Abstract
Crohn disease (CD) ileal lesions are colonized by adherent-invasive E. coli (AIEC) that locally induce inflammation. Hypoxia inducible factor (HIF)-1alpha protein is expressed in acute and chronically inflamed site; however the molecular basis of this expression is not fully understood. The aim of the study was to access whether AIEC induce HIF-1α expression and to study the consequence of HIF-1α expression on the onset of Crohn disease pathogenesis. We show that HIF-1α is maximally expressed in inflamed ileal epithelium of CD-patients. CEACAM6, a protein that acts as a receptor of AIEC, is expressed in this particular condition. Using CEABAC 10 transgenic mice that express CEACAM6, we show that AIEC bacteria, but not non-pathogenic E. coli K12, induce the production of HIF-1alpha protein and the activation of VEGF/VEGFR signaling. Downstream analyses on human intestinal epithelial cells silenced for hif-1α, highlight the crucial role of this protein in production of pro-angiogenic factors. This study highlights the crucial role of AIEC bacteria as promoter of inflammatory disorders of the gastrointestinal tract and provides clear evidence that HIF-1α protein plays a major role in mediating this effect.
Introduction
Mainly located in the gut, the microbiota is crucial to human life by influencing human physiology and nutrition.Citation1 In addition, it contributes to the shaping of healthy intestinal immune response.Citation2 Inflammatory bowel diseases (IBD) are human inflammatory disorders including Crohn disease (CD) and ulcerative colitis (UC) which represent a major public health problem. However, the etiology of IBD remains poorly understood and various genetic, environmental and infectious factors have been proposed.Citation3
Independent studies have reported the abnormal presence of intramucosal Escherichia coli or mucosa-associated E. coli with invasive properties in patients with CD or colorectal cancer.Citation3-Citation5 In particular, it was demonstrated that adherent-invasive E. coli (AIEC) isolated from the ileal mucosa of CD-patients induce expression of pro inflammatory cytokines (IL-1β, IL-6 and IL-17), decrease expression of anti-inflammatory cytokine (IL-10) and aggravate colitis in injured mouse colon. These cellular responses are mediated by type 1 pili expressed by AIEC bacteria which bound CEACAM6 receptor,Citation6,Citation7 protein overexpressed in 35% of CD patients having ileal involvement.Citation8
The Hypoxia-Inducible Factor (HIF-1), which orchestrates cellular metabolism and cellular adaptation to oxygen privation (hypoxia), is induced by growth factors or LPS challengeCitation9 and is known to regulate the angiogenic response.Citation10 Angiogenesis, which corresponds to the growth of new blood vessels in adults, occurs in physiological or in pathological situations. Vascular endothelial growth factor (VEGF) and interleukin 8 (IL-8) act as the major mediators of angiogenesis.Citation11,Citation12 Interestingly, VEGF levels and angiogenesis are increased in IBD and correlate with disease activity.Citation13
Since (1) increase in HIF-1α protein level induces VEGF production, (2) VEGF links angiogenesis to inflammation in IBD onsetCitation14 and (3) AIEC induce gut inflammation, we wonder whether AIEC were able to stabilize HIF-1α and trigger production of angiogenic factors. Our studies demonstrated that the ileum and colon of patients with CD contain high levels of the HIF1α protein, particularly in inflamed ileum, a tissue known to abnormally express CEACAM6 receptor. Further, CD-associated AIEC bacteria induce the production of HIF-1α and VEGF in both human intestinal epithelial cells (IEC) and transgenic mice model expressing human CEACAM6 (IEC). Interestingly, we demonstrated in human T84-IEC challenged with AIEC LF82 bacteria that HIF-1, via activation of the classical NFκB-associated pathway, triggers pro-angiogenic factor secretion.
Results
Inflamed ileum of CD-patients abnormally expressed HIF-1α
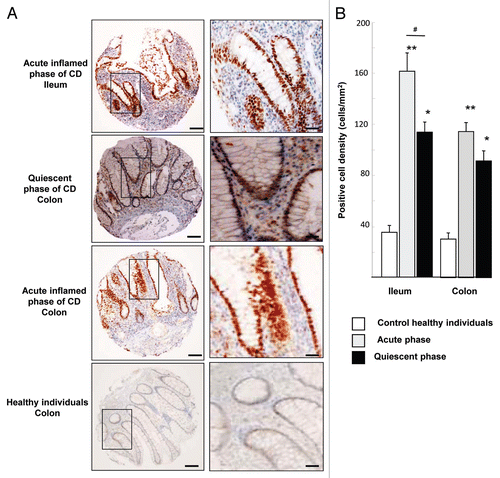
An increase in the level of the HIF-1α protein in IBD patients was previously reported,Citation15-Citation17 but the number of patients included in this previous study was low and the tissue location (ileum or colon) was not documented. Here we examined the presence of HIF-1α in ileal and colon biopsies of a large cohort of 180 CD-patients in acute and quiescent phases of the disease. As shown in , acute inflamed ileal and colon biopsies of CD-patients showed loss of glandular structures accompanied by strong inflammatory infiltrate whereas biopsies from quiescent patients retained glandular structures surrounded by inflammatory cells. Immunohistochemistry against HIF-1α using tissue micro arrays (TMA) revealed numerous stained nuclei, from the bottom of the crypts to the apical surface of villi, in the epithelia of the ileum and colon of CD-patients. In the acute phase of the disease, HIF-1α staining was observed in 86% of ileal biopsies and in 82% of colon biopsies. In the quiescent phase, fewer CD-patients expressed HIF-1α with 37% in ileal biopsies and 34% in colon biopsies whereas in healthy individuals staining was detected in 21% and 18% of biopsies from ileon and colon respectively. Quantification of immunostaining approach showed that the positive cell density for ileal biopsies of patients presenting an acute phase of CD was significantly higher (4-fold increase) than that observed in a quiescent phase of CD and in control healthy subjects (). In colon biopsies, we observed a 3-fold increase in the number of positive epithelial cells from patients presenting either an acute (inflamed) or a quiescent (less-inflamed) phase of CD. In contrast, a few positive epithelial cells were observed in colon biopsies of control healthy subjects. In conclusion, intestinal biopsies of CD-patients are positive for HIF-1α staining. Our studies highlight a difference in HIF-1α expression between ileum and colon. Indeed, HIF-1α expression is more intense in inflamed ileum (160 positive HIF-1α cells per mm2) than in inflamed colon (110 cells per mm2) or healthy individuals (cell density ≤ 40). Interestingly, we previously published that inflamed ileum overexpresses CEACAM6 protein and is colonized by AIEC.Citation8,Citation18
Figure 1. HIF-1α staining in intestinal biopsies of CD patients and healthy subjects. (A) Immunohistochemical (IHC) staining of tissues micro-array (TMA) from ileal and colonic biopsies of patients in acute inflamed and quiescent phase of CD. Each picture is representative of immunostaining for HIF-1α at low magnification (scale bar = 100 µm) and at high original magnification scale bar = 30 µm). IHC shows numerous HIF-1α-stained nuclei, from the bottom of the crypts to the apical surface of villi, in the epithelia of the ileum and colon of CD-patients. (B) Quantification using the Spot Browser software of HIF-1α immunostaining on TMA from colonic and ileal biopsies of 85 patients in the acute phase of CD, 95 patients in the quiescent phase of CD, and 48 control subjects *p < 0.01, **p < 0.001 CD patients vs. healthy subjects; # p < 0.01, ileum of acute inflamed phase vs. quiescent phase of CD patients. TMA quantification shows that HIF-1α positive cell density was increased in ileum and colon of CD-patients vs. control healthy individuals.
CD-associated AIEC LF82 bacteria increase HIF-1α protein levels in a CEABAC 10 mice model
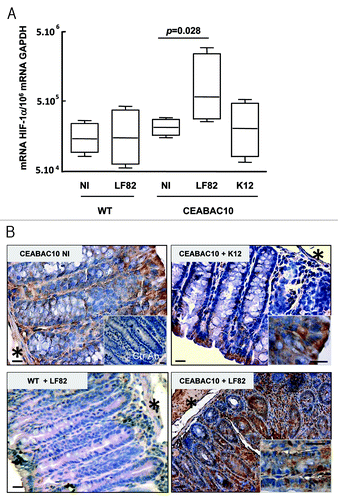
Intestinal biopsies of CD-patients expressed HIF-1α and we previously published that CD-associated AIEC LF82 bacteria are able to persist in the gut of CEABAC 10 transgenic mice that express human CEACAM6 protein and to induce severe colitis.Citation6 Since HIF-1α played a role in murine experimental colitis,Citation19 we investigated whether AIEC LF82 bacteria can induce HIF-1α stability in the intestinal mucosa of CEABAC 10 mice. Mice were orally challenged with 109 AIEC LF82 bacteria or non-pathogenic E. coli K12 bacteria. At 7 d post-infection, AIEC-LF82 bacteria were detected using anti-O83 antibody (directed against LPS O-core to detect AIEC strain LF82) in colon of CEABAC 10 mice, but not in the colon of AIEC infected-WT mice (Fig. S1). An increased level of HIF-1α mRNAs was observed in colonic tissue biopsies of CEABAC 10 mice infected with AIEC LF82 bacteria compared with that of non-infected CEABAC 10 mice or CEABAC 10 mice infected with non-pathogenic E. coli K12 (). At the protein level, we observed faint HIF-1α staining in the cytoplasm of mature enterocytes located on the luminal side of the crypts whereas cells located at the basal side were negative (). As described previously for CEABAC 10 mice, non-pathogenic E. coli K12 did not affect tissue organization and we reported here that HIF-1α staining is similar to that observed in non-infected CEABAC 10 mice. In contrast, AIEC LF82 bacteria infection in CEABAC 10 transgenic mice led to disappearance of glandular structures that correlate with an important inflammatory infiltrate and an increase in the number of HIF-1α positive cells. Staining within epithelial cells was nuclear and cytoplasmic; this cytoplasmic staining is specific, since it was not present when tissue sections were stained with a control antibody. Most of the immune cells were also positive for HIF-1α staining. In conclusion, AIEC LF82 infection induces HIF-1α mRNA and protein expression in transgenic CEABAC 10 mice mimicking ileal CD susceptibility.
Figure 2. AIEC LF82 bacteria increase HIF-1α expression in CEABAC 10 transgenic mice. (A) Quantification of the HIF-1α mRNA level was measured by RT-PCR in colonic biopsies of WT and CEABAC 10 mice non-infected (NI) or infected with AIEC LF82 or non-pathogenic E. coli K12 bacteria. HIF-1α mRNA level is only increased in colonic biopsies of CEABAC 10 mice infected with LF82 bacteria. Results shown are representative of two separate experiments (n = 7 mice per group). (B) Representative brown immunohistochemical staining for HIF-1α in sections of the colonic mucosa from WT or CEABAC 10 mice non-infected or infected with the non-pathogenic E. coli K12 or AIEC LF82 bacteria (scale bar = 20 µm). Inset in panel CEABAC 10 NI shows the absence of staining when biopsy is stained with control isotypic antibody. Inset in CEABAC 10 K12 panel shows HIF-1α cytoplasmic staining in mature enterocytes. Inset in CEABAC 10 LF82 panel highlights the HIF-1α nuclear staining within epithelial cells located along the crypts. *, muscularis mucosa
The VEGF pathway is induced by AIEC LF82 infection in CEABAC 10 mice
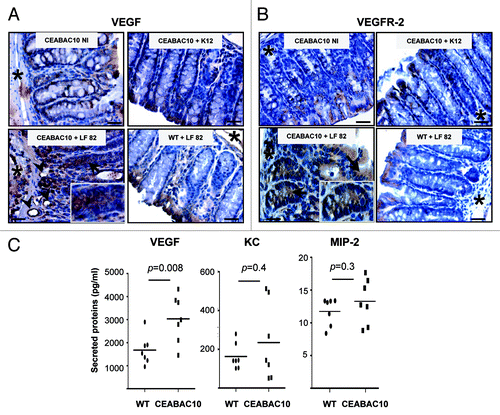
Since VEGF is a major factor in IBD pathogenesis,Citation14 we determined whether AIEC bacteria lead to production of angiogenic factor. To investigate this hypothesis, mucosal expression of VEGF was analyzed in CEABAC 10 mice challenged with AIEC LF82 bacteria. Immunohistochemistry revealed that VEGF was expressed at a low level and only located in the cytoplasm of mature enterocytes at the luminal side of the crypts in non-infected CEABAC 10 transgenic mice (). No increase in VEGF staining was observed in transgenic mice infected with the non-pathogenic E. coli K12 or in WT mice infected with AIEC LF82. In contrast, strong cytoplasmic staining in mature enterocytes but also in less mature epithelial cells located from the base to the luminal side of crypts were observed (). Moreover, in AIEC LF82 infected-CEABAC 10 mice the number of vessels was increased and endothelial cells that form vessels were stained with VEGF antibody. Finally, strong cytoplasmic staining in cells located within the lamina propria in areas of erosion was observed (Fig. S2). Next, we investigated the expression of VEGFR-2, the inducible form of VEGF receptor. VEGFR-2 was expressed at a very low level in mature enterocytes of non-infected CEABAC 10 mice (). As described for VEGF, its expression was not modulated in WT mice following either non-pathogenic K12 bacteria or by AIEC LF82 infection. However, AIEC LF82 infection in CEABAC 10 mice led to an upregulation of VEGFR-2 expression in epithelial cells located along the entire crypt and also in a few cells within the lamina propria (Fig. S2). Further, the production of VEGF was quantified. An 1.81-fold increase of VEGF was measured in colonic biopsies of AIEC LF82 infected-CEABAC 10 mice compared with those of WT mice (). Taken together, these data indicate that AIEC infection triggers production of VEGF.
Figure 3. AIEC LF82 bacteria induce production of VEGF in CEABAC 10 transgenic mice. Representative brown immunohistochemical staining for VEGF (A) and VEGFR-2 (B) in sections of the colonic mucosa from non-infected (NI) CEABAC 10 mice, or AIEC LF82- or non-pathogenic E. coli K12-infected CEABAC 10 and WT mice; (scale bar = 10 µm). Insets show cytoplasmic staining of VEGF (A) and VEGFR-2 (B) in epithelial cells located within crypts of CEABAC 10 mice infected with LF82 bacteria. Arrows point to cytoplasmic staining within epithelial cells and arrowheads indicate endothelial cell staining. *, muscularis mucosa. (C) Secretion of VEGF, KC and MIP-2 was measured in colon supernatants from AIEC LF82-infected WT (n = 7) or CEABAC 10 (n = 7) mice. An increase in secreted VEGF concentration in CEABAC 10 mice infected with LF82 bacteria was measured by ELISA. Results shown here are representative of two separate experiments.
Production of angiogenic factors in response to AIEC LF82 bacteria depends on HIF-1α in intestinal epithelial cells
We investigated the involvement of HIF-1α in VEGF production. For that purpose, we confirmed that AIEC LF82 infection led to an increase in the relative amount of HIF-1α protein in IEC. The amount of HIF-1α increased in AIEC LF82-infected T84 cells from a multiplicity of infection (MOI) of 10 (, left panel). To ensure that bacteria-induced HIF-1α expression and stability was not a general consequence of bacterial growth in close proximity to cells, T84 cells were infected with the 291071 clone of commensal E. coli. Whereas commensal bacteria induced only a slight increase in HIF-1α (, right panel), AIEC LF82 infection systematically gave higher levels. In addition, no change in oxygen concentration in cells infected with either AIEC LF82 or a non-adherent mutant was detected (Fig. S3A) and protein synthesis inhibitor led to a blockage of the HIF-1α increase in T84 cells (Fig. S3B). Taken together these results suggest that AIEC-induced HIF-1α protein levels in independent of hypoxia.
Figure 4. AIEC LF82-induced VEGF and IL-8 production require HIF-1α expression and NFκB signaling in human T84-IEC. (A) T84 cells were infected with increasing amounts of AIEC LF82 bacteria (from MOI of 1 to 100) or control commensal bacteria (clone 291071) for 2h. The HIF-1α protein was assayed by immunoblotting using an anti-HIF-1α antibody and the total actin level was monitored as a control for equal protein loading. We observed an increase in HIF-1α protein levels in cells infected with LF82 bacteria. (B) VEGF and IL-8 secretion by T84 and T84-Shhif-1α cells infected with AIEC LF82 or control commensal (clone 291071) bacteria was measured by ELISA as indicated in Materials and Methods. Data are expressed as the mean concentration of secreted proteins per 8 × 106 cells for 4h ± s.e.m. (n = 3). VEGF and IL-8 mRNA level from T84 and T84-Shhif-1α cells infected with AIEC LF82 (MOI = 10) were quantified by qPCR. Data are expressed as the mean of the relative amount of mRNA expressed per 8 × 106 cells ± s.e.m. (n = 4).). **p < 0.01 relative to values of non-infected T84 cells. ##p < 0.01 relative to values of LF82-infected vs. commensal (clone 291071)-infected T84 cells. ++p < 0.01 relative to values of LF82-infected T84 cells vs. LF82-T84 depleted for hif-1α expression (T84-Shhif-1α) cells. LF82 bacteria induced production of VEGF and IL-8 is dependent on HIF-1α.
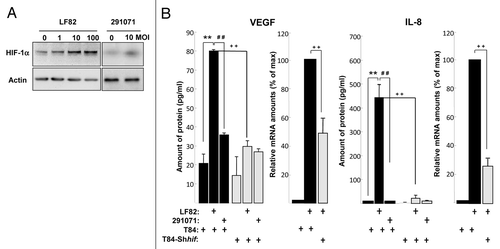
Since one of the consequence of an increase in the HIF-1α protein is the induction of angiogenic responses and because AIEC-infected CEABAC 10 transgenic mice presented an increase of HIF-1α protein expression and VEGF expression, we investigated whether AIEC LF82 bacteria induced VEGF production in human T84 IEC. The conditioned medium of T84 cells infected for a 2h period with LF82 bacteria contained up to 80 pg/ml of VEGF (). In contrast, the amount of VEGF produced by cells infected with the commensal 291071 E. coli clone is not different than that produced by non-infected T84 cells. In addition to VEGF, interleukin-8 (IL-8), cytokine associated with the promotion of neutrophil chemotaxis and degranulation, is also able to enhance angiogenesis.Citation12 We observed that both IL-8 mRNA and protein were upregulated in AIEC-LF82 infected T84 cells (). Interestingly, AIEC LF82-induced VEGF and IL-8 mRNA and proteins expression were significantly reduced in hif-1α-silenced T84 cells (T84-Shhif-1α) compared with wild-type T84 (, gray bars), whereas transfection with scramble siRNA or shRNA have no effect on VEGF and IL-8 production (data not shown). These results demonstrated the key role played by HIF-1 on the production of angiogenic factors in response to AIEC infection.
AIEC-induced production of angiogenic factors involves NFκB pathway
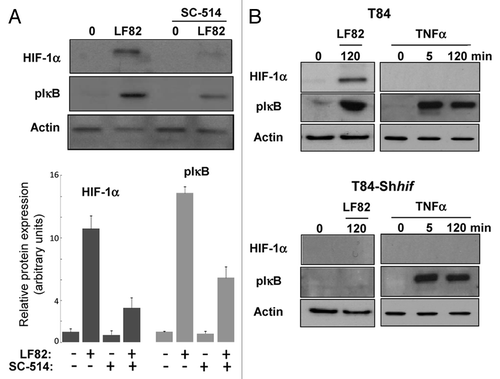
Recent studies highlighted an interdependent role for hypoxia-inducible factors and nuclear factor-κB (NFκB) in hypoxic inflammation.Citation20 Therefore, we analyzed the level of HIF-1α protein expression and the phosphorylation state of IκB (a key signature of NFκB signaling pathway activation) in response to bacterial infection. First, the impact of NFκB inhibition on HIF-1α expression in T84 cells was evaluated. For that purpose, cells were pre-treated with SC-514, a chemical inhibitor which blunts IKK-2 kinase activity. In that condition, inhibition of IKK-2 led to a 65% decreased in HIF-1α protein levels (). Further, the effect of HIF-1 on NFκB signaling was characterized. In cells depleted for HIF-1α expression, no phosphorylation of IκB was detected (). As control, the activation status of the NFκB signaling pathway in wild-type and HIF-1α silenced T84-IEC was checked by stimulating cells with Tumor Necrosis Factor α (TNFα). In both cell types (T84 and T84-Shhif-1α), TNFα induced phospho-IκB protein accumulation. In contrast, no HIF-1α expression was detected in T84 cells treated with TNFα. Together, these results highlight a complex interconnection between HIF-1 and NFκB signaling.
Figure 5. HIF-1α and NFκB signalings are interconnected. (A) protein gel blot analysis (upper panel) showed the effect of IKK-2 inhibitor (SC-514, 100 μM) added one hour prior the infection with AIEC LF82 (MOI of 10) on HIF-1α protein level. Quantification of immunoblot, using ImageJ software, (lower panel) demonstrated that blocking IKK-2 kinase activity results in a partial inhibition of HIF-1α protein expression. Data are expressed as the mean of amount of protein expression ± s.e.m. (n = 3). (B) Immunoblot showing HIF-1α and phosphorylation of IκB in T84 and T84-Shhif-1α cells infected for a 2 h period with AIEC LF82 bacteria (MOI = 10) or treated with TNFα for 45 and 120 min. The total actin level was monitored as a control for equal protein loading. This result highlights the ability of LF82 bacteria to induce phosphorylation of IκB in a HIF-1α dependent manner.
AIEC type 1 pili-mediated adhesion enhances TLR5 signaling
AIEC LF82 pathogenicity is due to expression of type 1 pili and flagellin that respectively bind to CEACAM6 and TLR5 receptors.Citation6,Citation7 However their respective roles in induction of signaling pathways are not fully resolved. To investigate the role of type 1 pili in AIEC-induced production of VEGF and IL-8, cells were treated with D-mannose, a sugar described as strong inhibitor of type 1-mediated adhesionCitation21 (Table S1). In this condition, the expression of both VEGF and IL-8 mRNA was impaired after AIEC infection (, gray bars), demonstrating that adhesion mediated by type 1 pili is needed for the angiogenic factors production. When cells were infected with AIEC LF82-ΔfimH isogenic mutant (unable to produce type 1 pili but still expressing flagella), the relative amount of VEGF and IL-8 angiogenic factors is decreased, reaching 18% and 40% respectively of those produced in response to WT bacteria (). Trans-complementation of AIEC LF82-ΔfimH isogenic mutant with fimH cloned gene resulted in restoration of VEGF and IL-8 production. Following infection with AIEC LF82-ΔfliC mutant that do not expressed flagella and type 1 pili,Citation22 VEGF production was decreased to the basal level and transformation of AIEC LF82-ΔfliC mutant with cloned fliC operon pPBI04 restored cellular responses. Finally, cytochalasin D, that inhibits 80% of cellular bacterial invasiveness (), has no significant effect on VEGF and IL-8 production (). Therefore, we conclude that induction of cell signaling depends on bacterial adhesion to extracellular proteins.
Figure 6. Type 1 pili and flagella expressed by AIEC LF82 bacteria enhance production of VEGF and IL-8 independently of cellular invasion. (A) Quantification of VEGF and IL-8 mRNA level by qPCR from LF82 or non-pathogen E. coli (clone 291071) infected T84 cells in the absence or presence of D-mannose (1%). D-mannose treatment partially inhibits LF82-induced VEGF and IL-8 mRNA expression. Data are expressed as the mean of the relative amount of mRNA expressed per 8 × 106 cells ± s.e.m. (n = 3). **p < 0.01 relative to values of LF82-infected vs. 291071-infected T84 cells. ##, p < 0.01 relative to values of LF82-infected T84 cells in presence vs. absence of D-mannose. (B) Quantification of VEGF and IL-8 mRNA level by qPCR of bacteria (MOI = 10)-infected T84 cells showing the effect of LF82, LF82-ΔfimH, LF82-ΔfliC and LF82-ΔfliC/pPBI04 (isogenic ΔfliC mutant complemented for flagella and type 1 pili expression) bacteria. Type 1 pili (ΔfimH) and flagella (ΔfliC) are necessary to mediate LF82-induced increase on VEGF and IL-8 mRNA level. Data are expressed as the mean of the relative amount of mRNA in 8 × 106 cells ± s.e.m. (n = 3). *p < 0.05, **p < 0.01 relative to values of LF82-infected vs. commensal-infected T84 cells. (C) Quantification of VEGF and IL-8 mRNA levels in LF82-infected T84 cells without or with cytochalasin D treatment (2µg/ml). This result indicates that cellular invasion is not necessary to mediate LF-82-induced production of angiogenic factors. Data are expressed as the mean of amount of relative mRNA expressed in 8 × 106 cells ± s.e.m. (n = 3). ns: non statistically significant.
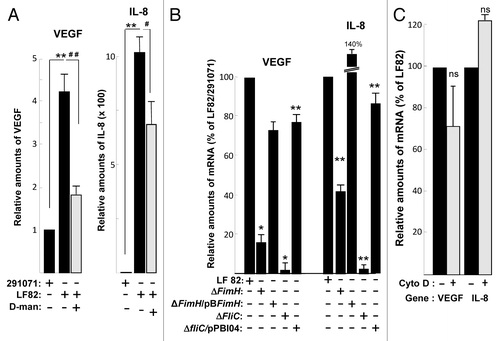
Table 1. Adherent and invasive characteristics of strains used in this study
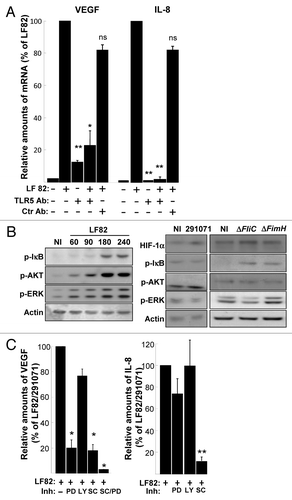
Flagella, acting via TLR5, lead to pro-inflammatory gene program activation in epithelial cells.Citation23 However involvement of TLR5 in AIEC-induced cellular responses is not fully resolved. To study the involvement of TLR5 in VEGF and IL-8 production, we blocked its activation by pre-treating T84-IEC with an anti-TLR5 antibody. This led to a decrease in the relative amount of VEGF and IL-8 mRNAs level (). TLR5-induced signaling pathways were described to activate transcription factors, such as NFκB and interferon regulatory factors, which in turn lead to induction of immune and inflammatory genes.Citation24 Because VEGF production is mainly known to be activated by the Ras > Raf > MEK > Erk as well as the PI3-kinase/Akt pathways,Citation25 the contribution of each signaling pathways was investigated in IEC. Immunoblot analysis were performed on total cell extracts and phosphorylations of the ERKs (ERK-1, ERK-2), AKT and IkB proteins were investigated, since phosphorylation of these proteins is a read out of MAP-kinases, PI-3K and NFκB signaling pathway activation respectively. Bands corresponding to phospho-ERKs, -AKT and -IkB appeared in a time dependent manner, with a maximum observed after 3h of AIEC infection (). In addition, these signaling pathways were barely activated in response to commensal E. coli or AIEC LF82-ΔfliC isogenic mutant. Finally, IEC were pre-treated with selective inhibitors of MEK (PD184352), PI-3K (Ly294002) and IKK-2 (SC-514), prior AIEC LF82 or commensal bacterial infection and VEGF and IL-8 production was measured by quantitative RT-PCR. Whereas MEK and NFκB inhibitors significantly inhibited the increase in VEGF mRNA level, only the NFκB signaling pathway is involved in increase in IL-8 mRNA level (). Together our results demonstrate that AIEC LF82 bacteria via TLR5 induce HIF-1α, VEGF and IL-8 production. In human IEC, the increase in pro-angiogenic factors is dependent on the expression of bacteria-induced HIF-1α, which cooperates with the NFκB-dependent signaling pathways.
Figure 7. AIEC LF82-induced cell signaling pathways require binding of flagella to TLR5. (A) Quantification of VEGF and IL-8 mRNA by qPCR in LF82 (MOI = 10)-infected T84 cells pre-treated with anti-TLR5 and non-relevant antibodies indicates that LF82 bacterial component should interact with TLR5 to induce VEGF and IL-8 mRNA increase levels. Antibodies are added 1h prior to infection at 10 μg/ml. Data are expressed as the mean of the relative amount of mRNA expressed by 8 × 106 cells ± s.e.m. (n = 3). *p < 0.05, **p < 0.01 relative to values of LF82-infected vs. commensal-infected T84 cells. (B) Immunoblot showing HIF-1α and phosphorylation of ERK, AKT and IκB in T84 cells before and after exposure to LF82 bacteria (MOI = 10) for indicated times (left panel) or indicated bacteria at a MOI of 10 for a 2 h period (right panel). The total actin level is monitored as a control for equal protein loading. Here we show that LF82 bacteria induce classical ERK, AKT and IκB signaling pathways. (C) Effect of cell signaling inhibitors on VEGF and IL-8 gene expression of LF82-infected T84 cells. MEK inhibitor (PD184352, 5 μM), PI-3 Kinase inhibitor (LY203580, 25 μM) and IKK inhibitor (SC-514, 100 μM) were added to the cells 1h prior to infection. ERK and IκB dependent signaling pathways are required to mediate LF82-induced production of angiogenic factors. Data are expressed as the mean of the relative amount of mRNA expressed by 8 × 106 cells ± s.e.m. (n = 4). *p < 0.05, **p < 0.01 relative to values of LF82-infected vs. commensal-infected T84 cells.
Discussion
It is now well admitted that intestinal inflammation arises from abnormal host-microbiota interactions. Indeed, dysbiosis toward selected microorganisms and decreased complexity of commensal bacteria have been observed in patients with Crohn disease; however search for a single pathogen that induces CD was not successful. Therefore it is likely that CD-associated bacteria act in association with hosts’ immune defects and/or mucosal barrier dysfunctions. Since (1) the interaction between AIEC and epithelial cells induces an upregulation of IL-8 expression, a marker of inflammation,Citation26 and (2) an increase in HIF-1α protein levels is observed under inflammatory conditions, we wondered whether AIEC might induce HIF-1α expression and what could be its consequence on CD pathogenesis. We demonstrated in this study that CD-associated AIEC bacteria induce the production of HIF-1α, VEGF and IL-8 in human IEC. Further, we showed that bacteria induce the production of HIF-1α and VEGF in a transgenic mice model. Then, we deciphered on a cell line model the mechanisms leading to HIF-1α production and the role of HIF-1 in AIEC LF82-induced production of VEGF and IL-8. Finally we showed that HIF-1 acts in combination with the classical NFκB-associated pathway to trigger the secretion of pro-angiogenic factors ().
Figure 8. AIEC LF82 bacteria induce inflammatory disorders via HIF-dependent responses. Binding of AIEC LF82’s type 1 pili and flagella to CEACAM-6 and TLR-5 receptors triggers induction of signaling pathways (IKK, MAPK). Consequently, NFκB and HIF-1α translocate to the nucleus where they cooperatively control VEGF and IL-8 transcription. Since AIEC are unable to induce phosphorylation of IκB in cells silenced for HIF-1α expression, we proposed that the expression of CEACAM-6 and TLR-5 receptors is under the control of the HIF-1 transcription factor.
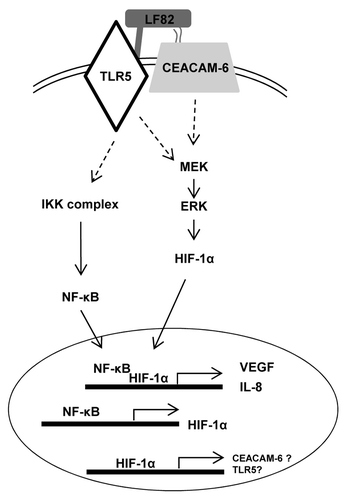
CEACAM-6 and TLR5 have been respectively involved in AIEC type 1 pili and flagella binding. Using mutants unable to produce either type 1 pili (AIEC LF82-ΔfimH) or both pili and flagella (AIEC LF82-ΔfliC) we confirmed the existence of a cooperative interaction between pili and flagella in mediating AIEC-induced cellular responses. Indeed, AIEC LF-ΔfimH is more potent than AIEC LF- ΔfliC to induce both HIF-1α expression and pro-angiogenic factor production. This could be explained by the fact that in the absence of bacterial adhesion, due to the lack of type 1 pili, less amount of flagella are in contact with transmembrane cellular receptors, such as Toll Like Receptor (TLR)-5. In agreement with this hypothesis, trans-complementation with fimH cloned gene led to an overexpression of IL-8, likely due to an overexpression of type 1 pili and consequently increased in TLR5 signaling.
Further, we and otherCitation26,Citation27 showed that AIEC LF82 bacteria induce expression of IL-8 in human cells. Expression of IL-8 has been described to be under the control of IκB/NFκB signaling, downstream of TLR5.Citation28 In agreement with this, we showed that the IKK-2 inhibitor efficiently blocks IL-8 mRNA induction. However, our results demonstrate that IL-8 expression, in response to AIEC bacteria, is also dependent on the HIF-1 transcription factor since cells silenced for HIF-1α produce less IL-8 when challenged with AIEC LF82. Such result is consistent with the existence of interdependence on HIF and NFκB.Citation20 It was shown that the NFκB pathway transcriptionally regulates HIF-1α mRNA which subsequently leads to an increase in HIF-1 activity and transcriptional regulation of HIF-dependent genes.Citation29,Citation30 Accordingly, when we treated IEC with the IKK-2 inhibitor we observed a decrease in HIF-1α protein levels suggesting that NFκB acts up-stream of HIF-1α. Further, we observed that AIEC LF82 were unable to induce phosphorylation of IκB in cells invalidated for HIF-1α expression. This result was surprising as we demonstrated in the same cells that the IKK complex was functional, since TNFα efficiently phosphorylated IκB. Reconsidering all these evidence we made the hypothesis that HIF-1 could transcriptionally regulate CEACAM-6 and TLR5. In such a scenario, in the absence of HIF-1α, the expression of CEACAM-6 and TLR5 would be decreased; in return, bacteria wouldn’t be able to induce the production of IL-8 and VEGF. Since the scanning of 5′-UTR of both CEACAM-6 and TLR5 genes using the “Tfsitescan” program highlights the presence of HIF-1 binding sites and since we noticed that cells depleted for HIF-1α produced less angiogenic factors when challenged with AIEC LF82 bacteria, we believe that this hypothesis may be correct. In addition, HIF-1α directly controls the transcription of IL-8, since a hypoxic responsive element has been identified on the IL-8 promoter.Citation31
In epithelial cells, modulation of HIF by either chemical agents or gene silencing suggested a protective role for HIF proteins in murine experimental colitis.Citation19 However, results obtained from mice in which hif-1α or vhl (the von Hippel-Lindau protein (VHL) were silenced in the intestinal epithelium showed contradictory results. In a study by Karhausen et al., hif-1α deletion in mature enterocytes (triggered by the Fabp promoter) resulted in worse trinitrobenzenesulfate acid (TNBS)-induced colitis whereas vhl deletion led to protection against TNBS-induced colitis. In contrast, Shah et al. reported that hif-1α deletion in progenitors of intestinal epithelial cells (triggered by the vilin promoter) had no effect on dextran sulfate sodium (DSS)-induced colitis, whereas deletion of vhl increased inflammatory response.Citation32 Whether these contradictory results could be explained by the chemical agents used in these two studies (TNBS vs. DSS), by the deleted cellular target (mature enterocytes vs. progenitors) or by the targeting of HIF-α subunits in vhl knockout mice (upregulation of HIF-1α and -2α in the report of Karhausen et al. and only HIF-2α in report by Shah et al.) requires additional experiments. Here, we sought to study the expression of HIF-1α in a more physiological colitis murine model; thus far we infected transgenic CEABAC 10 mice with the virulent AIEC LF82 strain and demonstrated, for the first time, that bacteria associated with CD increased the level of HIF-1α in epithelial cells. In both human and mice studies we also noted that some inflammatory cells are stained for HIF-1α. This finding is in agreement with the observation that the HIF-1α expressed by macrophages is an important regulator of innate immunity.Citation33 Our results suggest that HIF-1α expression in response to AIEC LF82 bacteria was independent of hypoxia. Because AIEC LF82 bacteria induced an increase in HIF-1α protein synthesis and because AIEC LF82 bacteria-increased gene expression was mediated by flagella/TLR5 signaling, we believe that CD-associated bacteria act through classical microbial sensing via pattern recognition receptors.
We demonstrate here that AIEC-infected mice and T84 cells secrete VEGF. This cellular response is, in part, mediated by HIF-1 signaling, since we noted a decrease in cells silenced for hif-1α. Given the fact that a combination of ERK and IKK-2 inhibitors efficiently blocks VEGF production, it is likely that NFκB signaling also participates in regulation. Interestingly, we demonstrate that the activation of the flagella/TLR5 signaling pathway accounted for enhanced VEGF production. In humans, expression of VEGF and its inducible receptor, VEGFR-2/KDR, are increased in IBD patients. Modulation of VEGF, or of VEGFR-2, expression in a DSS-induced colitis murine model demonstrates that VEGF links angiogenesis to inflammation.Citation14 Our results suggest that AIEC could participate in the pathogenesis of CD by increasing locally VEGF/VEGFR-2 signaling and IL-8 production.
The angiogenic switch results in the transition from dormant avascularized hyperplasia to outgrowing vascularized tumors and, eventually, to malignant tumor progression. In the present study, we demonstrated that AIEC LF82 bacteria isolated from a CD patient induce production of pro-angiogenic factors. Since angiogenesis is linked to carcinogenesis, it is tempting to propose that bacteria might favor the onset of colon cancer in CD patients. Indeed, increased numbers of mucosa-associated E. coli forming a biofilm on the surface of the gut mucosa are observed in patients with colorectal cancerCitation34 and a subset of mucosa-associated E. coli isolated from patients with colorectal cancer share pathogenicity islands with urinary pathogenic E. coli.Citation35 The observations that: (1) similar mucosa-associated E. coli are detected both in cancer and CD and (2) infection of CEABAC 10 mice by AIEC induced the production of angiogenic factors, raises the possibility that colorectal cancer might be the result of bacteria-induced inflammation.
Materials and Methods
Ethics statement
Human research: all study participants and/or their legal guardians provided written, informed consent and all study participants signed agreement for this study. The protocol was approved by the local ethics committee of the University of Nice (n° 77643).
Animal research: This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the University of Clermont Ferrand France. Animal protocol was approved by the committee for Research and Ethical Issues of Auvergne department (CEMEA, Auvergne) following international directive 86/609/CEE (n° CE16–09).
Patients and biopsy specimens
Patients included in this study were hospitalized in the Department of Gastroenterology (Archet II Hospital, Nice, France). Intestinal biopsies were obtained from macroscopically inflamed mucosa of the terminal ileum and of the colon in 85 CD patients (group 1) and from macroscopically non-inflamed mucosa of the terminal ileum and of the colon in 95 CD patients (group 2). There were 97 men and 83 women, with a mean age of 42 y (range 19–62) and mean disease duration of 15 y (range 2–25). Patients were all French Caucasian residents. In addition, biopsies were taken from the ileum and colon of 48 control patients consisting of individuals who had no significant pathological findings following endoscopic examination for changes in stool habits, abdominal pain, or upper gastrointestinal bleeding or cancer surveillance.
Bacterial strains and CEABAC 10 mice
E. coli LF82, isolated from a chronic ileal lesion of a CD patient, a non-flagellated isogenic mutant of AIEC LF82 deleted of the AIEC-LF82-ΔfliC gene and the corresponding mutant transcomplemented with pPBI04 harboring the entire fliC operon,Citation22 a non-piliated isogenic mutant, deleted for fimH gene and the corresponding mutant transcomplemented with fimH gene,Citation36 and a control commensal bacteria, strain 291071 which was isolated from stool of healthy individuals, were used in this study. Bacteria were cultured as previously described.Citation37
WT (FVB/N) mice were purchased from Charles River Laboratories and CEABAC 10 transgenic mice were obtained from Chan and Stanners.Citation38 All mice were housed and infected as previously described.Citation6
Cell culture and invasion assays
The human colon carcinoma cell line T84 was supplied by the ATCC (number CCL-248). Cells were cultured as a monolayer for 2–3 d to confluency in DMEM: F12 medium supplemented with 5% heat-inactivated fetal calf serum (FCS) without antibiotics. Human hif-1α-silenced T84 (Shhif-1α) T84 cells were transduced with lentiviral particles containing shRNA-HIF-1α from Sigma-Aldrich (TRCN0000003810) as described previously.Citation39 Prior to infection, cells were starved for 24 h in the medium without FCS and then infected with bacteria as indicated in figure legends.
Reagents and antibodies
Human TNFα was from PeproTech. The following inhibitors Ly203580 (Calbiochem), PD 184352 (M. Kohno, University of Nagasaki), SC-514 (Cayman Chemicals) were used. Anti-HIF-1α antibody (BD Transduction), anti-phospho-IkB-α (Ser 32/36) and anti-phospho-Akt (Ser 473) (Cell Signaling Technology), anti-actin (clone AC40) and anti-phospho-ERK (Sigma) were used for immunoblot analysis. Anti-mouse HIF-1α (NB100–105, Novus), VEGF (AF-493, R&D System) and VEGFR-2 (GTX73105, GeneTex) were used for immuhistochemistry. Anti-TLR5 (PAb-hTLR5-Cayla-InvivoGen-5 μg/ml) was incubated with cells for 30 min prior to bacterial infection.
Tissue microarray (TMA) construction and immunohistochemistry
Representative intestinal biopsies obtained for each individual in building TMAs were selected from hematoxylin and eosin stained sections. These sections were examined by two pathologists (VH, PH) who were blinded to the other experimental results. EDTA-pretreated sections were immunostained for HIF-1α (mouse, clone 54; diluted 1 to 10) by using an automated single-staining procedure (Benchmark XT, Ventana Medical Systems, Roche Group, Inc., Tucson, AR). HIF-1α staining was quantified with an image analysis workstation (Spot Browser version 7; Alphelys, Paris, France), as described previously.Citation40 Briefly, after antibody staining, images were acquired using automated quantitative analysis. The HIF-1α signal was measured on a grayscale of 0 (black) to 255 (white) and gray values ranged between 0 and 152, and a value superior or equal to 35 was arbitrarily defined as HIF-1α overexpression. Results were expressed as positive cell density per mm2.
Protein gel blotting
Cells seeded in a 12-well plate were washed twice with ice-cold PBS and immediately lysed in SDS sample buffer. Protein extracts were resolved by SDS-PAGE and transferred onto a polyvinylidene difluoride membrane (Immobilon-P; Millipore). Prior to incubation with the antibodies, membranes were saturated for 30 min in 5% non-fat milk diluted in 50 mM TRIS-HCl 150 mM NaCl (TN buffer). Membranes were incubated overnight in the same buffer containing the indicated antibody, washed in TN supplemented with 0.1% Triton X-100 (TNT) three times and finally with TN buffer. Membranes were then incubated with the secondary anti-rabbit or anti-mouse horseradish peroxidase conjugated antibody. Bound antibodies were revealed using an ECL system (Pierce).
VEGF, KC and IL-8 measurements
VEGF, KC and IL-8 secretion from mice colon was measured using a specie specific ELISA kits from PEPROTECH (Co, UK) according to the manufacturer’s protocol.
RT-qPCR procedure
Total RNA were prepared as previously described.Citation39 Each sample was run in duplicate. All results were normalized to the unaffected housekeeping GAPDH gene. For human IL-8 (Hs00174103-m1) and VEGF (Hs00173626-m1), cDNA were quantified using a 7500 Real-Time PCR system (Applied Biosystems). The relative expression of the indicated transcripts was quantified using the Taqman PCR Master Mix (Applied Biosystems) and specific primers using the 2[–ΔΔC(T)] method. According to this method, the C (T) values for the expression of each transcript in each sample was normalized to the C (T) values of the control mRNA (36B4; Hs99999902-m1) of the same sample. The values of untreated cell samples were then set to 1 and the fold increase was calculated. The sense and antisense oligonucleotides used were GAPDH, 5′-ATGGCCTTCCGTGTTCCTAC-3′ and 5′-CAGATGCCTGCTTCACCAC-3′; HIF-1α, 5′-GAAACGACCACTGCTAAGGCA-3′ and 5′-GGCAGACAGCTTAAGGCTCCT-3′ respectively.
Statistical analysis
Statistical analysis was performed using a one-tailed Mann Whitney test. TMA assays were compared using the Student’s t-test. The association of HIF-1α expression with categorical pathological features was made using XCitation2 analysis. Calculations and analyses were performed with SPSS 11.5 for Windows (SPSS, Inc.), and when appropriate, were two-tailed. P values less than 0.05 were considered statistically significant.
Acknowledgments
We thank Dr V. Hofman for pathological expertise, Agnès Loubat for flow cytometry analysis and Dr Chan and Pr Stanners (McGill University) for the CEABAC 10 mice. Studies in the research unit INSERM ERI21/EA4319 were supported by the «Institut National de la Santé et de la Recherche Médicale», the Institut National du Cancer (Grant Vancol n° R07129AA), the Centre National de la Recherche Scientifique, the Ministère de l’Education, de la Recherche et de la Technologie, and by grants from Association pour la Recherche sur le Cancer (ARC, subvention 1142), Association François Aupetit and Fondation Infectiopole Sud. Studies in the research unit JE2526 Université d'Auvergne were supported by the Ministère de la Recherche et de la Technologie, by the Institut National de la Recherche Agronomique (USC 2018) and by grants from Association F. Aupetit (AFA). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 2006; 124:837 - 48; http://dx.doi.org/10.1016/j.cell.2006.02.017; PMID: 16497592
- Inagaki H, Suzuki T, Nomoto K, Yoshikai Y. Increased susceptibility to primary infection with Listeria monocytogenes in germfree mice may be due to lack of accumulation of L-selectin+ CD44+ T cells in sites of inflammation. Infect Immun 1996; 64:3280 - 7; PMID: 8757865
- Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007; 448:427 - 34; http://dx.doi.org/10.1038/nature06005; PMID: 17653185
- Podolsky DK. Inflammatory bowel disease. N Engl J Med 2002; 347:417 - 29; http://dx.doi.org/10.1056/NEJMra020831; PMID: 12167685
- Sands BE. Inflammatory bowel disease: past, present, and future. J Gastroenterol 2007; 42:16 - 25; http://dx.doi.org/10.1007/s00535-006-1995-7; PMID: 17322989
- Carvalho FA, Barnich N, Sivignon A, Darcha C, Chan CH, Stanners CP, et al. Crohn's disease adherent-invasive Escherichia coli colonize and induce strong gut inflammation in transgenic mice expressing human CEACAM. J Exp Med 2009; 206:2179 - 89; http://dx.doi.org/10.1084/jem.20090741; PMID: 19737864
- Carvalho FA, Barnich N, Sauvanet P, Darcha C, Gelot A, Darfeuille-Michaud A. Crohn's disease-associated Escherichia coli LF82 aggravates colitis in injured mouse colon via signaling by flagellin. Inflamm Bowel Dis 2008; 14:1051 - 60; http://dx.doi.org/10.1002/ibd.20423; PMID: 18338780
- Barnich N, Carvalho FA, Glasser AL, Darcha C, Jantscheff P, Allez M, et al. CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J Clin Invest 2007; 117:1566 - 74; http://dx.doi.org/10.1172/JCI30504; PMID: 17525800
- Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006; 441:437 - 43; http://dx.doi.org/10.1038/nature04871; PMID: 16724055
- Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol 1996; 16:4604 - 13; PMID: 8756616
- Risau W. Mechanisms of angiogenesis. Nature 1997; 386:671 - 4; http://dx.doi.org/10.1038/386671a0; PMID: 9109485
- Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res 2008; 14:6735 - 41; http://dx.doi.org/10.1158/1078-0432.CCR-07-4843; PMID: 18980965
- Danese S, Sans M, Spencer DM, Beck I, Donate F, Plunkett ML, et al. Angiogenesis blockade as a new therapeutic approach to experimental colitis. Gut 2007; 56:855 - 62; http://dx.doi.org/10.1136/gut.2006.114314; PMID: 17170016
- Scaldaferri F, Vetrano S, Sans M, Arena V, Straface G, Stigliano E, et al. VEGF-A links angiogenesis and inflammation in inflammatory bowel disease pathogenesis. Gastroenterology 2009; 136:585 95; http://dx.doi.org/10.1053/j.gastro.2008.09.064; PMID: 19013462
- Mariani F, Sena P, Marzona L, Riccio M, Fano R, Manni P, et al. Cyclooxygenase-2 and Hypoxia-Inducible Factor-1alpha protein expression is related to inflammation, and up-regulated since the early steps of colorectal carcinogenesis. Cancer Lett 2009; 279:221 - 9; http://dx.doi.org/10.1016/j.canlet.2009.02.001; PMID: 19268443
- Giatromanolaki A, Sivridis E, Maltezos E, Papazoglou D, Simopoulos C, Gatter KC, et al. Hypoxia inducible factor 1alpha and 2alpha overexpression in inflammatory bowel disease. J Clin Pathol 2003; 56:209 - 13; http://dx.doi.org/10.1136/jcp.56.3.209; PMID: 12610101
- Vannay A, Sziksz E, Prokai A, Veres G, Molnar K, Nagy Szakal D, et al. Increased expression of hypoxia inducible factor 1alpha in coeliac disease. Pediatr Res 2010; 68:118 22; http://dx.doi.org/10.1203/PDR.0b013e3181e5bc96; PMID: 20453713
- Rolhion N, Barnich N, Bringer MA, Glasser AL, Ranc J, Hebuterne X, et al. Abnormally expressed ER stress response chaperone Gp96 in CD favours adherent-invasive Escherichia coli invasion. Gut 2010; 59:1355 - 62; http://dx.doi.org/10.1136/gut.2010.207456; PMID: 20587550
- Cummins EP, Seeballuck F, Keely SJ, Mangan NE, Callanan JJ, Fallon PG, et al. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology 2008; 134:156 - 65; http://dx.doi.org/10.1053/j.gastro.2007.10.012; PMID: 18166353
- Taylor CT. Interdependent roles for hypoxia inducible factor and nuclear factor-kappaB in hypoxic inflammation. J Physiol 2008; 586:4055 - 9; http://dx.doi.org/10.1113/jphysiol.2008.157669; PMID: 18599532
- Krogfelt KA, Bergmans H, Klemm P. Direct evidence that the FimH protein is the mannose-specific adhesin of Escherichia coli type 1 fimbriae. Infect Immun 1990; 58:1995 - 8; PMID: 1971261
- Barnich N, Boudeau J, Claret L, Darfeuille-Michaud A. Regulatory and functional co-operation of flagella and type 1 pili in adhesive and invasive abilities of AIEC strain LF82 isolated from a patient with Crohn's disease. Mol Microbiol 2003; 48:781 - 94; http://dx.doi.org/10.1046/j.1365-2958.2003.03468.x; PMID: 12694621
- Smith KD, Ozinsky A. Toll-like receptor-5 and the innate immune response to bacterial flagellin. Curr Top Microbiol Immunol 2002; 270:93 - 108; http://dx.doi.org/10.1007/978-3-642-59430-4_6; PMID: 12467246
- O'Neill LA, Bryant CE, Doyle SL. Therapeutic targeting of Toll-like receptors for infectious and inflammatory diseases and cancer. Pharmacol Rev 2009; 61:177 - 97; http://dx.doi.org/10.1124/pr.109.001073; PMID: 19474110
- Pages G, Pouyssegur J. Transcriptional regulation of the Vascular Endothelial Growth Factor gene–a concert of activating factors. Cardiovasc Res 2005; 65:564 - 73; http://dx.doi.org/10.1016/j.cardiores.2004.09.032; PMID: 15664382
- Eaves-Pyles T, Allen CA, Taormina J, Swidsinski A, Tutt CB, Jezek GE, et al. Escherichia coli isolated from a Crohn's disease patient adheres, invades, and induces inflammatory responses in polarized intestinal epithelial cells. Int J Med Microbiol 2008; 298:397 - 409; http://dx.doi.org/10.1016/j.ijmm.2007.05.011; PMID: 17900983
- Semiramoth N, Gleizes A, Turbica I, Sandre C, Gorges R, Kansau I, et al. Escherichia coli type 1 pili trigger late IL-8 production by neutrophil-like differentiated PLB-985 cells through a Src family kinase- and MAPK-dependent mechanism. J Leukoc Biol 2009; 85:310 - 21; http://dx.doi.org/10.1189/jlb.0608350; PMID: 19015376
- Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol 2001; 167:1882 - 5; PMID: 11489966
- Frede S, Stockmann C, Freitag P, Fandrey J. Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-kappaB. Biochem J 2006; 396:517 - 27; http://dx.doi.org/10.1042/BJ20051839; PMID: 16533170
- Jung Y, Isaacs JS, Lee S, Trepel J, Liu ZG, Neckers L. Hypoxia-inducible factor induction by tumour necrosis factor in normoxic cells requires receptor-interacting protein-dependent nuclear factor kappa B activation. Biochem J 2003; 370:1011 - 7; http://dx.doi.org/10.1042/BJ20021279; PMID: 12479793
- Kim KS, Rajagopal V, Gonsalves C, Johnson C, Kalra VK. A novel role of hypoxia-inducible factor in cobalt chloride- and hypoxia-mediated expression of IL-8 chemokine in human endothelial cells. J Immunol 2006; 177:7211 - 24; PMID: 17082639
- Shah YM, Ito S, Morimura K, Chen C, Yim SH, Haase VH, et al. Hypoxia-inducible factor augments experimental colitis through an MIF-dependent inflammatory signaling cascade. Gastroenterology 2008; 134:2036 48; http://dx.doi.org/10.1053/j.gastro.2008.03.009; PMID: 18439915
- Imtiyaz HZ, Williams EP, Hickey MM, Patel SA, Durham AC, Yuan LJ, et al. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest 2010; 120:2699 - 714; http://dx.doi.org/10.1172/JCI39506; PMID: 20644254
- Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, Singh R, et al. Enhanced Escherichia coli adherence and invasion in Crohn's disease and colon cancer. Gastroenterology 2004; 127:80 - 93; http://dx.doi.org/10.1053/j.gastro.2004.03.054; PMID: 15236175
- Bronowski C, Smith SL, Yokota K, Corkill JE, Martin HM, Campbell BJ, et al. A subset of mucosa-associated Escherichia coli isolates from patients with colon cancer, but not Crohn's disease, share pathogenicity islands with urinary pathogenic E. coli. Microbiology 2008; 154:571 - 83; http://dx.doi.org/10.1099/mic.0.2007/013086-0; PMID: 18227261
- Boudeau J, Barnich N, Darfeuille-Michaud A. Type 1 pili-mediated adherence of Escherichia coli strain LF82 isolated from Crohn's disease is involved in bacterial invasion of intestinal epithelial cells. Mol Microbiol 2001; 39:1272 - 84; http://dx.doi.org/10.1111/j.1365-2958.2001.02315.x; PMID: 11251843
- Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, et al. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn's disease. Gastroenterology 1998; 115:1405 - 13; http://dx.doi.org/10.1016/S0016-5085(98)70019-8; PMID: 9834268
- Chan CH, Stanners CP. Novel mouse model for carcinoembryonic antigen-based therapy. Mol Ther 2004; 9:775 - 85; http://dx.doi.org/10.1016/j.ymthe.2004.03.009; PMID: 15194045
- Cane G, Ginouves A, Marchetti S, Busca R, Pouyssegur J, Berra E, et al. HIF-1alpha mediates the induction of IL-8 and VEGF expression on infection with Afa/Dr diffusely adhering E. coli and promotes EMT-like behaviour. Cell Microbiol 2010; 12:640 - 53; http://dx.doi.org/10.1111/j.1462-5822.2009.01422.x; PMID: 20039880
- Ilie M, Mazure NM, Hofman V, Ammadi RE, Ortholan C, Bonnetaud C, et al. High levels of carbonic anhydrase IX in tumour tissue and plasma are biomarkers of poor prognostic in patients with non-small cell lung cancer. Br J Cancer 2010; 102:1627 - 35; http://dx.doi.org/10.1038/sj.bjc.6605690; PMID: 20461082