide further evidence that biasing can increase the sensitivity to detect small changes in cytosine content, it could also indicate that the number of CpGs within a target region influence sensitivity.
We then applied the L1 assay to various tissue samples isolated from two different mouse strains that have been shown to have differences in genomic 5-mdC levels. Our assay revealed tissue and gender specific differences in L1 methylation consistent with the literatureCitation20,Citation47 where spleen is reported to be hypomethylated when compared with other tissues, in two inbred laboratory mouse strains (Balb/c and CBA) that are commonly used in global DNA methylation studies.Citation20,Citation24,Citation25,Citation57-Citation61 The L1 DNA methylation levels of murine peripheral blood have not previously been reported, however it is commonly used to measure methylation changes in humans.Citation30,Citation56,Citation62,Citation63 The L1-HRM assay is amenable to the analysis of the small volumes of blood obtained during tail vein bleeds allowing repeated blood sampling and peripheral blood methylation levels to be monitored at various time points from the same mouse.
In summary, the assay provides the ability to quantitatively measure L1 DNA methylation changes where individual CpG methylation data or mean percent (%) methylation is not sought. The assay will provide a useful tool for the ongoing investigation of the transcriptional control of DNA repeat elements and their impact on genomic stability. Furthermore, we propose that the L1-HRM assay is a high throughput and inexpensive screening method for the investigation and identification of small but potentially disease-initiating aberrant methylation changes in in vivo mouse models of diseases such as cancer. The assay can also be used to investigate changes that occur with aging, or in response to treatment with or exposure to genotoxins for comparison with human studies, which often utilize serial blood sampling to monitor changes to L1 methylation at multiple concentrations and time points.
Methods and Materials
Cell Culture
A11 murine hybridoma cells were originally derived by fusion of C57Bl/6-pKZ1 transgenic spleen cells with P3653 murine myeloma cells.Citation64 A11 cells were grown in suspension in RPMI 1640 media supplemented with 5% FCS (cat # 10099–141 Invitrogen), 50 IU/mL penicillin and 50 µg/mL streptomycin (cat # 15070–063, Invitrogen) at 37°C with 5% CO2. Cells were passaged twice weekly.
5-aza-2’-deoxycytidine treatment
A11 cells were cultured at a concentration of 2 × 106 during exponential growth phase in 10 mL RPMI 1640 (Flinders University and Medical Centre) in a 25 cm3 tissue culture flask (Nunc). Cells were treated for 24 h with 0.125 µM or 0.5 µM 5-aza-2’-deoxycytidine (5-aza; cat #A3656, Sigma-Aldrich) in DMSO (cat # D8418, Sigma-Aldrich) or vehicle control (0.1% DMSO) (n = 5 flasks per treatment group), and then centrifuged for 5 min at 1200 x g, the media removed by aspiration, and washed with 10 mL RPMI 1640. Cells were resuspended at a density of 2 x 104 cells per mL in RPMI 1640 and cultured for a further 72 h before pelleting for DNA extraction.
Mice
Three-month old male and female Balb/c and CBA mice (Animal Resource Centre, Perth, Australia) were barrier maintained in micro-isolators at the Flinders University Animal House with a 12 h light/dark cycle. Food and water were given ad libitum. All experiments were approved by the Flinders University and Institute of Medical and Veterinary Science Animal Welfare Committees.
Tissue Collection
Immediately prior to euthanasia, mice were placed in a holding restraint. The tails were swabbed with ethanol and a small incision was made to the lateral tail vein using a GoldenRod lancet (MEDIpoint Inc.). No more than 100 µL of peripheral blood (PB) was collected in EDTA-collection tubes (Bectin-Dickinson and Company). Pressure was applied to the wound until bleeding ceased and animals were returned to their cages. Mice were then euthanized by CO2 asphyxiation and tissues were collected and fresh frozen in OCT cryoprotectant (Tissue-Tek). Tissues were stored at -80°C and PB samples were stored at -20°C until DNA extraction.
DNA samples
Genomic DNA was extracted from cells and tissues using the QIAmp DNA Mini extraction kit (Qiagen). DNA was extracted from three fresh frozen tissue sections (25 µM) cut using a cryostat (Reichert-Jung Cryocut 1800) or from the frozen blood samples, as per manufacturer’s instructions except that DNA was eluted with 2 x 100 µL aliquots of Buffer AE. For PB samples, DNA was eluted in 1 x 100 µL aliquot of Buffer AE. DNA was quantified using a Nanodrop 8000 (Thermo Scientific).
Bisulphite Modification
Genomic DNA (200 ng for all samples; 40 ng for PB) was bisulphite modified using the Zymo Research EZ DNA Methylation-Gold kit (Zymo Research) as per manufacturer’s instructions. Modified DNA was diluted with water (Qiagen, Hilden, Germany) to 10 ng/µL (theoretical amount based on genomic DNA concentration input into the bisulphite modification reaction). Unless stated, all DNA underwent two separate bisulphite modification reactions.
Primers
Primers (Geneworks) for LINE1 (Long Interspersed Nucleic Elements – 1), B1_Mm (Mm family of SINE, Short Interspersed Nucleic Elements) and IAP_LTR (Intracisternal-A-Particle Long Terminal Repeat element) specific for bisulphite modified DNA were designed using MethPrimer.Citation33 Pyrosequencing primers and assays were designed using the PSQ Assay Design program (Qiagen). outlines the primer sequences and amplicon sizes. Each primer that was designed specifically for bisulphite modified DNA failed to amplify unmodified genomic DNA template.
Table 1. Primer sequences
PCR
PCR cycling and HRM were performed on a Rotor-Gene Q (Qiagen). A 20 µL reaction mix consisted of 20 ng equivalent of bisulphite modified DNA (10 ng of bisulphite modified PB DNA) and a final concentration of: 1 x EpiTect HRM PCR Master Mix (cat # 59445, Qiagen), 0.75 µM forward primer and 0.75 µM reverse primer, and water (Qiagen). Cycling conditions for LINE1 were as follows: 95°C for 5 min followed by 35 cycles of 95°C for 20 sec, 60°C for 30 sec, and 72°C for 20 sec. Melt curve analysis occurred from 73°C to 84°C rising by 0.1°C/2 sec. Cycling conditions for B1_Mm and IAP_LTR were: 95°C for 5 min followed by 35 cycles of 95°C for 10 sec, 52°C for 15 sec, and 72°C for 10 sec, for 35 cycles. Melt analysis occurred from 65°C - 80°C rising by 0.1°C/2 sec. Each sample was amplified in duplicate per PCR reaction. Unless stated, all samples underwent two separate PCR reactions.
Control DNA
Universal Methylated Mouse DNA (cat # D5012, Zymo Research) was used as a methylated genomic DNA control. Mean methylation of the methylated control was determined to be 86% (± 1.3) by pyrosequencing and contain a 5-mdC content of 5% (± 1.44) by LC-MS. A LINE1 unmethylated control DNA was made as follows. LINE1 sequences from unmodified Universal Methylated DNA were amplified using F_unmod_mLINE1 and R_unmod_mLINE1 (see ). The PCR products were purified using the QIAquick PCR purification kit as per manufacturer’s instructions (Qiagen) and 200 ng of the purified PCR products were subjected to bisulphite modification and purification as described above for genomic DNA. Pyrosequencing analysis was performed to confirm that each CpG was unmethylated, and was determined to have a mean methylation of 3% representing background noise level. The final product was diluted in water to a concentration at which the Cq (quantification cycle) range was within that of test samples. The unmethylated control DNA was included in each HRM-PCR as a quality control sample.
Sequencing Analysis
HRM PCR products were purified using the QIAquick PCR purification kit (Qiagen) and quantified on a Nanodrop 8000 (Thermo Scientific). Sequencing primers are outlined in . Sequencing reactions were performed by the SouthPath and Flinders Sequencing Facility (Flinders University and Medical Centre, Adelaide, Australia). For pyrosequencing, HRM PCR reaction replicates for each sample (n = 5 per treatment group) were combined and aliquoted into 96 well plates in duplicate in a volume of 10 µL and sent to EpigenDx (MA, USA) for analysis on a Qiagen-Pyrosequencing PSQ-MD.
Liquid Chromatography- Mass Spectrometry (LC-MS)
DNA hydrolysis
Genomic DNA was hydrolyzed as outlined in Song et al.Citation65 Briefly, 250 ng of DNA in a volume of 20 µL was denatured at 100°C for 3 min and placed on ice. To the DNA, 2 µL of 0.1 M ammonium acetate (cat # A7330, Sigma-Aldrich), pH 5.3, containing 2 units Nuclease P1 from Penicillium citrinum (cat #N8630, Sigma-Aldrich) was added and samples were incubated at 45°C for 2 h. Following this, 2.2 µL of 1 M ammonium bicarbonate (cat # 09830, Sigma-Aldrich) containing 0.002 units of Phosphodiesterase I from Crotalus adamanteus venom (cat # P3243, Sigma-Aldrich) was added and samples were incubated at 37°C for 2 h. Finally, 1 unit of calf intestinal phosphatase (cat # F201-S, Finnzymes) was added and the reaction was incubated at 37°C for 1 h. Hydrolyzed samples were stored at -20°C. Each sample underwent two separate hydrolysis reactions.
LC-MS Procedure
LC-MS was performed by Flinders Analytical (Flinders University, Adelaide, Australia). Briefly, Liquid chromatography (LC) separation was performed on a Waters 2695 HPLC (Milford), at a flow rate of 0.25 mL/min with a column temperature of 22°C. The LC column was a Waters Atlantis T3, 2.1 mm x 150 mm, 5 µm particle (cat # 186003736, Milford). Two buffers were used: mobile phase A - 0.1% aqueous formic acid (cat # 06440, Fluka); and mobile phase B - 0.1% formic acid in acetonitrile. The LC program was 100% solvent A for 3 min, a linear gradient to 85% solvent A at 15 min, then back to 100% solvent A at 15.5 min where it was held for 6.5 min for re-equilibration. A 20 µL volume of hydrolyzed sample was diluted with 100 µL of type 1 water (cat # SYNSV00WW, Millipore Synergy System) and transferred to a 250 µL volume insert inside a 2 mL LC-MS vial and 10 µL of this solution was injected into the LC. Mass Spectrometry was performed on a Waters Quattro micro, triple quadruple mass spectrometer (Milford) fitted with an electrospray source. Positive ion electrospray and multiple reaction monitoring conditions were determined manually using ribonucleoside and deoxyribonucleoside standards at a concentration of 10 µg/mL in water (dC, cat # D3897; C, cat # C122106; U, cat # U3750; 5mC, cat # M4254; T, cat # T9250; dA, cat # D7400; A, cat # A9251; dG, cat # D7145; and G, cat # G6752; Sigma-Aldrich; 5-mdC, cat # N-1044, Jena Bioscience). Quantitation for 5-mdC was monitored with a precursor ion of 242, fragment ion of 126 with a dwell time of 0.1 sec. Cone voltage was 12 V and collision voltage was 8 V. For dG; the precursor ion was 267.8, the fragment ion was 152, dwell time was 0.1 sec, cone voltage was 12 V and collision voltage was 12 V. Each sample was injected in duplicate. Percent methylation was calculated as the ratio of 5-mdC to dG [5-mdC/dG].
Calculation of methylation differences and statistical analyses
Differences in DNA methylation as detected by HRM were calculated using the average difference between the melt curves of a test sample and the methylated control, termed the Net Temperature Shift (NTS), such that a negative NTS results from a shift of a sample’s normalized melt curve to the left of the normalized melt curve of the methylated control and indicates that the sample is less methylated. Calculation of the NTS is as follows: within the Rotor-Gene Q program, two normalization regions (at temperatures before and after melt peak temperature) were nominated for HRM analysis. For LINE1, normalization region 1 was between 74°C-75°C, while normalization region 2 was between 82°C-84°C. For B1_Mm the regions were: 66°C-68°C and 79°C-80°C; for IAP_LTR: 70°C-72°C, 78°C-79°C. The subtraction of the methylated control normalized curve from each test normalized curve was performed automatically within the Rotor-Gene Q program and the summed difference of the fluorescence value at each temperature point (0.1°C intervals) within the entire melt range was divided by 100 to obtain the average distance between the curves, or the NTS. Data were analyzed using the statistical program SPSS Statistics (version 17, SPSS Inc.). The effect of treatment group and the analysis of methylation differences between mouse tissues was first tested by ANOVA, with Bonferroni post-hoc analysis used to compare means between groups. Linear bivariate correlations were analyzed using Pearson correlation. In all cases p < 0.05 was considered significant. All means are displayed with error bars representing ± 1 standard error of the mean.
| Abbreviations: | ||
| HRM | = | high resolution melt |
| PCR | = | polymerase chain reaction |
| 5-aza | = | 5-aza-2’-deoxycytidine |
| L1 | = | LINE1, Long Interspersed Elements |
| NTS | = | net temperature shift |
| LC-MS | = | liquid chromatography–mass spectrometry |
| PB | = | peripheral blood |
Additional material
Download Zip (137.1 KB)Acknowledgments
We thank Ami–Louise Cochrane, Mark Lawrence, Alex Staudacher and Katrina Bexis for assistance with animal procedures; Dr. Tanya Day for the 5-aza cell culture experiments; and Dr. Tina Bianco-Miotto for critically reading this manuscript. This work was supported by the Low Dose Radiation Research Program, Biological and Environmental Research, US. Department of Energy (DE-FG02–05ER64104); and the Flinders Medical Centre Foundation and BHP Billiton Low Dose Radiation Research Scholarship (to M.N.).
Related Research Data
References
- Cropley JE, Suter CM, Beckman KB, Martin DI. CpG methylation of a silent controlling element in the murine Avy allele is incomplete and unresponsive to methyl donor supplementation. PLoS ONE 2010; 5:e9055; http://dx.doi.org/10.1371/journal.pone.0009055; PMID: 20140227
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature 2001; 409:860 - 921; http://dx.doi.org/10.1038/35057062; PMID: 11237011
- Fedorov A. Regulation of mammalian LINE1 retrotransposon transcription. Cell Tissue Biol 2009; 3:1 - 13; http://dx.doi.org/10.1134/S1990519X09010015
- Ostertag EM. Biology of Mammalian L1 Retrotransposons. Annu Rev Genet 2001; 35:501; http://dx.doi.org/10.1146/annurev.genet.35.102401.091032; PMID: 11700292
- Scott AF, Schmeckpeper BJ, Abdelrazik M, Comey CT, O'Hara B, Rossiter JP, et al. Origin of the human L1 elements: Proposed progenitor genes deduced from a consensus DNA sequence. Genomics 1987; 1:113 - 25; http://dx.doi.org/10.1016/0888-7543(87)90003-6; PMID: 3692483
- Furano AV, Robb SM, Robb FT. The structure of the regulatory region of the rat L1 (L1Rn, long interspersed repeated) DNA family of transposable elements. Nucleic Acids Res 1988; 16:9215; http://dx.doi.org/10.1093/nar/16.19.9215; PMID: 2845369
- Howard G, Eiges R, Gaudet F, Jaenisch R, Eden A. Activation and transposition of endogenous retroviral elements in hypomethylation induced tumors in mice. Oncogene 2008; 27:404 - 8; http://dx.doi.org/10.1038/sj.onc.1210631; PMID: 17621273
- Estécio MR, Gharibyan V, Shen L, Ibrahim AE, Doshi K, He R, et al. LINE-1 hypomethylation in cancer is highly variable and inversely correlated with microsatellite instability. PLoS ONE 2007; 2:e399; http://dx.doi.org/10.1371/journal.pone.0000399; PMID: 17476321
- Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, et al. Induction of tumors in mice by genomic hypomethylation. Science 2003; 300:489 - 92; http://dx.doi.org/10.1126/science.1083558; PMID: 12702876
- Igarashi S, Suzuki H, Niinuma T, Shimizu H, Nojima M, Iwaki H, et al. A novel correlation between LINE-1 hypomethylation and the malignancy of gastrointestinal stromal tumors. Clin Cancer Res 2010; 16:5114 - 23; http://dx.doi.org/10.1158/1078-0432.CCR-10-0581; PMID: 20978145
- Ogino S, Kawasaki T, Nosho K, Ohnishi M, Suemoto Y, Kirkner GJ, et al. LINE-1 hypomethylation is inversely associated with microsatellite instability and CpG island methylator phenotype in colorectal cancer. Int J Cancer 2008; 122:2767 - 73; http://dx.doi.org/10.1002/ijc.23470; PMID: 18366060
- Ogino S, Nosho K, Kirkner GJ, Kawasaki T, Chan AT, Schernhammer ES, et al. A cohort study of tumoral LINE-1 hypomethylation and prognosis in colon cancer. J Natl Cancer Inst 2008; 100:1734 - 8; http://dx.doi.org/10.1093/jnci/djn359; PMID: 19033568
- Yamamoto E, Toyota M, Suzuki H, Kondo Y, Sanomura T, Murayama Y, et al. LINE-1 hypomethylation is associated with increased CpG island methylation in Helicobacter pylori-related enlarged-fold gastritis. Cancer Epidemiol Biomarkers Prev 2008; 17:2555 - 64; http://dx.doi.org/10.1158/1055-9965.EPI-08-0112; PMID: 18842996
- Wolff EM, Byun H-M, Han HF, Sharma S, Nichols PW, Siegmund KD, et al. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet 2010; 6:e1000917; http://dx.doi.org/10.1371/journal.pgen.1000917; PMID: 20421991
- Saito K, Kawakami K, Matsumoto I, Oda M, Watanabe G, Minamoto T. Long interspersed nuclear element 1 hypomethylation is a marker of poor prognosis in stage IA non-small cell lung cancer. Clin Cancer Res 2010; 16:2418 - 26; http://dx.doi.org/10.1158/1078-0432.CCR-09-2819; PMID: 20371677
- Sauer J, Jang H, Zimmerly EM, Kim KC, Liu Z, Chanson A, et al. Ageing, chronic alcohol consumption and folate are determinants of genomic DNA methylation, p16 promoter methylation and the expression of p16 in the mouse colon. Br J Nutr 2010; 104:24 - 30; http://dx.doi.org/10.1017/S0007114510000322; PMID: 20236555
- Finnell RH, Spiegelstein O, Wlodarczyk B, Triplett A, Pogribny IP, Melnyk S, et al. DNA methylation in Folbp1 knockout mice supplemented with folic acid during gestation. J Nutr 2002; 132:2457S - 61S; PMID: 12163711
- Vanhees K, Coort S, Ruijters EJB, Godschalk RWL, van Schooten FJ. Barjesteh van Waalwijk van Doorn-Khosrovani S. Epigenetics: prenatal exposure to genistein leaves a permanent signature on the hematopoietic lineage. FASEB J 2011; 25:797 - 807; http://dx.doi.org/10.1096/fj.10-172155; PMID: 21048042
- Koturbash I, Scherhag A, Sorrentino J, Sexton K, Bodnar W, Swenberg JA, et al. Epigenetic mechanisms of mouse interstrain variability in genotoxicity of the environmental toxicant 1,3-Butadiene. Toxicol Sci 2011; 122:448 - 56; http://dx.doi.org/10.1093/toxsci/kfr133; PMID: 21602187
- Giotopoulos G, McCormick C, Cole C, Zanker A, Jawad M, Brown R, et al. DNA methylation during mouse hemopoietic differentiation and radiation-induced leukemia. Exp Hematol 2006; 34:1462 - 70; http://dx.doi.org/10.1016/j.exphem.2006.06.008; PMID: 17046565
- Pogribny I, Koturbash I, Tryndyak V, Hudson D, Stevenson SM, Sedelnikova O, et al. Fractionated low-dose radiation exposure leads to accumulation of DNA damage and profound alterations in DNA and histone methylation in the murine thymus. Mol Cancer Res 2005; 3:553 - 61; http://dx.doi.org/10.1158/1541-7786.MCR-05-0074; PMID: 16254189
- Pogribny I, Raiche J, Slovack M, Kovalchuk O. Dose-dependence, sex- and tissue-specificity, and persistence of radiation-induced genomic DNA methylation changes. Biochem Biophys Res Commun 2004; 320:1253 - 61; http://dx.doi.org/10.1016/j.bbrc.2004.06.081; PMID: 15249225
- Raiche J, Rodriguez-Juarez R, Pogribny I, Kovalchuk O. Sex- and tissue-specific expression of maintenance and de novo DNA methyltransferases upon low dose X-irradiation in mice. Biochem Biophys Res Commun 2004; 325:39 - 47; http://dx.doi.org/10.1016/j.bbrc.2004.10.002; PMID: 15522198
- Tawa R, Kimura Y, Komura J, Miyamura Y, Kurishita A, Sasaki MS, et al. Effects of X-ray irradiation on genomic DNA methylation levels in mouse tissues. J Radiat Res (Tokyo) 1998; 39:271 - 8; http://dx.doi.org/10.1269/jrr.39.271; PMID: 10196782
- Barbot W, Dupressoir A, Lazar V, Heidmann T. Epigenetic regulation of an IAP retrotransposon in the aging mouse: progressive demethylation and de-silencing of the element by its repetitive induction. Nucleic Acids Res 2002; 30:2365; http://dx.doi.org/10.1093/nar/30.11.2365; PMID: 12034823
- Singhal RP, Mays-Hoopes LL, Eichhorn GL. DNA methylation in aging of mice. Mech Ageing Dev 1987; 41:199 - 210; http://dx.doi.org/10.1016/0047-6374(87)90040-6; PMID: 3431172
- Ririe KM, Rasmussen R, Wittwer C. Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal Biochem 1997; 245:154 - 60; http://dx.doi.org/10.1006/abio.1996.9916; PMID: 9056205
- Stanzer S, Balic M, Strutz J, Heitzer E, Obermair F, Hauser-Kronberger C, et al. Rapid and reliable detection of LINE-1 hypomethylation using high resolution melting analysis. Clin Biochem 2010; 43:1443 - 8; http://dx.doi.org/10.1016/j.clinbiochem.2010.09.013; PMID: 20883681
- Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res 2004; 32:e38; http://dx.doi.org/10.1093/nar/gnh032; PMID: 14973332
- Bollati V, Schwartz J, Wright R, Litonjua A, Tarantini L, Suh H, et al. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev 2009; 130:234 - 9; http://dx.doi.org/10.1016/j.mad.2008.12.003; PMID: 19150625
- Kato Y, Kaneda M, Hata K, Kumaki K, Hisano M, Kohara Y, et al. Role of the Dnmt3 family in de novo methylation of imprinted and repetitive sequences during male germ cell development in the mouse. Hum Mol Genet 2007; 16:2272 - 80; http://dx.doi.org/10.1093/hmg/ddm179; PMID: 17616512
- Lane N, Dean W, Erhardt S, Hajkova P, Surani A, Walter J, et al. Resistance of IAPs to methylation reprogramming may provide a mechanism for epigenetic inheritance in the mouse. Genesis 2003; 35:88 - 93; http://dx.doi.org/10.1002/gene.10168; PMID: 12533790
- Li L-C, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics 2002; 18:1427 - 31; http://dx.doi.org/10.1093/bioinformatics/18.11.1427; PMID: 12424112
- Wittwer CT, Reed G, Gundry C, Vandersteen J, Pryor R. High-resolution genotyping by amplicon melting analysis using LCGreen. Clin Chem 2003; 49:853; http://dx.doi.org/10.1373/49.6.853; PMID: 12765979
- Gundry CN, Vandersteen J, Reed G, Pryor R, Chen J, Wittwer C. Amplicon melting analysis with labeled primers: a closed-tube method for differentiating homozygotes and heterozygotes. Clin Chem 2003; 49:396; http://dx.doi.org/10.1373/49.3.396; PMID: 12600951
- Candiloro IL, Mikeska T, Hokland P, Dobrovic A. Rapid analysis of heterogeneously methylated DNA using digital methylation-sensitive high resolution melting: application to the CDKN2B (p15) gene. Epigenetics Chromatin 2008; 1:7; http://dx.doi.org/10.1186/1756-8935-1-7; PMID: 19014416
- Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, et al. MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res 2000; 28:E32; http://dx.doi.org/10.1093/nar/28.8.e32; PMID: 10734209
- Rand KN, Molloy P. Sensitive measurement of unmethylated repeat DNA sequences by end-specific PCR. Biotechniques 2010; 49:xiii - xvii; http://dx.doi.org/10.2144/000113494; PMID: 20964632
- Fandy TE, Herman JG, Kerns P, Jiemjit A, Sugar EA, Choi S-H, et al. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood 2009; 114:2764 - 73; http://dx.doi.org/10.1182/blood-2009-02-203547; PMID: 19546476
- Gebhard C, Benner C, Ehrich M, Schwarzfischer L, Schilling E, Klug M, et al. General transcription factor binding at CpG islands in normal cells correlates with resistance to de novo DNA methylation in cancer cells. Cancer Res 2010; 70:1398; http://dx.doi.org/10.1158/0008-5472.CAN-09-3406; PMID: 20145141
- Suzuki S, Ono R, Narita T, Pask AJ, Shaw G, Wang C, et al. Retrotransposon silencing by DNA methylation can drive mammalian genomic imprinting. PLoS Genet 2007; 3:e55; http://dx.doi.org/10.1371/journal.pgen.0030055; PMID: 17432937
- Zhang D, Wang Y, Bai Y, Ge Q, Qiao Y, Luo J, et al. A novel method to quantify local CpG methylation density by regional methylation elongation assay on microarray. BMC Genomics 2008; 9:59; http://dx.doi.org/10.1186/1471-2164-9-59; PMID: 18237388
- Weeks RJ, Morison I. Detailed methylation analysis of CpG islands on human chromosome region 9p21. Genes Chromosomes Cancer 2006; 45:357 - 64; http://dx.doi.org/10.1002/gcc.20297; PMID: 16372313
- Wojdacz TK, Dobrovic A. Methylation-sensitive high resolution melting (MS-HRM): a new approach for sensitive and high-throughput assessment of methylation. Nucleic Acids Res 2007; 35:e41; http://dx.doi.org/10.1093/nar/gkm013; PMID: 17289753
- Wojdacz TK, Hansen L. Reversal of PCR bias for improved sensitivity of the DNA methylation melting curve assay. Biotechniques 2006; 41:274; http://dx.doi.org/10.2144/000112240; PMID: 16989087
- Kristensen LS, Mikeska T, Krypuy M, Dobrovic A. Sensitive melting analysis after real time-methylation specific PCR (SMART-MSP): high-throughput and probe-free quantitative DNA methylation detection. Nucleic Acids Res 2008; 36:e42; http://dx.doi.org/10.1093/nar/gkn113; PMID: 18344521
- Gama-Sosa MA, Midgett RM, Slagel VA, Githens S, Kuo KC, Gehrke CW, et al. Tissue-specific differences in DNA methylation in various mammals. Biochimica et Biophysica Acta (BBA) -. Gene Structure and Expression 1983; 740:212 - 9; http://dx.doi.org/10.1016/0167-4781(83)90079-9
- Smith E, Jones ME, Drew PA. Quantitation of DNA methylation by melt curve analysis. BMC Cancer 2009; 9:123; http://dx.doi.org/10.1186/1471-2407-9-123; PMID: 19393074
- Malentacchi F, Forni G, Vinci S, Orlando C. Quantitative evaluation of DNA methylation by optimization of a differential-high resolution melt analysis protocol. Nucleic Acids Res 2009; 37:e86; http://dx.doi.org/10.1093/nar/gkp383; PMID: 19454604
- Lorente A, Mueller W, Urdangarín E, Lázcoz P, Von Deimling A, Castresana J. Detection of methylation in promoter sequences by melting curve analysis-based semiquantitative real time PCR. BMC Cancer 2008; 8:61; http://dx.doi.org/10.1186/1471-2407-8-61; PMID: 18298842
- Jüttermann R, Li E, Jaenisch R. Toxicity of 5-aza-2'-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc Natl Acad Sci USA 1994; 91:11797 - 801; http://dx.doi.org/10.1073/pnas.91.25.11797; PMID: 7527544
- Mund C, Hackanson B, Stresemann C, Lübbert M, Lyko F. Characterization of DNA demethylation effects induced by 5-aza-2 -deoxycytidine in patients with myelodysplastic syndrome. Cancer Res 2005; 65:7086; http://dx.doi.org/10.1158/0008-5472.CAN-05-0695; PMID: 16103056
- Wojdacz TK, Hansen LL, Dobrovic A. A new approach to primer design for the control of PCR bias in methylation studies. BMC Res Notes 2008; 1:54; http://dx.doi.org/10.1186/1756-0500-1-54; PMID: 18710507
- Wojdacz TK, Dobrovic A, Hansen LL. Methylation-sensitive high-resolution melting. Nat Protoc 2008; 3:1903 - 8; http://dx.doi.org/10.1038/nprot.2008.191; PMID: 19180074
- Mund C, Hackanson B, Stresemann C, Lübbert M, Lyko F. Characterization of DNA Demethylation Effects Induced by 5-Aza-2'-Deoxycytidine in Patients with Myelodysplastic Syndrome. Cancer Res 2005; 65:7086 - 90; http://dx.doi.org/10.1158/0008-5472.CAN-05-0695; PMID: 16103056
- Irahara N, Nosho K, Baba Y, Shima K, Lindeman N, Hazra A, et al. Precision of pyrosequencing assay to measure LINE-1 methylation in colon cancer, normal colonic mucosa, and peripheral blood cells. J Mol Diagn 2010; 12:177; http://dx.doi.org/10.2353/jmoldx.2010.090106; PMID: 20093385
- Akagi K, Li J, Stephens RM, Volfovsky N, Symer DE. Extensive variation between inbred mouse strains due to endogenous L1 retrotransposition. Genome Res 2008; 18:869 - 80; http://dx.doi.org/10.1101/gr.075770.107; PMID: 18381897
- Boulton E, Cole C, Knight A, Cleary H, Snowden R, Plumb M. Low-penetrance genetic susceptibility and resistance loci implicated in the relative risk for radiation-induced acute myeloid leukemia in mice. Blood 2003; 101:2349 - 54; http://dx.doi.org/10.1182/blood-2002-08-2394; PMID: 12411293
- Carole L, Yauk AP. Andrea Rowan-Carroll, Igor Kortubash, Andrew Williams, Olga Kovalchuk, Tandem repeat mutation, global DNA methylation, and regulation of DNA methyltransferases in cultured mouse embryonic fibroblast cells chronically exposed to chemicals with different modes of action. Environ Mut Mutag 2008; 49:26 - 35
- Plumb M. Genetic instability in radiation-induced leukaemias: mouse models. Int J Radiat Biol 1998; 74:711 - 20; http://dx.doi.org/10.1080/095530098140989; PMID: 9881716
- Yu Y, Okayasu R, Weil MM, Silver A, McCarthy M, Zabriskie R, et al. Elevated breast cancer risk in irradiated BALB/c mice associates with unique functional polymorphism of the Prkdc (DNA-dependent protein kinase catalytic subunit) gene. Cancer Res 2001; 61:1820 - 4; PMID: 11280730
- El-Maarri O, Walier M, Behne F, van Üüm J, Singer H, Diaz-Lacava A, et al. Methylation at global LINE-1 repeats in human blood are affected by gender but not by age or natural hormone cycles. PLoS ONE 2011; 6:e16252; http://dx.doi.org/10.1371/journal.pone.0016252; PMID: 21311577
- Zhu Z-Z, Hou L, Bollati V, Tarantini L, Marinelli B, Cantone L, et al. Predictors of global methylation levels in blood DNA of healthy subjects: a combined analysis. Int J Epidemiol 2010; http://dx.doi.org/10.1093/ije/dyq154; PMID: 20846947
- Hooker AM, Home R, Morley A, Sykes P. Dose-dependent increase or decrease of somatic intrachromosomal recombination produced by etoposide. Mutat Res 2002; 500:117 - 24; http://dx.doi.org/10.1016/S0027-5107(02)00007-6; PMID: 11890941
- Song L, James S, Kazim L, Karpf A. Specific method for the determination of genomic DNA methylation by liquid chromatography-electrospray ionization tandem mass spectrometry. Anal Chem 2005; 77:504 - 10; http://dx.doi.org/10.1021/ac0489420; PMID: 15649046