Abstract
Synaptic homeostasis is a form of neuronal plasticity that stabilizes activity of neural networks. Both presynaptic and postsynaptic effects are well documented in response to activity changes. The electrical signaling machinery of individual neurons, or intrinsic properties, have also been implicated in this plasticity. How synaptic and intrinsic changes are coordinated, however, is still a puzzle. A recent study by Ping and Tsunoda shows both synaptic and intrinsic changes in Drosophila central neurons in response to prolonged inactivity.Citation1 Changes include the upregulation of Dα7 nicotinic acetylcholine receptors (nAChRs) and Shal (Kv4) potassium channels. This work has two noteworthy findings. First, although mediated by different receptors, synaptic homeostasis in the central nervous system (CNS) is conserved across species. This is perhaps the most direct demonstration that nAChRs mediate synaptic homeostasis. Changes in the expression of nAChRs have long been noted during development, as well as during pathological conditions, such as nicotine addictionCitation2 and Alzheimer disease.Citation3 The second interesting finding is the relationship between synaptic and intrinsic plasticity: nAChRs are upregulated immediately, subsequently triggering a rapid increase in Shal K+ channels. This novel mechanism regulates synaptic homeostasis to stabilize synaptic potentials. This study sets the stage for Drosophila central neurons as a model for cholinergic synaptic homeostasis, its regulation and role in disease.
Glutamatergic Homeostatic Plasticity
Synaptic homeostatic plasticity is generally viewed as a negative feedback mechanism that adjusts neuronal synaptic strength, preventing under- or over-activity of circuits.Citation4-Citation6 Typically, studies of synaptic homeostasis report that inactivity induces compensatory changes in postsynaptic receptors, presynaptic release of neurotransmitters, or intrinsic electrical properties of individual neurons.Citation7,Citation8 Chronic elevation of synaptic strength has also been shown to induce homeostatic responses. For example, elevated neural activity leads to AMPAR endocytosis and degradation by a proteasome-dependent pathway.Citation9,Citation10 Since Hebbian plasticity is a positive-feedback phenomenon, a compensatory negative-feedback mechanism such as synaptic homeostasis is likely to be critical for preventing runaway excitation and maintaining stability. Indeed, synaptic homeostasis is widely proposed as a mechanism to counter-balance the Hebbian plasticity induced, for example, during memory formation.Citation11
In the last decade, researchers have characterized glutamatergic homeostasis and begun to unravel the signaling pathways leading to an increase in synaptic strength following synaptic inactivity. In brief, studies have shown that signaling molecules, such as Arc/Arg3.1,Citation12 CaMKII,Citation13 CaMKIV,Citation14,Citation15 BDNFCitation16,Citation17 and retinoic acid,Citation16 are likely involved. Most of these signaling molecules are required for postsynaptic synaptic scaling, except for BDNF, which in cooperation with the voltage-dependent calcium channel CaV2.1,Citation17 are involved in increasing transmitter release at presynaptic sites. While most studies have focused on the signaling components involved in scaling-up synaptic strength in response to inactivity, little is known about how pathways are precisely regulated such that over-compensation is prevented. Interestingly, a recent study has shown that the monomeric G-protein, Rab3, binds a repressor that blocks presynaptic compensation to inactivity, making it a candidate for such a role.Citation18 In our recent work,Citation1 we find that the homeostatic response triggers an acute increase in Shal K+ channels that functions to stablize synaptic potentials.
Cholinergic Homeostatic Plasticity
Although homeostatic plasticity has been intensively studied, relatively little of this work has focused on non-glutamatergic synapses. A proper excitation-inhibition balance, however, is important for all neural circuits. Thus, other fast excitatory, as well as inhibitory and G-protein coupled receptor-mediated synapses in the brain must harbor a similar homeostatic capability. Understanding the underlying mechanisms at other synapses is likely to give insight into the commonalities and scope of this plasticity. Further, dysfunction of compensatory response mechanisms may point to important pathological conditions. For example, cholinergic synaptic homeostasis has been implicated in nicotine addiction as well as Alzheimer disease (AD) (see final section).
We used central neurons in Drosophila as a model for studying homeostatic mechanisms at cholinergic synapses. The major fast excitatory and inhibitory currents in Drosophila central neurons are mediated by nAChRs and GABA receptors (GABARs), respectively.Citation19,Citation20 In the Drosophila genome, there are ten nAChR subunit genes: seven of the α subtype, Dα1–7, and three of the β subtype, Dβ1–3. Work in vertebrates has shown that five subunits form a functional receptor, where at least one α subunit is required.Citation21 Receptors with different subunit combinations have been shown to have different biophysical properties and expression patterns, and are likely to play distinct physiological roles. For example, homomeric α7 nAChRs, which are one of the most abundant nAChR subtypes in the vertebrate brain, exhibit a higher Ca2+ permeability and faster desensitization kinetics than other subtypes.Citation22,Citation23 The α4β2 subtype is also abundant in the CNS, and exhibits clear upregulation following prolonged nicotine exposure.Citation2 Since nAChRs underlie the vast majority of fast excitatory synaptic transmission in the Drosophila CNS, they likely mediate Hebbian and homeostatic plasticity mechanisms similar to glutamate receptors in the mammalian CNS.
To test this, we pharmacologically blocked neural activity using the nAChR antagonist curare. We found that after 24 h of curare treatment, the Dα7 nAChR was selectively increased. Although little is known about the functional properties of the different nAChR subunits in Drosophila, the high sequence identity (~60%) between the vertebrate α7 and Drosophila Dα5–7 subunits suggests that Dα5–7 are orthologs of α7, likely encoding similar Ca2+ permeable homomers. It is interesting that GluA2 lacking AMPA receptors, which are Ca2+ permeable (Cp-AMPARs), have also been shown to play an important role in glutamatergic homeostatic plasticity.Citation24-Citation26 Studies have shown that blocking AMPARs induces increased surface expression of both Ca2+-impermeable (Ci) and Cp-AMPARs. Ca2+ permeable receptors, such as α7 nAChRs or Cp-AMPARs, likely mediate Ca2+-dependent pathways involved in homeostasis, regulation of homeostasis, and/or additional forms of plasticity. Our study indicates that the upregulation of Dα7 nAChRs is due to increased translation of new receptors. The increase in receptor number was observed both biochemically and functionally, as an increase in miniature excitatory synaptic current (mEPSC) amplitude. An mEPSC is the smallest, unitary, post-synaptic response, corresponding to the effect of a single vesicle of neurotransmitter released by a presynaptic cell. Thus, an increase in mEPSC amplitude is consistent with an increase in synaptic strength mediated by the postsynaptic cell. It remains to be seen whether it is the blockage of Dα7 nAChRs, and the Ca2+ influx through these receptors, in particular, that is required for their homeostatic upregulation. If decreased Ca2+ influx is a trigger for increased translation, an open and interesting question will be whether this signaling occurs locally, in dendrites, and perhaps at single synapses.
We additionally observed an increase in the mEPSC frequency in response to prolonged inactivity, suggesting that homeostatic changes are also mediated by the presynaptic cell. These results are similar to mammalian studies.Citation24,Citation27,Citation28 These results suggest the existence of presynaptic changes following inactivity that lead to a higher vesicle release probability. Thus, it is possible that decreased Ca2+ influx during synaptic blockade, or perhaps increased Ca2+ influx immediately following synaptic blockade may contribute to retrograde signaling to pre-synaptic terminals. Homeostatic plasticity in Drosophila and mammalian central neurons indicates the preservation of an important phenomenon in the CNS, and differences may be significant for understanding how glutamatergic and cholinergic synaptic homeostasis are distinctly implemented.
Intrinsic Plasticity and Roles in Synaptic Homeostasis
Intrinsic properties also play important roles in homeostatic plasticity. Studies have shown that both Na+ and K+ currents are often altered in response to inactivity.Citation29 The coordination between synaptic and intrinsic changes, however, is still a mystery. For example, how do synaptic changes trigger intrinsic changes, or vice versa? We show that prolonged inhibition nAChRs not only increased the expression of Dα7 nAChRs (discussed above), but also induced the rapid upregulation of Shal K+ (Kv4) channels during the 30–60 min following the recovery of synaptic activity (). Shal channels carry a rapidly activating, and rapidly inactivating, A-type K+ current in somato-dendritic regions of neurons. The upregulation of Shal channels following synaptic blockade brought up several interesting questions. For example, does the upregulation of Shal K+ channels contribute to synaptic homeostasis, or does it serve to downregulate the homeostatic response? Also, how is this intrinsic response signaled—by way of the synaptic response? And does it lead to a transcriptional/translational regulation of the channels? Our data shows that the increase in Shal channels is blocked with any one of the following: no recovery of synaptic transmission following inactivity, the absence of Dα7 receptors, Ca2+ chelation during the recovery period, CaMKII inhibition during the recovery period, inhibition of transcription or translation during the recovery period. These results suggest that the increase in Shal channels is signaled during recovery of synaptic activity, by Dα7 nAChRs, and presumably, the Ca2+ influx from the increased number of Dα7 receptors induced by inactivity. Further studies are needed to confirm an increase in Ca2+ influx through these receptors, and direct activation of CaMKII.
Figure 1. A model of homeostatic plasticity in Drosophila central neurons. Twenty-four hour block of nAChRs results in a translational upregulation of Dα7 nAChRs, through an unknown signal pathway (indicated by dotted arrow). During the block, Shal K+ channel levels are unchanged. Briefly after washout of the antagonist, an increased influx of Ca2+ ions through newly translated Dα7 nAChRs activate CaMKII, leading to enhanced transcription of Shal K+ channels. mEPSCs are larger due to the increased number of nAChRs, whereas mEPSPs are stabilized to control levels because of regulation by newly expressed Shal K+ channels.
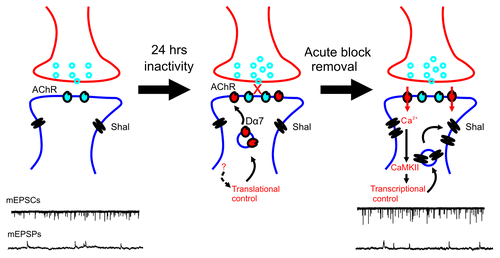
One of our most intriguing findings is that, unlike most other intrinsic changes reported, the upregulation of Shal K+ channels appears to downregulate the homeostatic response. That is, despite the increase in mEPSCs, mEPSPs were stabilized to control levels by the increase in Shal current. Our interpretation is that the increase in Shal channels, which is triggered by the homeostatic increase in Dα7 nAChRs, serves to modulate or tune the homeostatic response. It is not surprising that regulatory mechanisms would be needed to prevent over-compensation. No such mechanism, however, has been proposed until now. Since other reported intrinsic changes have been shown to function homeostatically, it will be critical to understand how neurons signal intrinsic changes to play these opposing roles. Action potential (AP) firing, for example, has been implicated as a signal for some intrinsic changes, even the upregulation of K+ (delayed rectifier type) currents, induced by inactivity.Citation30 Blocking AP firing, however, did not prevent the upregulation of Shal channels, supporting the idea that distinct pathways regulate intrinsic targets.
Another noteworthy feature of our model () is that the increase in Dα7 nAChRs is slow and regulated at the translational level, while the subsequent increase in Shal K+ channels is fast and transcriptionally controlled. Since excessive activity could be toxic to neurons, it makes sense that the upregulation of nAChRs is slower than the increase Shal channels, and the faster negative feedback by Shal channels may be a good safeguard to prevent over-activity. Indeed, chronic activation of AMPARs has been shown to induce AMPAR endocytosis within minutes, whereas compensatory responses to inactivity take hours.Citation9,Citation26,Citation30,Citation31 The translational upregulation of Dα7 nAChRs parallels the translational increase reported for AMPARs.Citation32 It was, however, somewhat surprising to us that the more rapid upregulation of Shal channels appeared to be transcriptionally regulated. We used the transcriptional inhibitor actinomycin-D in whole-brain cultures, as well as in single cells, monitored by immunoblot analysis and electrophysiologically, respectively. Additional studies, however, should be performed to test these results. For example, while the lack of upregulation of Shal from an exongenous transgene supported this result, it would be more convincing to see the upregulation of a reporter gene inserted at the Shal locus, and/or the identification of transcriptional activators and their corresponding binding sites in the Shal gene. Since it is commonly believed that translational regulation allows for more rapid changes in protein level than transcriptional regulation, the upregulation of Shal following inactivity may suggest that transcription is a more versatile option than previously assumed.
Implications for Human Diseases
Synaptic homeostatic plasticity acts to maintain a proper balance of neural activity. With such an important function, it has, not surprisingly, been implicated in human disease and dysfunction. nAChRs and cholinergic synaptic homeostasis have also been implicated in multiple pathologies, in both the peripheral and central nervous systems. For example, at the vertebrate NMJ, nAChRs mediate fast excitatory synaptic transmission. These muscular nAChRs are the target for curare-mimetic muscle relaxants. Interestingly, blocking activity at the NMJ with curare results in the permanent loss of nAChRs in muscle cells,Citation33 which may explain the paralytic syndrome often observed in patients after treatment with reversible nAChR blocker.Citation34 In the mammalian CNS, homomeric α7 and heteromeric α4β2 nAChRs are among the most abundant and ubiquitously expressed nAChRs, with various other subunit combinations expressed in more restricted regions of the brain. Changes in nAChR function and number have been associated with a wide variety of disorders, including epilepsy, dementia, nicotine dependence, and Alzheimer disease (AD).Citation21,Citation35
Prolonged exposure to nicotine has long been known to lead to an upregulation of nAChRs.Citation36-Citation38 While some reports have implicated that this may be a homeostatic response to the desensitization of nAChRs, very little is known about cholinergic synaptic homeostasis, and whether nAChRs can mediate synaptic homeostasis. We suggest that inactivation of nAChRs triggers the upregulation of nAChRs in a homeostatic pathway. Desensitization of nAChRs by nicotine may trigger a homeostatic signaling pathway for upregulating receptors. Work in Drosophila central neurons may give important insights for mechanisms contributing to nicotine addiction.
Cholinergic synaptic homeostasis has also been implicated in AD. Toxic Aβ deposits in the early AD brain have been shown to decrease AMPARs and suppress synaptic function, reducing overall neural activity.Citation39-Citation43 This downregulation of activity is likely to induce a homeostatic response which provides initial protection of circuit function. In the AD brain, nAChRs, and in particular, the α7 receptor, has been implicated in mediating synaptic homeostasis.Citation3,Citation44-Citation49 While cholinergic synaptic scaling likely protects neural circuit function initially,Citation46 it has been suggested to ultimately promote neurogeneration.Citation44 In this model, homeostatic upregulation of the α7 nAChR, with its high Ca2+ permeability, eventually renders cells more susceptible to Aβ toxicity, thereby promoting neurodegeneration. Much remains to be understood, however, about cholinergic synaptic scaling and its involvement in AD. Our results indicate that inactivity can indeed induce an increase in Dα7 nAChR expression. The increase in Dα7 leads to upregulation of Shal K+ channels, which modulate synaptic potentials, suggesting that intrinsic mechanisms also contribute to the stabilization of neural activity, and perhaps even the progression of AD. Despite its potentially critical roles, cholinergic synaptic homeostasis has been surprisingly understudied. We hope that our recent work will help launch Drosophila as a model for studying cholinergic homeostasis.
References
- Ping Y, Tsunoda S. Inactivity-induced increase in nAChRs upregulates Shal K(+) channels to stabilize synaptic potentials. Nat Neurosci 2012; 15:90 - 7; http://dx.doi.org/10.1038/nn.2969; PMID: 22081160
- Fenster CP, Whitworth TL, Sheffield EB, Quick MW, Lester RA. Upregulation of surface alpha4beta2 nicotinic receptors is initiated by receptor desensitization after chronic exposure to nicotine. J Neurosci 1999; 19:4804 - 14; PMID: 10366615
- Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer’s disease. J Neurosci 2001; 21:4125 - 33; PMID: 11404397
- Thiagarajan TC, Lindskog M, Malgaroli A, Tsien RW. LTP and adaptation to inactivity: overlapping mechanisms and implications for metaplasticity. Neuropharmacology 2007; 52:156 - 75; http://dx.doi.org/10.1016/j.neuropharm.2006.07.030; PMID: 16949624
- Turrigiano G. Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol 2012; 4:a005736; http://dx.doi.org/10.1101/cshperspect.a005736; PMID: 22086977
- Davis GW. Homeostatic control of neural activity: from phenomenology to molecular design. Annu Rev Neurosci 2006; 29:307 - 23; http://dx.doi.org/10.1146/annurev.neuro.28.061604.135751; PMID: 16776588
- Pozo K, Goda Y. Unraveling mechanisms of homeostatic synaptic plasticity. Neuron 2010; 66:337 - 51; http://dx.doi.org/10.1016/j.neuron.2010.04.028; PMID: 20471348
- Turrigiano G. Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu Rev Neurosci 2011; 34:89 - 103; http://dx.doi.org/10.1146/annurev-neuro-060909-153238; PMID: 21438687
- Patrick GN, Bingol B, Weld HA, Schuman EM. Ubiquitin-mediated proteasome activity is required for agonist-induced endocytosis of GluRs. Curr Biol 2003; 13:2073 - 81; http://dx.doi.org/10.1016/j.cub.2003.10.028; PMID: 14653997
- Hou Q, Gilbert J, Man HY. Homeostatic regulation of AMPA receptor trafficking and degradation by light-controlled single-synaptic activation. Neuron 2011; 72:806 - 18; http://dx.doi.org/10.1016/j.neuron.2011.10.011; PMID: 22153376
- Bushey D, Tononi G, Cirelli C. Sleep and synaptic homeostasis: structural evidence in Drosophila. Science 2011; 332:1576 - 81; http://dx.doi.org/10.1126/science.1202839; PMID: 21700878
- Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, et al. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron 2006; 52:475 - 84; http://dx.doi.org/10.1016/j.neuron.2006.08.034; PMID: 17088213
- Thiagarajan TC, Piedras-Renteria ES, Tsien RW. alpha- and betaCaMKII. Inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron 2002; 36:1103 - 14; http://dx.doi.org/10.1016/S0896-6273(02)01049-8; PMID: 12495625
- Goold CP, Nicoll RA. Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron 2010; 68:512 - 28; http://dx.doi.org/10.1016/j.neuron.2010.09.020; PMID: 21040851
- Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron 2008; 57:819 - 26; http://dx.doi.org/10.1016/j.neuron.2008.02.031; PMID: 18367083
- Lindskog M, Li L, Groth RD, Poburko D, Thiagarajan TC, Han X, et al. Postsynaptic GluA1 enables acute retrograde enhancement of presynaptic function to coordinate adaptation to synaptic inactivity. Proc Natl Acad Sci U S A 2010; 107:21806 - 11; http://dx.doi.org/10.1073/pnas.1016399107; PMID: 21098665
- Jakawich SK, Nasser HB, Strong MJ, McCartney AJ, Perez AS, Rakesh N, et al. Local presynaptic activity gates homeostatic changes in presynaptic function driven by dendritic BDNF synthesis. Neuron 2010; 68:1143 - 58; http://dx.doi.org/10.1016/j.neuron.2010.11.034; PMID: 21172615
- Müller M, Pym EC, Tong A, Davis GW. Rab3-GAP controls the progression of synaptic homeostasis at a late stage of vesicle release. Neuron 2011; 69:749 - 62; http://dx.doi.org/10.1016/j.neuron.2011.01.025; PMID: 21338884
- Gu H, O’Dowd DK. Cholinergic synaptic transmission in adult Drosophila Kenyon cells in situ. J Neurosci 2006; 26:265 - 72; http://dx.doi.org/10.1523/JNEUROSCI.4109-05.2006; PMID: 16399696
- Wilson RI, Laurent G. Role of GABAergic inhibition in shaping odor-evoked spatiotemporal patterns in the Drosophila antennal lobe. J Neurosci 2005; 25:9069 - 79; http://dx.doi.org/10.1523/JNEUROSCI.2070-05.2005; PMID: 16207866
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 2009; 89:73 - 120; http://dx.doi.org/10.1152/physrev.00015.2008; PMID: 19126755
- Ballivet M, Alliod C, Bertrand S, Bertrand D. Nicotinic acetylcholine receptors in the nematode Caenorhabditis elegans. J Mol Biol 1996; 258:261 - 9; http://dx.doi.org/10.1006/jmbi.1996.0248; PMID: 8627624
- Séguéla P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. J Neurosci 1993; 13:596 - 604; PMID: 7678857
- Lindskog M, Li L, Groth RD, Poburko D, Thiagarajan TC, Han X, et al. Postsynaptic GluA1 enables acute retrograde enhancement of presynaptic function to coordinate adaptation to synaptic inactivity. Proc Natl Acad Sci U S A 2010; 107:21806 - 11; http://dx.doi.org/10.1073/pnas.1016399107; PMID: 21098665
- Man HY. GluA2-lacking, calcium-permeable AMPA receptors--inducers of plasticity?. Curr Opin Neurobiol 2011; 21:291 - 8; http://dx.doi.org/10.1016/j.conb.2011.01.001; PMID: 21295464
- Thiagarajan TC, Lindskog M, Tsien RW. Adaptation to synaptic inactivity in hippocampal neurons. Neuron 2005; 47:725 - 37; http://dx.doi.org/10.1016/j.neuron.2005.06.037; PMID: 16129401
- Mitra A, Mitra SS, Tsien RW. Heterogeneous reallocation of presynaptic efficacy in recurrent excitatory circuits adapting to inactivity. Nat Neurosci 2012; 15:250 - 7; http://dx.doi.org/10.1038/nn.3004; PMID: 22179109
- Jakawich SK, Nasser HB, Strong MJ, McCartney AJ, Perez AS, Rakesh N, et al. Local presynaptic activity gates homeostatic changes in presynaptic function driven by dendritic BDNF synthesis. Neuron 2010; 68:1143 - 58; http://dx.doi.org/10.1016/j.neuron.2010.11.034; PMID: 21172615
- Turrigiano G. Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu Rev Neurosci 2011; 34:89 - 103; http://dx.doi.org/10.1146/annurev-neuro-060909-153238; PMID: 21438687
- Nataraj K, Le Roux N, Nahmani M, Lefort S, Turrigiano G. Visual deprivation suppresses L5 pyramidal neuron excitability by preventing the induction of intrinsic plasticity. Neuron 2010; 68:750 - 62; http://dx.doi.org/10.1016/j.neuron.2010.09.033; PMID: 21092863
- Hou Q, Gilbert J, Man HY. Homeostatic regulation of AMPA receptor trafficking and degradation by light-controlled single-synaptic activation. Neuron 2011; 72:806 - 18; http://dx.doi.org/10.1016/j.neuron.2011.10.011; PMID: 22153376
- Sutton MA, Schuman EM. Dendritic protein synthesis, synaptic plasticity, and memory. Cell 2006; 127:49 - 58; http://dx.doi.org/10.1016/j.cell.2006.09.014; PMID: 17018276
- Lichtman JW, Colman H. Synapse elimination and indelible memory. Neuron 2000; 25:269 - 78; http://dx.doi.org/10.1016/S0896-6273(00)80893-4; PMID: 10719884
- Beecher HK, Todd DP. A study of the deaths associated with anesthesia and surgery: based on a study of 599, 548 anesthesias in ten institutions 1948-1952, inclusive. Ann Surg 1954; 140:2 - 35; http://dx.doi.org/10.1097/00000658-195407000-00001; PMID: 13159140
- Steinlein OK, Bertrand D. Neuronal nicotinic acetylcholine receptors: from the genetic analysis to neurological diseases. Biochem Pharmacol 2008; 76:1175 - 83; http://dx.doi.org/10.1016/j.bcp.2008.07.012; PMID: 18691557
- Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ. A subtype of nicotinic cholinergic receptor in rat brain is composed of alpha 4 and beta 2 subunits and is up-regulated by chronic nicotine treatment. Mol Pharmacol 1992; 41:31 - 7; PMID: 1732720
- Nashmi R, Xiao C, Deshpande P, McKinney S, Grady SR, Whiteaker P, et al. Chronic nicotine cell specifically upregulates functional alpha 4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J Neurosci 2007; 27:8202 - 18; http://dx.doi.org/10.1523/JNEUROSCI.2199-07.2007; PMID: 17670967
- Quick MW, Lester RA. Desensitization of neuronal nicotinic receptors. J Neurobiol 2002; 53:457 - 78; http://dx.doi.org/10.1002/neu.10109; PMID: 12436413
- Zhang C, Wu B, Beglopoulos V, Wines-Samuelson M, Zhang D, Dragatsis I, et al. Presenilins are essential for regulating neurotransmitter release. Nature 2009; 460:632 - 6; http://dx.doi.org/10.1038/nature08177; PMID: 19641596
- Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science 2002; 298:789 - 91; http://dx.doi.org/10.1126/science.1074069; PMID: 12399581
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 2003; 39:409 - 21; http://dx.doi.org/10.1016/S0896-6273(03)00434-3; PMID: 12895417
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, et al. APP processing and synaptic function. Neuron 2003; 37:925 - 37; http://dx.doi.org/10.1016/S0896-6273(03)00124-7; PMID: 12670422
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, et al. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci U S A 1999; 96:3228 - 33; http://dx.doi.org/10.1073/pnas.96.6.3228; PMID: 10077666
- Small DH. Network dysfunction in Alzheimer’s disease: does synaptic scaling drive disease progression?. Trends Mol Med 2008; 14:103 - 8; http://dx.doi.org/10.1016/j.molmed.2007.12.006; PMID: 18262842
- Nordberg A. Nicotinic receptor abnormalities of Alzheimer’s disease: therapeutic implications. Biol Psychiatry 2001; 49:200 - 10; http://dx.doi.org/10.1016/S0006-3223(00)01125-2; PMID: 11230871
- Frölich L, Kayed R, Zheng H, Sweatt JD, Dineley KT. The cholinergic pathology in Alzheimer’s disease--discrepancies between clinical experience and pathophysiological findings. J Neural Transm 2002; 109:1003 - 13; http://dx.doi.org/10.1007/s007020200083; PMID: 12111437
- DeKosky ST, Ikonomovic MD, Styren SD, Beckett L, Wisniewski S, Bennett DA, et al. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol 2002; 51:145 - 55; http://dx.doi.org/10.1002/ana.10069; PMID: 11835370
- Bednar I, Paterson D, Marutle A, Pham TM, Svedberg M, Hellström-Lindahl E, et al. Selective nicotinic receptor consequences in APP(SWE) transgenic mice. Mol Cell Neurosci 2002; 20:354 - 65; http://dx.doi.org/10.1006/mcne.2002.1112; PMID: 12093166