Abstract
Recent studies demonstrated that cytosine methylation in the genome can be reversed without DNA replication by enzymatic mechanisms based on base excision-repair pathways. Both enzymatic methylation and demethylation mechanisms are active in the cell nucleus at the same time. One can hypothesize that the actual level of CpG methylation could be the result of a balance between the two antagonistic processes with a rapid turnover. In the present study, we used mass spectrometry to measure the total methyl-cytosine content of the genome in cultured human cells after short incubation with the known methyltransferase inhibitor 5-deoxy-azacytidine. A significant decrease of the DNA methylation was observed. Indeed, the inhibition of the methylation can only result in a rapid reduction of the overall methyl-cytosine level if the process of demethylation is simultaneous. These observations suggest that the enzymatic mechanisms responsible of the opposing reactions of DNA methylation and demethylation act simultaneously and may result in a continuous and rapid turnover of methylated cytosines. This conclusion is supported by the observation that 5-deoxy-azacytidine was incorporated in the genomic DNA of non-dividing cells and could be detected as soon as after two hours of incubation, hence providing a mechanistic explanation to the inhibition of methyltransferases. The observations are compatible with the idea that the enzymatic mechanisms that bring together of the opposing reactions of DNA methylation and demethylation act simultaneously and may result in a continuous and unsuspected rapid turnover of DNA methylation. This conclusion is at odds with the generally accepted view of high stability of cytosine methylation where the role of enzymatic demethylation is considered as limited to some special situations such as transcription. It places DNA methylation in the same category as other epigenetic modifications with covalent modifications dynamically added to and removed from the chromatin with high turnover rate.
Keywords: :
Background
Methylation of the genomic DNA is one of the key epigenetic mechanisms involved in the regulation of the chromatin structure in eukaryotic cells. Gene expression and inactivation, recombination and replication of the genome are all dependent on the chromatin structure, hence influenced by DNA methylation. The enzymatic mechanisms of DNA methylation are fairly well characterized. Most of the methylation is found on cytosines in CpG dinucleotides. The methylation reaction of the cytosines is performed by a family of methyltransferase enzymes. DNMT1 is the main enzyme that is present in virtually all cell types analyzed and responsible for the maintenance of methylation. The other members of the DNMT1 enzyme family are involved in de novo and maintenance methylation in various cell types and physiological conditions.
Until recently, CpG methylation has been viewed as a stable epigenetic modification that could only be reversed passively, over several cell cycles, by the effect of dilution in the absence of maintenance methylation (reviewed in ref. Citation1). For a long time, the possibility of enzymatic demethylation was considered controversial because of the absence of reproducible experimental evidence.Citation2,Citation3 This situation has recently changed. An increasing number of reports revealed that cytosine methylation can decrease rapidly in the genomic DNA under the action of enzymatic mechanisms without DNA replication. Demethylation of gene promoters,Citation4 plasmid constructs,Citation5 imprinted gene sequencesCitation6 or of the whole genomeCitation7 has been described. More detailed studies have shown that cyclic DNA demethylation/methylation may occur during gene transcription.Citation5,Citation8,Citation9 Several recent studies on the molecular mechanisms of enzymatic demethylation in animals and plants have revealed the direct involvement of DNA repair mechanisms, mainly base excision-repair pathways that act by replacing the methylated cytosine into a non-methylated.Citation1
Although different cell types may use different mechanisms, it is likely that one or several of these mechanisms are permanently active in the cell nucleus. The simultaneous presence of DNA methylating and demethylating enzymatic mechanisms within the same nucleus raises the possibility that the methylation pattern of any sequence at any moment reflects the actual balance of the two opposing processes.Citation6,Citation10 Although this hypothesis is usually not considered, in the light of the recent progress in the characterization of active demethylation mechanisms we felt necessary to formally examine its validity using a direct approach. We measured the 5-methyl-cytosine (mC) content of the genome with high precision before and after a short-term inhibition of the DNA methyltransferases by deoxy-azacitidine (5-Aza-dC) in non-dividing cells. If the working hypothesis is correct, the inhibition of DNA methyltransferases should lead to a detectable decrease of the overall methyl-cytosine level. The results show that this is indeed the case.
Results and Discussion
First, we set up a new mass spectrometry-based method for the measurement of the total methyl-cytosine content of the genome. We used formic acid hydrolysis followed by nucleobase detection. The resulting nucleobases were analyzed by MS (). The precision and reproducibility of the method was determined by the analysis of 12 independent fractions of the same genomic DNA preparation. Each fraction was measured three times. The measurement produced highly reproducible results with a coefficient of variation σ/µ = 0.0227 (). When DNA samples isolated from 12 independent cell cultures were processed together the coefficient of variation varied typically between σ/µ = 0.08 to 0.06 indicating that most of the variation was due to the differences between the cultures and/or the DNA extraction procedure. The high precision and sensitivity of our method makes it suitable for the detection of very small differences in the total methyl-cytosine content between two samples that remain undetectable by any other method.
Next, we have analyzed the effect of a short 5-Aza-dC treatment on the overall mC content in the genome. First, we used a myogenic cell line of human origin (CHQhT).Citation11 In order to decrease the potential contribution of passive demethylation during DNA replication to the final result we reduced the proportion of cells in S phase by allowing the cultures to grow to close to 100% confluence. The CHQhT cells have the property to exit the cell cycle to G0 phase at confluence and enter the myogenic differentiation phase. Indeed, the cytometric analyses showed that only 2–3% of the cells were in S phase (). The cells were treated with 5-Aza-dC for 2 h at the concentration of 10 or 100 µM. The mC/C ratio was determined in the genomic DNA of the cells. The total mC content in normal cells was between 3.5 to 4% of the total C content. After only 2 h of 5-Aza-dC treatment this ratio decreased in both cell types by around 10% (). Such a decrease cannot be the consequence of a passive DNA demethylation during DNA replication since only an insignificant fraction of the cells was in S phase (see estimations of the passive demethylation below).
In order to verify that the observed 10% decrease is not a specific feature of the myogenic cell line, we repeated the analysis in confluent HepG2 cells. Since this is a cancer cell line, the proportion of the cells in S phase was expected to be higher than in the myogenic cells. We have quantified this fraction by measuring BrdU incorporation and the DNA content. We found that 6.5% of the cells were in S phase at 95% of confluence (). The cells were treated with 5-Aza-dC as above. The result of the mC content measurement was very similar seen in the myogenic cells: a 2 h treatment with 10 µM of 5-Aza-dC resulted in approximately 10% decrease in the mC content of the genomic DNA ().
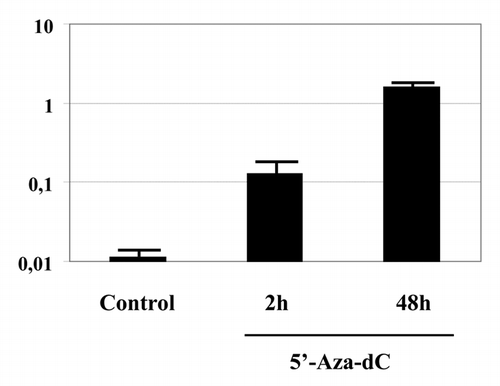
5-Aza-dC is a cytidine nucleoside analog that acts on DNA methyltransferases only after incorporation into the DNA.Citation12,Citation13 Therefore, we investigated if the presence of the inhibitor can be detected in the genomic DNA after a short treatment. A confluent culture of HepG2 cells was treated with tritiated 5-Aza-dC(6-3H) either for 2 or 48 h. DNA was prepared from the cells and the incorporated radioactivity was measured. As shown on , the genomic DNA showed a significant incorporation of the radiolabeled inhibitor even after 2 h. Importantly, the incorporation of the radiolabeled 5-Aza-dC was proportional with the duration of the incubation and reached a one order of magnitude higher level after 48 h compared with the 2 h treatment.
The work presented here aimed to test the hypothesis that the actual level of DNA methylation is the result of a balance between the rapidly and simultaneously acting methylating and demethylating enzymatic activities in the absence of DNA replication. The most straightforward way to do this is to inhibit one of these activities and assess the effect on the final balance. If the hypothesis of rapid methylation turnover is correct, the inhibition of the maintenance methylation should lead to a decrease of the total genomic mC content. Such decrease was indeed observed. We used 5-Aza-dC, a well-known DNA methyltransferase inhibitor to block DNA methylation. As short as 2 h of treatment was sufficient to generate a significant decrease in the mC/C ratio in two different cell types. It is unlikely that this decrease resulted from the incorporation of 5-Aza-dC into the replicating DNA. Assuming that the 5-Aza-dC treatment is 100% efficient in inhibiting the maintenance methylation, the contribution of the passive demethylation to the decrease in mC/C ratio can be estimated as follows: if all cells were in S phase and the S phase is 10 h long, then after 2 h the overall decrease in mC content due to the absence of maintenance methylation would be 20% in the whole population. However, only 2.5% of CHQhT cells and 6.5% of HepG2 cells are in S phase under our experimental conditions. Therefore, passive demethylation can occur only in these cells and its contribution to the decrease in mC content of the whole cell population including the cells that are in G1, G2 or M phase is around 0.5 and 1.3% in CHQhT and HepG2 cell respectively. However, the detected decrease in the mC content of the genome was approximately 10% in both cell types. This is significantly higher than what could be explained by passive demethylation. Interestingly, the observed decrease was very close to that obtained after 48 h treatment suggesting that the new dynamic equilibrium between demethylation/methylation is reached rapidly. It is possible is that stronger overall loss of methylation or demethylation may not be tolerated by the cells. This idea is supported by the observation that after 4 d of 5-Aza-dC treatment non-genic regions undergo stronger demethylation than genic ones.Citation14
The action of 5-Aza-dC on DNA methylation is based on its incorporation into the DNA during the replication.Citation12,Citation13 The incorporated nucleoside analog covalently binds the DNMT molecule resulting in depletion of the methylating enzyme activity in the nucleusCitation15 that leads to the reduction of the overall mC level in the genome. Growing evidence suggests that very rapid enzymatic demethylation of DNA is possible in the absence of replication by the action of base excision and/or nucleotide excision repair mechanisms (reviewed in ref. Citation1). In all cases the original methylated cytosine is replaced by an unmethylated by the action of DNA repair polymerases and ligases. Our observations of the incorporation of the 5-Aza-dC into the genomic DNA even after 2 h incubation suggest that the short-term action of the inhibitor may be mediated by a similar mechanism of action based on the replacement of the methyl-cytosines presumably by the action of excision/repair mechanisms even in the non-replicating genome. Contrary to the decrease of the mC content of the genomic DNA, the incorporation of the 5-Aza-dC is proportional with the incubation time. This suggests that the incorporation of the 5-Aza-dC is a continuous process that occurs independently of the inhibition of DNA methyltransferases and unmethylated cytosines can also be replaced.
Since we measured the global mC content on the whole genome, it is impossible to deduce on the exact nature of sequences that are subject to demethylation. As an enzymatic process involving interactions with the DNA, the rate of demethylation/methylation also have to depend on the accessibility of the DNA, mainly of the chromatin structure. Therefore, coding and non-coding sequences, actively transcribed or silenced regions are expected to have a different rate of mC/C exchange. Indeed, it has been shown that active demethylation can be linked to transcriptional activity.Citation5,Citation8,Citation9 Actively transcribed genomic regions are certainly more accessible to the action of enzymes, including those that catalyze demethylation. However, gene promoters, enhancers and high CpG density regions (islands) are usually unmethylated; it is unlikely therefore that transcriptional activity over these regions can account for the demethylation of 10% of all methylated cytosines in the genome.
The presented observations are consistent with the idea that the enzymatic mechanisms that bring together of the opposing reactions of DNA methylation and demethylation act simultaneously and may result in a continuous and unsuspected rapid turnover of DNA methylation. More importantly, these observations cannot be explained on the basis of the prevailing view on DNA methylation that links enzymatic demethylation to some specific event triggered by specific signals. Although this finding might be surprising at the first glance, we should note that all other epigenetic marks are also dynamically added to and removed from the chromatin with high turnover rate. Altogether the results reported here suggest that DNA methylation behaves similarly to other epigenetic modifications rather than having very different stability.
Materials and Methods
Cell culture.
Two different human cell lines were used: a myoblast cell line, CHQhTCitation11 and HepG2 cells. CHQhT cells were cultured in F-10 (Gibco) supplemented with 20% of heat-inactivated fetal bovine serum. HepG2 cells were also cultured in D-MEM (Gibco) supplemented with 10% of heat-inactivated fetal bovine serum, 100 IU/ml penicillin and 100 mg/ml streptomycine (Gibco). The cells were incubated at 37C°, 95% air and 5% CO2 and grown to over 90% confluence. 5-Aza-dC (Sigma) was solubilised in water at a stock concentration of 10 mM and stored at −80C° before use. 5-Aza-dC (10 µM) was used for 2 h treatment in the same medium. Mock-treatment with an identical volume of water was used as a control. After treatment cells were frozen at −80°C until DNA extraction.
Cell cycle analysis by flow cytometry.
CHQhT cells were cultured as described above until over 95% of confluency then harvested. 1 × 106 cells were washed in PBS, resuspended in 100 µl of PBS, and 1 ml of 70% ethanol was added. Cells were then stored at −20°C until analyzed. For analysis, cells were washed twice in PBS and then resuspended in fresh 1 ml of PBS containing 0.1 mg/ml propidium iodide (Invitrogen) and 0.25 mg/ml RNase (Sigma). Following incubation for 15 min in dark, cells were analyzed by flow cytometry for propidium iodide fluorescence.
For BrdU incorporation analysis the cells were cutured for 15 min in 10 mM BrDU (Sigma) then harvested and fixed in 70% ethanol. The cells were permeabilized with 2 N HCl/0.5% Triton X-100 for 30 min, neutralized in 0.1 M Na Tetraborate pH 8.5 for 1 min, stained with anti-BrDU-FITC antibody for 30 min in 100 µl of PBS/0.5% Tween 20/1% BSA/1 µl for BrDU detection, incubated with propidium iodide and analyzed by flow cytometry.
DNA treatment for the analysis of the 5-methyl-cytosine (mC) content.
DNA from the cultured cells was prepared using Nucleospin genomic DNA extraction kit (Clontech) according to the protocol provided by the manufacturer. In order to eliminate all contaminating RNA, the DNA was digested with RNase A.
Sample Preparation and HPLC-MS Measurements
We hydrolyzed the genomic DNA to nucleobases by chemical means using formic acid as described previously.Citation16 Briefly, 100 µl of formic acid (100%) was added to 20 µl of samples and pipetted into a 2 ml glass vial. The tightly crimped vial was kept at 130°C for 90 min. After nitrogen evaporation the samples were reconstituted in 100 µl of 10 mM ammonium-formate solution and pipetted to a 200 µl microvial. To optimize the MS procedure (3200 QTrap) chemical standards were used for cytosine (C), methyl-cytosine (mC), adenine (A).
Perkin Elmer Series200 system (consisting of binary pump, autosampler and column oven) was used for separation. Ten millimolar ammonium formate (eluent A) and acetonitrile (eluent B) was used for separation using gradient elution: A/B 90/10(1)-1.80/20-2-50/50(1.5)-0.5.10/90(2)-1.90/10(5). The flow rate on the RX-Sil column was 1 ml/min. Twenty microliters of samples were injected. The oven temperature was 60°C.
For mass spectrometric measurements TurboV electrospray source was used in positive ion detection mode. Spray voltage was 5,000 V, evaporation temperature: 500°C, curtain, evaporation and drying gases were 25, 50 and 50 instrument units, respectively. 150 msec dwell time and 5 msec pause times were used for each MRM transitions. Collision energy was set to 30eV.
Incorporation of radiolabeled 5-Aza-dC into the genomic DNA.
Tritiated 5-Aza-dC(6-3H) (1 mCi/ml, 20.6 Ci/mmol, 11.1 µg/ml) was purchased from Moravek Biochemicals, Inc. The HepG2 cells were incubated with labeled 5-Aza-dC at 7.5 µM or 1 µM concentration for 2 or 48 h, respectively. Genomic DNA was prepared and treated by RNase to eliminate co-purified RNA as described above. The radioactivity incorporated into the DNA was measured in a Beckman scintillation counter.
Statistical analysis.
One-tailed t-test was used to compare mC content in treated cells and the control.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Figures and Tables
Figure 1 (A) Mass spectrometry detection of the mC content in the genomic DNA. Different peaks on the MRM scan represent different transitions of the related different compounds. The smallest peak corresponds to methyl-cytosine. The insets on the right side show the higher magnification of the four peaks. (B) Reproducibility of the measurement. The same DNA preparation was divided into 12 fractions and each fraction was measured 3 times.
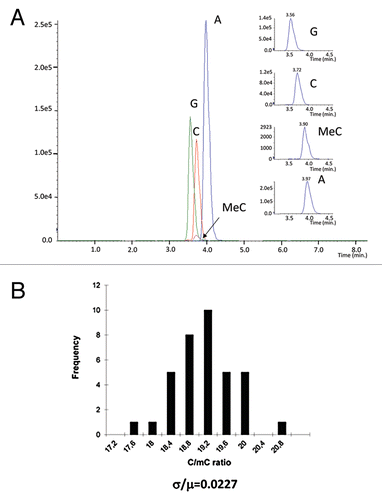
Figure 2 (A) Upper part: Flow cytometric cell cycle analysis of a typical CHQ-HT cell culture used in this study. Note the low (2.2%) proportion of cells in S phase. Lower part: Flow cytometric cell cycle analysis of BrDU incorporation in HepG2 cells at high confluence used in this study. 6.5% of cells are in S phase. (B) Decrease of the methyl-cytosine content in the genomic DNA of the myogenic (CHQ-HT) cells treated with 100 µM of 5′-Aza-dC for 1 and 2 h and 10 µM for 2 h compared with untreated or 48 h treated cells. (*) indicates significant difference (p < 0.05). (C) HepG2 cells treated with 10 µM of 5′-Aza-dC for 2 h.
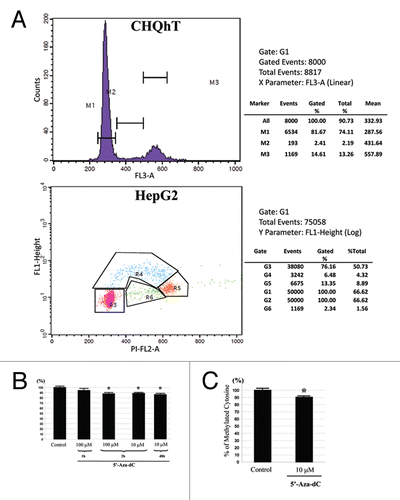
Figure 3 Incorporation (cpm) of radio labeled 5′-Aza-dC(6-3H) in the genomic DNA of HepG2 cells after 2 h and 48 h treatment.
Acknowledgments
We authors are grateful to Maria Bihari, Andras Varadi, Jorg Tost and Anne Galy for continuous discussions and to Gyorgy Varadi for technical assistance.
Funding
This work was supported by Genethon and by the grant PD79183 from the National Research Fund of Hungary. T.A. is a Bolyai Janos Foundation fellow.
Related Research Data
References
- Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol 2010; 11:607 - 620; PMID: 20683471; http://dx.doi.org/10.1038/nrm2950
- Ooi SK, Bestor TH. The colorful history of active DNA demethylation. Cell 2008; 133:1145 - 1148; PMID: 18585349; http://dx.doi.org/10.1016/j.cell.2008.06.009
- Ramchandani S, Bhattacharya SK, Cervoni N, Szyf M. DNA methylation is a reversible biological signal. Proc Natl Acad Sci USA 1999; 96:6107 - 6112; PMID: 10339549; http://dx.doi.org/10.1073/pnas.96.11.6107
- Bruniquel D, Schwartz RH. Selective, stable demethylation of the interleukin-2 gene enhances transcription by an active process. Nat Immunol 2003; 4:235 - 240; PMID: 12548284; http://dx.doi.org/10.1038/ni887
- D'Alessio AC, Weaver IC, Szyf M. Acetylation-induced transcription is required for active DNA demethylation in methylation-silenced genes. Mol Cell Biol 2007; 27:7462 - 7474; PMID: 17709385; http://dx.doi.org/10.1128/MCB.01120-07
- Imamura T, Kerjean A, Heams T, Kupiec JJ, Thenevin C, Paldi A. Rapid CpG and non-CpG methylation turnover of Peg/Mest in the mouse oocyte and pre-implantation embryo. J Biol Chem 2005; 280:20171 - 20175; PMID: 15778220; http://dx.doi.org/10.1074/jbc.M501749200
- Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. Demethylation of the zygotic paternal genome. Nature 2000; 403:501 - 502; PMID: 10676950; http://dx.doi.org/10.1038/35000656
- Kangaspeska S, Stride B, Métivier R, Polycarpou-Schwarz M, Ibberson D, Carmouche RP, et al. Transient cyclical methylation of promoter DNA. Nature 2008; 452:112 - 115; PMID: 18322535; http://dx.doi.org/10.1038/nature06640
- Métivier R, Gallais R, Tiffoche C, Le Péron C, Jurkowska RZ, Carmouche RP, et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature 2008; 452:45 - 50; PMID: 18322525; http://dx.doi.org/10.1038/nature06544
- Arányi T, Páldi A. The constant variation: DNA methylation changes during preimplantation development. FEBS Lett 2006; 580:6521 - 6526; PMID: 17134704; http://dx.doi.org/10.1016/j.febslet.2006.11.018
- Zhu CH, Mouly V, Cooper RN, Mamchaoui K, Bigot A, Shay JW, et al. Cellular senescence in human myoblasts is overcome by human telomerase reverse transcriptase and cyclin-dependent kinase 4: consequences in aging muscle and therapeutic strategies for muscular dystrophies. Aging Cell 2007; 6:515 - 523; PMID: 17559502; http://dx.doi.org/10.1111/j.1474-9726.2007.00306.x
- Glover AB, Leyland-Jones B. Biochemistry of azacitidine: a review. Cancer Treat Rep 1987; 71:959 - 964; PMID: 2443243
- Veselý J, Cihák A. Incorporation of a potent antileukemic agent, 5-aza-2′-deoxycytidine, into DNA of cells from leukemic mice. Cancer Res 1977; 37:3684 - 3689; PMID: 71199
- Lim HW, Iwatani M, Hattori N, Tanaka S, Yagi S, Shiota K. Resistance to 5-aza-2′-deoxycytidine in genic regions compared to non-genic repetitive sequences. J Reprod Dev 2010; 56:86 - 93; PMID: 19996551; http://dx.doi.org/10.1262/jrd.20247
- Zhou L, Cheng X, Connolly BA, Dickman MJ, Hurd PJ, Hornby DP. Zebularine: a novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases. J Mol Biol 2002; 321:591 - 599; PMID: 12206775; http://dx.doi.org/10.1016/S0022-2836(02)00676-9
- Kok RM, Smith DE, Barto R, Spijkerman AM, Teerlink T, Gellekink HJ, et al. Global DNA methylation measured by liquid chromatography-tandem mass spectrometry: analytical technique, reference values and determinants in healthy subjects. Clin Chem Lab Med 2007; 45:903 - 911; PMID: 17617036; http://dx.doi.org/10.1515/CCLM.2007.137