Abstract
The structure of the human gut microbial community is determined by host genetics and environmental factors, where alterations in its structure have been associated with the onset of different diseases. Establishing a defined human gut microbial community within inbred rodent models provides a means to study microbial-related pathologies, however, an in-depth comparison of the established human gut microbiota in the different models is lacking. We compared the efficiency of establishing the bacterial component of a defined human microbial community within germ-free (GF) rats, GF mice and antibiotic-treated specific pathogen-free mice. Remarkable differences were observed between the different rodent models. While the majority of abundant human-donor bacterial phylotypes were established in the GF rats, only a subset was present in the GF mice. Despite the fact that members of the phylum Bacteriodetes were well established in all rodent models, mice enriched for phylotypes related to species of Bacteroides. In contrary to the efficiency of Clostridiales to populate the GF rat in relative proportions to that of the human-donor, members of Clostridia cluster IV only poorly colonize the mouse gut. Thus, the genetic background of the different recipient rodent systems (that is, rats and mice) strongly influences the nature of the populating human gut microbiota, determining each model’s biological suitability.
Introduction
The structure of the human gut microbial community is determined by host genetics and environmental factors,Citation1,Citation2 where the complex communication between the gut microbiota and the innate immune system governs metabolic homeostasis.Citation3 Alterations in the gut microbial community have been associated with the onset of bowel diseases,Citation4,Citation5 obesity,Citation6,Citation7 allergies and asthmaCitation8,Citation9 and autoimmune diseases such as diabetes.Citation10,Citation11 However, the study and manipulation of the in situ human gut microbial community is constrained by sampling difficultiesCitation12 as well as ethical issues and the immense inter-individual differences in the microbial composition.Citation13,Citation14 Alternatives have been to use in vitro model bioreactors simulating the human gastrointestinal tractCitation15 or the establishment of the human gut microbiota into germ-free (GF) surrogate animal models such as zebrafish,Citation16 pigs,Citation13 miceCitation17-Citation19 and rats.Citation14,Citation20,Citation21 Although GF animals lack intestinal microbiota which is important for educating the immune system,Citation22 microbial colonization of GF animals has been reported to restore the mucosal and systemic functionality of the immune system.Citation23 The use of transplanted microbial community models has been decisive for the elucidation of key processes involved in host-microbeCitation16,Citation24 or microbe-microbe interactions,Citation25 as well as for determining the influence of host factors, dietary manipulations and therapeutics such as probiotics, prebiotics and antibiotics on bacterial community structure and function.Citation17,Citation18,Citation24,Citation26
The analysis of genetically obese and lean mice revealed obese animals to have a 50% reduction in the abundance of Bacteroidetes and a proportional increase in Firmicutes,Citation27 a finding also reported in obese and lean humansCitation6 which correlated with an increased capacity to harvest energy from the diet.Citation7,Citation28 Importantly, the obese phenotype could be transferred to GF recipient mice by colonization with the “obese microbiota” from an obese donor, thus identifying the gut microbiota as an important factor contributing to the pathophysiology of obesity.Citation7
Because of their commercial availability, small size, high reproductive rate, minimal costs of purchase and maintenance as well as their well-characterized and easily manipulated anatomy, physiology and genetic background rats and mice are the most used and perhaps best understood laboratory animal species. While, previous studies have reported that the dominant human bacterial groups can be transplantable and stable in both GF miceCitation17,Citation19 and GF rats,Citation20 a comparison between animal models has yet to be made. Previous studies have reported successful colonization when either the dominant groups can be establishedCitation20,Citation21 or when most of the diversity can be established.Citation19 However, due to the complex interactions between gut species and reports on significant changes to the balance between bacterial groups, successful colonization may be regarded appropriate when a significant fraction of the diversity can be established into the surrogate host at levels of abundance similar to that of the original donor. Nevertheless, as stated by SilleyCitation29 the primary concern is a proper understanding of the limitations of a model, and in the case where this is accepted, the model can be useful even if it fails to be a true representation of the gastrointestinal tract.
Analysis of either GF mice or GF rats have revealed that the intestinal microbiota of animals being inoculated with human microbiota may represent a limited sample of bacteria from the human source which are selected by yet unknown interactions between the host and the bacteria.Citation21,Citation30 A more recent study in reference Citation19, reported on the engraftment of the majority of genera of the human gut microbiota into GF mice, but where diet had a significant effect on community structure and function. However, a comparison of the fecal microbiota of 59 mammalian species to humans showed that host phylogeny also significantly influences bacterial diversity.Citation31 More recently, evidence for the importance of host genetics was supported through the analysis of gut microbial communities of 645 mice of an advanced intercross lineCitation2 where correlations between host genotype and the relative abundances of specific microbial taxa were observed.
Despite the numerous studies on human-microbiota associated rodent models, the spectra of human bacterial species that can be successfully established in the different models (in their relative proportions), and thus, in how closely they mimic the human intestinal microbiota remains to be determined. Here, we compare the establishment efficiency, quality and stability of a transplanted human gut microbial community in different strains of GF recipient rats, GF recipient mice and antibiotic-treated specific pathogen-free (SPF) recipient mice to evaluate the influence of host phylogeny and genetic background on such establishment, across two independent but reproducibly designed experiments.
Results
Global patterns in establishment.
The efficiency of establishing a human gut microbial community derived from feces into different rodent models was determined by inoculating the bacterial community of a healthy human-donor into GF WKY rats, GF C57BL/6 and antibiotic-treated SPF C57BL/6 mice (). Similarities between fecal community structures and stability over time were assessed by T-RFLP followed by ordination. The cecal community was compared with the fecal community after 32 d (GF WKY rats) or 41 d (GF C57BL/6 and antibiotic-treated SPF C57BL/6 mice). In the latter case, the comparison of fecal samples showed gut communities to be stable between day 32 and day 41 (Fig. S1A).
Table 1. Experimental design
The established bacterial community of both the feces and the ceca of GF WKY recipient rats was more similar to the inoculating bacterial community of the human-donor in respect to both the percent abundance of the individual bacterial groups () and their presence/absence () in comparison to the communities established in GF C57BL/6 and antibiotic-treated SPF C57BL/6 recipient mice. This observation was supported by statistical significance testing by comparing all fecal/cecal samples collected for each rodent group over time to the human-donor using analysis of similarity (ANOSIM). Although all groups were significantly different, the pairwise comparison between the bacterial communities of the human-donor and the GF WKY rat resulted in a lower ANOSIM R value than those delivered for comparisons between the human-donor and the mice groups ( and Fig. S1A). Comparison of the different mouse systems sharing the same genetic background showed that the gut bacterial community established in the antibiotic-treated SPF C57BL/6 mice was more similar to that of the autochthonous SPF C57BL/6 mice bacterial community (R = 0.292) than to that established in the GF C57BL/6 recipient mice (R = 0.792, ; Fig. S1A). This indicates that only a minor portion of the human gut bacterial community could be established (Fig. S2).
Figure 1. Comparison of the global bacterial communities between the human-donor and the recipient animals using non-metric multidimensional scaling (nMDS) ordination. (A–D) Similarity between the global bacterial community of the human-donor, antibiotic-treated SPF C57BL/6 recipient mice, non-recipient SPF C57BL/6 mice, GF C57BL/6 recipient mice and GF WKY recipient rats in the first experiment where similarities in bacterial structures were assessed using the Bray-Curtis similarity algorithm on (A) abundance T-RFLP data comprising 168 T-RFs, (B) presence/absence T-RFLP data comprising 168 T-RFs, (C) RT-qPCR data, (D) pyrosequencing data comprising 290 phylotypes. (E–G) Similarity between the global bacterial community of the human-donor, GF WKY recipient rats, GF SPRD recipient rats and GF NMRI recipient mice in the second experiment, using (E) abundance T-RFLP data comprising 58 T-RFs, (F) presence/absence T-RFLP data comprising 58 T-RFs, (G) RT-qPCR data. All symbols represent the bacterial community at the time of being sacrificed. The number above the symbol indicates the individual animal (), while the Roman numeral above the black dots indicates the human-donor bacterial community on each day the respective community was administered. The multiple human-donor points represent the community structure of the stored sample over several days. Stress values between 0 and 0.1 indicate very good ordination power with little chance of misinterpretation.
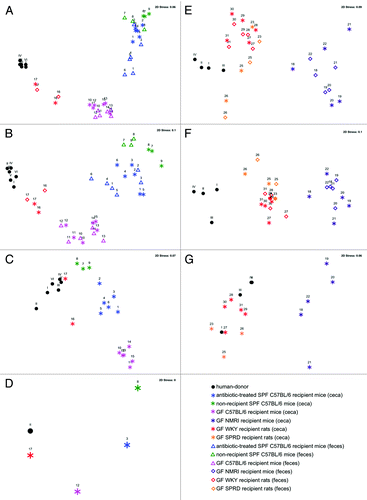
Table 2. Comparison of the global bacterial communities between recipient rodent models, the SPF non-recipient mice and the human-donor using analysis of similarity (ANOSIM)
To further investigate whether variations in the genetic background of the recipient species could influence the efficiency of human microbiota establishment, a second experiment using GF recipient rats with two different genetic backgrounds, namely GF WKY and GF SPRD rats compared with GF NMRI mice was performed. These animals were housed, fed and treated in an identical manner. The findings from this experiment reiterated the results from the first in that GF SPRD and GF WKY recipient rats were able to establish a bacterial community more similar to that of the human-donor than the GF NMRI recipient mice (; Fig. S1B), supported by lower R values of 0.364 and 0.499, respectively, as opposed to R = 0.994 for the mice (). In fact, the bacterial communities established in both GF SPRD and GF WKY recipient rats also showed a significant difference to each other even though their bacterial communities were less distinguishable as indicated by a low R value (R = 0.238, ), where the communities established in the GF SPRD recipient rats were more closely related to that of the human-donor. As housing and feeding factors were maintained for these groups of mice and rats, this further suggests that the genetic background of the recipient species can indeed influence the establishment of the transplanted human gut microbial community.
All bacterial communities of GF animal groups remained stable after the first week following inoculation with the human-donor community (Fig. S1B). At this level of resolution, there were no significant differences between the fecal and cecal bacterial communities in each model system after 4 weeks (Table S1). The differences between the active communities (as assessed by 16S rRNA profiling) and global communities (as assessed by 16S rRNA gene profiling) of both feces and ceca were always higher than those differences between fecal and cecal communities (as indicated by R values of 0.228–0.812), even though statistically significant differences were only evident for fecal samples from WKY recipient rats and NMRI recipient mice and cecal samples from WKY rats; likely due to the small sample size of the 3–5 animals per group and thus the low statistical power. Furthermore, rRNA profiling of communities from the GF SPRD and WKY recipient rats revealed them to be again more similar to that of the human-donor (R = 0.676 and 0.857, respectively) than the NMRI recipient mice communities (R = 1) (Table S1), confirming that profiling both the active and global communities, deduces the same pattern between animal models (Fig. S3). Incidentally, the transplantation efficiency between both experiments was reproducible, where the level of establishment within the GF rats comparing to the GF mice was akin and maintained at two independent experiments using inocula from the same human-donor, which were highly similar despite the 10 mo interval between collection of the donor material (Fig. S4).
Specific establishment.
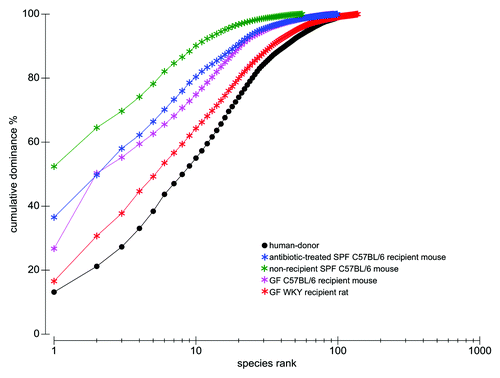
To further determine which human-donor bacterial species were established in the different recipient rodents, the fine-scale diversity (phylotype-level taxa defined at > 98% similarity) of representative recipient communities was analyzed by 454-pyrosequencing and compared with that of the human-donor (Table S2). The bacterial community of the human-donor (Fig. S5) comprised bacterial phylotypes which have been typically found in healthy adult human feces and belonging to the Clostridia clusters I, IV, XI, XIVa (represented by members of the families Clostridiaceae, Ruminococcaceae, Peptostreptococcaceae and Lachnospiraceae, respectively), and Bacteroides and Parabacteroides genera. The determined Firmicutes/Bacteroidetes (F/B) ratio of 6.3 was in the range of the published values for healthy adult humans.Citation32 The GF WKY recipient rat established most of the representative phylotypes of each of the human-donor’s bacterial phyla (). In contrast, neither of the mouse systems was capable of retaining the full spectra ( and ). This is also evident in the k-dominance plot () where the bacterial community in the human-donor and in the GF WKY recipient rat comprised a higher diversity and more balanced dominance than the autochthonous microbiota of the SPF mouse or the GF recipient mouse. Nineteen of the 25 most abundant human-donor bacterial phylotypes, which accounted for 80% of the total bacterial abundance in the community, were present in the GF recipient rat while only 5–11 of those were found in the recipient mouse models. Eight abundant phylotypes all belonging to Clostridia were present in the rat model in almost relative proportions, but excluded in the mouse models. Six of these abundant phylotypes belonging to the Clostridia clusters I, IV, XI and XIVa were excluded in all rodent models (Table S2).
Figure 2. Taxonomic distribution of the 16S rRNA gene sequence phylotypes from the human-donor bacterial community which were detected by 454-pyrosequencing analysis within the GF WKY recipient rat. Branching orders correspond to that of the human-donor bacterial community (Fig. S5) although the type strain/isolate names were removed for clarity. Those human-donor derived phylotypes detected in this rat model are denoted in red, while those human-donor derived phylotypes not detected are denoted in gray. Corresponding dots following each colored phylotype indicate its relative percent abundance contribution to the whole community where ●, < 1%; ●●, 1–2%; ●●●, 2–5%; ●●●●, 5–20% and ●●●●●, > 20%. Scale bars represent 5% nucleotide sequence divergence.
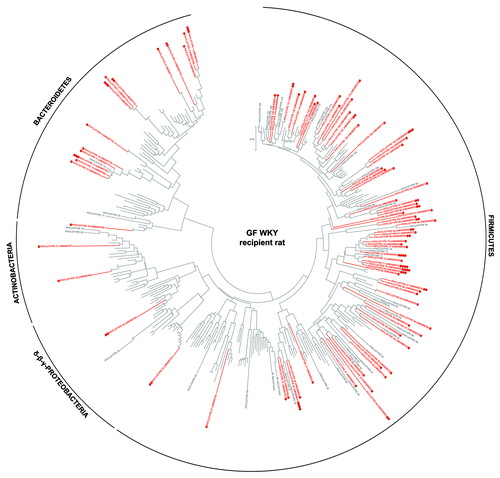
Figure 3. Taxonomic distribution of the 16S rRNA gene sequence phylotypes from the human-donor bacterial community which were detected by 454-pyrosequencing analysis within the GF C57BL/6 recipient mouse. Branching orders correspond to that of the human-donor bacterial community (Fig. S5) although the type strain/isolate names were removed for clarity. Those human-donor derived phylotypes detected in this mouse model are denoted in pink, while those human-donor derived phylotypes not detected are denoted in gray. Corresponding dots following each colored phylotype indicate its relative percent abundance contribution to the whole community where ●, < 1%; ●●, 1–2%; ●●●, 2–5%; ●●●●, 5–20% and ●●●●●, > 20%. Scale bars represent 5% nucleotide sequence divergence.
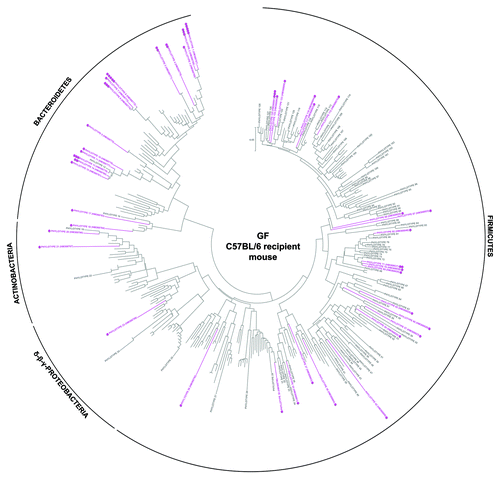
Figure 4. Taxonomic distribution of the 16S rRNA gene sequence phylotypes from the human-donor bacterial community which were detected by 454-pyrosequencing analysis within the antibiotic-treated SPF C57BL/6 recipient mouse. Branching orders correspond to that of the human-donor bacterial community (Fig. S5) although the type strain/isolate names were removed for clarity. Those human-donor derived phylotypes detected in the mouse model are denoted in blue, while those human-donor derived phylotypes not detected are denoted in gray. Corresponding dots following each colored phylotype indicate its relative percent abundance contribution to the whole community where ●, < 1%; ●●, 1–2%; ●●●, 2–5%; ●●●●, 5–20% and ●●●●●, > 20%. Scale bars represent 5% nucleotide sequence divergence.
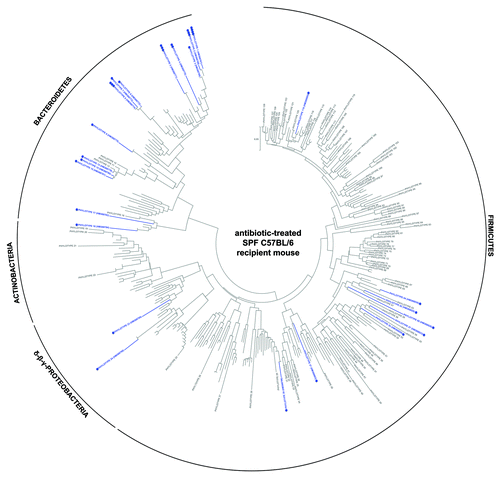
Figure 5. k-dominance plots presenting cumulative ranked abundances plotted against species rank depicting bacterial phylotype diversity and dominance, comparing the human-donor inoculum to the ceca of the 4 representative animals in experiment I (using 454-pyrosequencing data comprising 290 phylotypes); antibiotic-treated SPF C57BL/6 recipient mouse, non-recipient SPF C57BL/6 mouse, GF C57BL/6 recipient mouse and GF WKY recipient rat. These dominance curves are based on ranking species in decreasing order of their importance in terms of abundance. The cumulative curves are used for comparing the biodiversity between the different samples, where the most elevated curves have the lowest diversity, while the lower curves have the highest diversity (in this case, the human-donor and the recipient rat). A quickly rising line represents a sample showing high dominance of abundant species, while samples with a more even distribution of species will be flatter.
The success of human microbiota establishment at the genus-level was similar to that reported by Turnbaugh et al.Citation19 where 33 out of 42 genus-level human-donor taxa (determined using RDP level 6)Citation33 could be established at an abundance level > 0.02% of the total community in previously GF C57BL/6 mice, while here, 22 out of 35 genus-level human-donor taxa (determined using RDP level 6) could be established at an abundance level > 0.02% of the total community in the GF recipient C57BL/6 mouse (Table S3).
A further 86 phylotypes were detected across the three recipient animals which were not detected in the human-donor (Table S4). As these phylotypes were related to those observed in high abundance in the human-donor and comprised phylotypes typically associated with human gut species, it can be argued that they were in too low abundance to be detected by pyrosequencing with a detection limit of 0.02% of the total community, so thus are considered the “rare” fraction of the human-donor. This rare fraction contributed approximately 20% to the GF C57BL/6 mouse recipient community, 5% to the GF rat and 3% to the antibiotic-treated SPF mouse recipient community, indicating that the rare fraction of the human-donor was enriched in the GF mouse cecum.
Establishment of Bacteroidetes.
Members representing this phylum were present in all animal models, but seemed to be particularly enriched in the mice (– and ). All three representative recipient animals were capable of establishing 14–16 of the 18 abundant Bacteriodetes human-donor phylotypes. In particular, organisms related to Bacteroides spp (PT1 and PT6) were established in greater relative abundance to the whole community in mice (– and Table S2). Furthermore, the rare fraction of the Bacteroidetes community (Table S4) was also enriched in both mouse systems, contributing 2–4% to the total community, where in particular organisms related to Alistipes shahii (PT184) having been previously described as a human gut microbe,Citation34 were established in both mouse systems at an abundance > 1%.
Table 3. Summary of RT-qPCR data using group-specific 16S rDNA-targeted primers to target all bacteria, C. leptum subgroup members, Bacteroides group members and Clostridia cluster XIVa group members
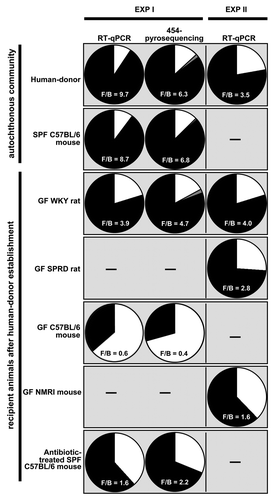
Overall, Bacteroidetes accounted for 17%, 67% and 29% of total abundance in the GF WKY recipient rat, GF C57BL/6 recipient mouse and the SPF C57BL/6 recipient mouse, respectively, as determined after pyrosequencing (Table S2). RT-qPCR targeting the Bacteroides group in the ceca of all 31 animals confirmed their specific enrichment in the mice ceca (61% in GF C57BL/6 recipient mice and 44% in GF NMRI recipient mice) ( and S5).
Establishment of Firmicutes.
Considerable differences were observed when assessing the efficiency of establishing Firmicutes in the different rodent models by pyrosequencing representative cecal communities. While the GF WKY recipient rat established 31/48 Clostridia cluster IV phylotypes (), the GF and SPF C57BL/6 recipient mice were only able to each establish ≤ 9/48 phylotypes ( and ). Some rare human-donor Clostridia cluster IV phylotypes were, however, enriched in the GF mouse, while not being detected in the rat. Similarly, Ruminococcaceae (Cluster IV Clostridia members) were reported to contribute only a low-level percent of the total community established in human-microbiota associated mice under different dietary regimes.Citation19
Of particular interest were the phylotypes of the C. leptum subgroup (Fig. S5) which comprise >18% of the total abundance in the human-donor (as determined by both RT-qPCR and pryosequencing, and Fig. S5). The most striking finding was that the GF WKY recipient rat was able to establish 12/15 phylotypes, while the GF C57BL/6 recipient mouse was only able to establish four of these. Furthermore, the antibiotic-treated SPF C57BL/6 recipient mouse was unable to establish any of the C. leptum subgroup phylotypes (). RT-qPCR targeting organisms of the C. leptum subgroup across the ceca of all 31 animals confirmed that GF recipient rats were able to establish more C. leptum than the GF recipient and antibiotic-treated recipient mice ( and S5) resembling the relative quantities observed in the human-donor.
The human-donor comprised 42 Clostridia cluster XIVa phylotypes accounting for upto 50% of the total abundance in the human-donor ( and Fig. S5). RT-qPCR targeting members of Clostridia cluster XIVa (also typically referred to as the C. coccoides group) revealed that all recipient animals established a significant amount of this group. However, while pyrosequencing revealed that the GF WKY recipient rat was able to establish a significant proportion of these phylotypes (29/42), the GF and antibiotic-treated C57BL/6 mice established relatively few phylotypes (8/42 and 1/42, respectively, –). Although the GF C57BL/6 recipient mice comprised only 9% of the predominant human-donor Firmicutes (Table S2), a further > 20% belonged to the rare fraction of the human-donor Firmicutes (Table S4), mostly belonging to Clostridia cluster XIVa (16%). So although the total abundance of members of this cluster resemble that of the human-donor, the GF mouse model enriched for the rare component of this community and could not establish the abundant members of this very donor.
Clostridia cluster XI was the third most abundant cluster observed in the human-donor (eight phylotypes predominantly related to members of the Peptostreptococcus) could also be much better established in the GF WKY recipient rat (5/8 phylotypes), compared with their establishment in the mice models (≤ 3/8 phylotypes, –). Some additional rare phylotypes, mainly belonging to the Incertae sedis family XIII were enriched to some extent in the mice models (0.5% total abundance).
The similarity between the global bacterial communities of samples generated using RT-qPCR data () illustrates that same pattern between samples using both T-RFLP data () and pyrosequencing data of representative samples (). This shows good agreement between all three bacterial profiling methods in respect to illustrating establishment of the human-donor community into the different animals.
Firmicutes to bacteroidetes ratio.
While the balance between Firmicutes and Bacteroidetes (F/B) varies between human individualsCitation35 with published F/B ratios ranging between 1.06–19.75, Bacteroidetes usually do not outnumber the Firmicutes in healthy humans or in most mammalian species.Citation31,Citation36 Inbred SPF mice had a F/B ratio of 6.8–9.6 () and Wistar rats had a F/B ratio of 1.5.Citation31 In fact, the balance between Firmicutes and Bacteroidetes in the mouse autochthonous community reflected that of the human. While the GF recipient rat maintained such a balance after human-donor inoculation with F/B ratios between 2.8–4.7 (using data generated by both RT-qPCR and pyrosequencing), the recipient GF mouse systems did not maintain such a balance (F/B ratios between 0.4–1.6, ). RT-qPCR gave comparable F/B ratios to those generated using pyrosequencing ().
Figure 6. Firmicutes/Bacteriodetes ratio as determined by either RT-qPCR or 454-pyrosequencing of each group of animals or representative animal of the recipient rodent models, respectively. Black wedges represent Firmicutes; white wedges represent Bacteroidetes and gray wedges collectively represent Proteobacteria and Actinobacteria. The Firmicutes/Bacteroidetes (F/B) ratio is given. However, when the RT-qPCR data are presented, a modified F/B ratio is given as this data only considers those Firmicutes and Bacteriodetes targeted here by these primer sets and not necessarily all possible Firmicutes and Bacteriodetes in the sample.
Discussion
In the current work, the establishment of a human microbial community in different rodent recipients was assessed. Typically, in an adult human gut microbial community, as that used in the current study, members of the phylum Firmicutes are the most numerous and diverse component, where approximately 95% of phylotypes belong to the phylogenetically and metabolically diverse class of Clostridia.Citation35 Clostridia cluster IV comprising the Clostridium leptum subgroup and Clostridia cluster XIVa comprising C. coccoides group members each make-up approximately one quarter of bacterial abundance of the human colon.Citation37 The phylum Bacteroidetes also comprises a prominent fraction typically accounting for 10–30% of the human colonic microbiotaCitation36 where a recent study indicated the mean abundance and inter-individual variation to be 27.8% ± 16.6%.Citation38 Overall, compared with the human-donor inoculum, Bacteroidetes were enriched for in the mouse ceca. A similar enrichment of Bacteroidetes after inoculation of human microbiota into GF mice was also observed by Turnbaugh and colleagues,Citation19 where Bacteriodetes had been enriched from 34.2% in the donor to 52.6% in C57BL/6 mice when fed a low fat, polysaccharide-rich diet. Of the members of the phylum Firmicutes, significant differences in the success of establishment were observed regarding Clostridia cluster IV, and specifically regarding members of the C. leptum subgroup. Members of these are fibrolytic and butyrate producers, which contribute to colonic healthCitation39 as butyrate is one of the major carbon sources for the epithelium and exhibits immunomodulatory and anti-inflammatory properties.Citation39,Citation40 C. leptum subgroup members have also been associated with playing a crucial role in gut homeostasis, as reductions in their observed abundance have been indicated in the onset of IBD.Citation39 The poor establishment of members of this subgroup in mice as reported here is in accordance with previous data on establishment of members of the Faecalibacterium genus into GF C57BL/6 mice.Citation19 Clostridia cluster XIVa are also deemed an important group comprising butyrate producers.Citation41 Although the total abundance of members of this cluster resemble that of the human-donor, the GF mouse model, in contrast to the rat model enriched for the rare component of this community and could not establish the abundant members of this donor. Considering that the predominant intestinal species belong mainly to Clostridia clusters XIVa and IV, and also that these clusters comprise both important pathogens,Citation42 saccharolytic and/or proteolytic species and species responsible for metabolizing a wide variety of substrates,Citation43 it is imperative that the recipient animals are able to establish a significant diversity and proportion of these species in order to be considered a surrogate “human” model.
The poor establishment of Firmicutes members in the mouse model and the enrichment of Bacteroidetes resulted in Firmicutes to Bacteroidetes (F/B) ratios < 1.6 in the human microbiota associated mice, which were neither observed in SPF mice (> 6.8), the human microbiota associated rats (> 2.4) nor in the human donor (6.3). A similar predominance of Bacteroidetes over Firmicutes (ratio of 0.78) has recently also been observed in human microbiota associated mice on a low-fat, polysaccharide-rich chow.Citation19 These authors also reported a significant effect of diet on the gut microbiota of such mice, where a high-fat western diet (18.7% protein, 40.7% carbohydrates, 40.6% fat) resulted in an increase in the relative abundance of Firmicutes, resulting in an F/B ratio of 4.3. However, such a diet also resulted in a significant increase in the relative abundance of Erysipelotrichi (previously misannotated as MollicutesCitation44) and Bacilli community members, which are rare in the human-donors of both studies (< 1%). The increase of Erysipelotrichi, in that case Clostridium ramosum, was also recently observed in a gnotobiotic rat model where seven bacterial species were introduced into germfree rats.Citation14 It should be noted that the Erysipelotrichi were also slightly enriched (to 3%) in previous GF C57BL/6 mice on the low fat diet of Turnbaugh et al.Citation19 compared with the results reported here, which may be due to the higher level of fat in the chow supplied by the authors. In contrast, members of the predominant Faecalibacterium genus of the C. leptum subgroup comprising between 17–19% of the total community in both human-donors in these two independent studies (see our Table S3 and Turnbaugh et al.Citation19), could only be established to low levels in mice under the low fat, polysaccharide-rich diets supplied and where a western diet resulted in a further decrease of its abundance.Citation19 However, rats were able to establish an adequate proportion of members of the C. leptum subgroup comprising Faecalibacterium, despite being on a low fat, polysaccharide diet (which was the same diet as for our mice).
Thus, the imbalance between Firmicutes and Bacteriodetes in the mouse host is a caveat of these human microbiota-associated models since they may not reflect what is currently considered the typical ratio within of a healthy human gut microbial community. Interestingly, such distortions in the composition of the Firmicutes phylum have been reported in IBD patients,Citation39 where in particular, a restriction of biodiversity in the fecal microbiota of Crohn’s disease, ulcerative colitis and infectious colitis patients and in the C. leptum subgroup has been observedCitation39,Citation45 resulting in F/B ratios as low as shown here in the GF recipient mouse models. The underlying reasons for this low F/B ratio in GF recipient mice remains to be elucidated.
Trials to establish the human-donor community into antibiotic-treated SPF mice showed poor success. Similarly among others, Manichanh et al.Citation24 reported that antibiotic intake prior to transplantation in rats did not facilitate the establishment of another rat’s exogenous microbiota. Thus, even though indigenous gut microbial composition shows some degree of plasticity,Citation24,Citation46 microbial groups have been found to develop in parallel with the host and depend on the physiological environment in unity with their host. Hence, the interaction between the host and the host-adapted commensal microbiota plays an important role not only for the maintenance of homeostasis but also for the recognition of allochthonous microorganisms, and thus ensuring a rapid elimination of invading microbes.Citation23,Citation47,Citation48 It should be noted, that although GF animals lack a trained immune systemCitation22 it has also been reported that they rapidly develop immunological capacity upon bacterial colonization.Citation23
It is clear that the establishment of the gut microbiota is mediated by a variety of factors, and while diet and processes associated with microbial adhesion are important for colonization, host genetics is now regarded as a principal factor in determining the specific profile of the commensal microbiota in the human gut.Citation2,Citation49 More specifically, as a primary barrier and an essential component of the innate immune system, the intestinal epithelium and its complex network of immune cells elicit an integrated response that dictates the host response to the intestinal microbiota.Citation50 The characterization of microbial-epithelial interactions has further suggested that the innate epithelial response can recognize and discriminate between different indigenous bacteria.Citation50 At the forefront of the mucosal immune system are constitutively secreted antimicrobial peptides (defensins) and immunoglobulins (IgA),Citation50,Citation51 for which, even among the few species and orders of mammals that have been studied, significant differences exist,Citation52,Citation53 possibly as each animal species evolves its antimicrobial arsenal under pressure from distinct pathogens.Citation53 As an example, the abundant neutrophil defensins of humans are similar to those of rabbits, guinea pigs, rats and hamsters. Interestingly, mice and pigs lack neutrophil defensins, but their neutrophils have antimicrobial peptides that belong to the cathelicidin family.Citation53 Accordingly, it was speculated that these neutrophil defensins play an important role in determining host specificity for Bordetella across different mammals.Citation54 Thus, the observed differences in the colonizing bacterial species may be attributed to such variations in the gut mucosa of these animals.
Recent work comparing the indigenous microbiota of 645 mice (crossed between C57BL/6J and an ICR-derived outbred line) found that the individual host genotype had a measurable contribution to the observed variation in the core measurable microbiota across animals in the population.Citation2 They and others in the literature have suggested that the adaptive immune system has specifically evolved in vertebrates to regulate and maintain beneficial microbial communities, and Benson et al. concluded that insights to this effect will clearly emerge after analyses across multiple host species. Although the sample sizes of the experimental groups in this current work are admittedly low, thus limiting extensive conclusions, the results reported here support this suggestion, by showing that the rat was able to be relatively better colonized by the human bacterial phylotypes of the donor used in the present study. However, the precise physiological mechanisms that permit certain members of the human gut consortia to colonize the rat in relative proportions and not the mouse, remain to be elucidated. While it is argued that a range of different factors such as diet, housing conditions, inoculating factors, age, gender, antibiotics and physiological status can have different levels of impact on colonization, this work shows that the host itself is an important determinant for colonization.
However, it should be noted that the microbial community structure of the adult human gut is also highly variable between individuals, where it has been suggested that this is the result of restricted migration of microorganisms between hosts, strong ecological interactions within hosts, as well as host variability in terms of diet, genotype and colonization history.Citation55 A more recent report by Arumugam et al. revealed that the intestinal community structure of humans are not continuous, but can be classified into one of three clusters, termed “enterotypes.” These clusters, which neither clustered due to continent nor specific nations were differentiated by the abundance of either Bacteroides species, Prevotella species and/or Ruminococcus species. Thus, it is reasonable to assume that the human-donor of this current study belongs to enterotype 3 based on the classification of Arumugam et al. In contrast, the human-donor used in the experiments by Turnbaugh et al.Citation19 can be tentatively grouped into enterotype 2, mainly based on the high abundance of Prevotella spp. Independent of the deduced enteroptype used in both experimental set-ups, a significant enrichment of Bacteroidetes and a poor establishment of members of the C. leptum subgroup after inoculation of human microbiota into GF mice was observed, indicating a general selection of the mouse intestine for specific members of the human microbiota.
While it was beyond the scope of this work, future directions should assess the colonization efficiency and fitness of the different microbial communities of human-donors on establishment across various mammal models. Nevertheless, this current work demonstrated that the different components of the human gut microbial community could be transferred to different recipient rodents with differing degrees of efficiency, providing further evidence that the genetic background of the recipient may strongly influence the nature of the populating human gut microbiota.
Material and Methods
Animals.
A total of 31 animals across different strains of mice and rats were used: GF C57BL/6JZtm mice, GF NMRI/MaxZtm mice, SPF C57BL/6J mice as well as GF WKY/Ztm (Wistar Kyoto) and GF Ztm:SPRD (Sprague-Dawley) rats (), across two independent but reproducibly designed experiments. Animals were either housed at the Helmholtz Centre for Infection Research (HZI) in Braunschweig, Germany in sterile IVC cages or at the Institute for Laboratory Animal Science and Central Animal Facility, Hannover Medical School (MHH), Hannover, Germany in sterile static micro-isolators (gnotocages) or sterile filter top cages placed in a sterilized laminar flow cabinet. Animals were fed an autoclaved low-fat polysaccharide-rich chow diet comprising 53–58% carbohydrates, 33–36% protein, 9–11% fat and 16.3–16.8 MJ/kg gross energy (V1124 or V1534, Ssniff Spezialdiäten), ad libitum (Table S7). This study was conducted in accordance with German law for animal protection and with the European Communities Council Directive 86/609/EEC for the protection of animals used for experimental purposes. All experiments were approved by the Local Institutional Animal Care and Research Advisory committee and permitted by the local government.
Antibiotic treatment of SPF C57BL/6 mice.
A group of SPF C57BL/6 mice were treated with ciprofloxacin orally via the drinking water ad libitum at a dose of 30 mg per kg body mass per day for four consecutive days prior to the inoculation of the human-donor fecal inoculum, as previously described in reference Citation56. Fresh drinking water replaced the ciprofloxacin-drinking water 3 h prior to the first administration of the human-donor inoculum. This same inoculum was repeatedly given a further 24, 48, 72 and 96 h after the arrest of antibiotic treatment to ensure that any effect of the remaining antibiotic on the establishing community was negligible. Antibiotic treatment initially reduced the mouse’s autochthonous gut microbial load by 1–2 orders of magnitude (data not shown).
Collection and treatment of the human-donor bacteria.
Fecal material was obtained from a healthy 32 y-of-age female volunteer who had not taken antibiotics for up to 12 mo prior to fecal deposition. For each experiment, 65 g of fresh stool was collected immediately upon defecation and suspended into sterile nitrogen-sparged PBS buffer, centrifuged, washed and re-suspended several times in the sterile nitrogen-sparged PBS buffer as previously described in reference Citation57. One millileter aliquots at a final bacterial concentration of 9 × 109 cells per ml, as determined by direct microscopic countsCitation58 were stored at 4°C in nitrogen-sparged PBS buffer until its first inoculation into the animals within 12 h. This same inoculum (stored at 4°C) was also used in subsequent inoculations in the following days (). At each time of inoculation, an aliquot was sampled for extraction of DNA (in both experiments) and RNA (in the second experiment).
Treatment with human-donor bacteria.
Mice in groups A, C and E () were given 100 ∝l (equating to 9 × 108 cells) of human-donor bacterial inoculum along with 100 ∝l 3% bicarbonate administered intragastrically via a gavage needle without sedation. This inoculum was administered to mice in groups A and C daily for five consecutive days and mice in groups E thrice (every second day). Mice in group B were treated as those of group A, but human-donor bacterial inoculum was replaced with sterile PBS. Rats (groups D, F and G) were treated as mice in group E, however using a 5-fold inoculum volume to adjust for the difference in intestinal surface area (and thus available colonizing space) between the rats and mice. This difference was calculated according to the Brody-Kleiber relationship equated as body weight3/4,Citation59 which assumes the relationship of mucosal intestinal surface area to body weight is a constant.Citation60 However, the surface area is not a uniform structure and numerous Peyer’s patches and follicles/microvilli also line the walls of the intestine, adding to the overall surface area and thus the overall numbers of potential colonization sites. Both rats and mice have similar mean numbers of follicles per cm-1 of their small intestine and colon, and Peyer’s patches in their small intestine, where only in the colon a slight difference in the number of Peyer’s patches per cm-1 has been reported in reference Citation61. It can thus be estimated that a 5-fold inoculum size to the rats results in the application of equivalent amounts of bacteria per available surface area of the GI tract corresponding to approximately 5–10 bacterial cells per ∝m2 of colonizable intestinal surface area of both rats and mice.
Sampling of animal feces and ceca.
Fecal samples were collected from the different animals after the first dose of the human-donor inoculum at days 0, 4, 7, 11, 18, 25, 32 and 41. At either day 32 (all animals housed at MHH) or day 41 (for animals housed at HZI), animals were sacrificed by CO2 inhalation and their cecal and fecal contents collected. All fecal and cecal samples (50–100 mg material) were immediately stored frozen at -70°C. Those samples for RNA extraction were supplemented with 1 ml of RNAprotectTM Bacterial Reagent (Qiagen), homogenized with a sterile pestal, incubated for 15 min at room temperature, pelleted, supernatant removed and pellets stored at -70°C.
Nucleic acid extraction—DNA extraction.
DNA was extracted using the FastDNA Spin Kit for Soil (MP Biomedicals) following manufacturers instructions. DNA was quantified using the NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA USA).
Nucleic acid extraction—RNA extraction and preparation of cDNA.
RNA was extracted using the RNeasy Mini Kit (Qiagen) from 5 × 108 bacterial cells of the fecal/cecal material. These cells were then placed into Lyzing Matrix B tubes (MP Biomedicals) containing 700 ∝l of buffer RLT (reconstituted with 7 ∝l ®-mercaptoethanol) and lyzed in a Fast Prep®-24 instrument (30 sec, intensity 5.5). Possibly contaminating DNA was eliminated by digesting with RNase-Free DNase (Qiagen). RNA was quantified using the NanoDrop 2000 spectrophotometer (Thermo Scientific). The 16S rRNA was converted to cDNA and amplified using OneStep RT-PCR kit (Qiagen) and the 5' end fluorescent-labeled primers FAM-63F and VIC-1389R (Applied Biosystems) (see T-RFLP section below). Reverse transcription PCR (50 ∝l) was performed at an annealing temperature of 55°C using 100 ng of template.
Terminal restriction fragment length polymorphism (T-RFLP).
For PCR amplification, the 5' end fluorescent-labeled 63F-FAM and 1389R-VIC primers targeting the 16S rRNA gene (Applied Biosystems) were applied (see Table S8). Duplicate 50 ∝l PCR reactions were performed at an annealing temperature of 55°C using 2.5 ng DNA as template. Duplicates for each sample were pooled and purified using the QIAquick individual spin columns PCR purification kit (Qiagen) or the Macherey-Nagel 96-well plate purification kit (Macherey-Nagel). Some PCR products were further purified using the Qiagen Gel extraction kit (Qiagen). Exo-Klenow fragment treatment was performed, where 10 U of exo-Klenow was incubated with 80 ng of amplicons in a volume of 40 ∝l (1 h, 20°C) followed by inactivation (20 min, 75°C). Digestion was then performed with 2.5 U of AluI (3 h, 37°C) followed by heat inactivation (20 min, 65°C) and purification through DyeEx gel-filters (Qiagen) with re-elution into 40 ∝l of water. Triplicate 10 ∝l aliquots containing 20 ng of digested fluorescent-labeled DNA fragments for each sample were dried, resuspended in 9.75 ∝l HiDi formamide (Applied Biosystems) and 0.25 ∝l of GeneScan 500 LIZ size standard (Applied Biosystems), and denatured (3 min, 95°C). DNA fragments were separated on an ABI 3130xl Genetic Analyzer (Applied Biosystems). A terminal restriction fragment (T-RF) was defined as any peak with a fluorescence area of greater than 0.1% of the total fluorescence within that profile. Any fragment of 35 bp in size or less was excluded. T-align (http://inismor.ucd.ie/~talign) was used to normalize total peak area across replicates and align peaks across samples using a ± 0.5 bp cut-off.Citation62 The T-RFLP profile generated using community DNA is herein referred to as the “global” bacterial community, while the T-RFLP profile generated using community RNA (converted to cDNA) is herein referred to as the “active” bacterial community. In total, 181 discrete T-RFs (peaks) were distinguishable across all experiments including those generated from both the active and global communities.
Real-time quantitative PCR of the dominant bacterial groups.
Real-time quantitative PCR (RT-qPCR), used to quantify the dominant bacterial groups-of-interest against the total bacterial population was performed on a LightCycler® 480 Real-Time PCR System with the LightCycler® 480 Software (Roche Applied Science). Group-specific 16S rDNA-targeted primers targeting eubacteria, members of the C. leptum subgroup, Bacteroides and Clostridia cluster XIVa (also typically referred to as the C. coccoides group) have been described and used previously (listed in Table S8).Citation32,Citation39,Citation63-Citation66 Primers were purchased from Eurofins MWG-Operon. Amplification and detection were performed in 96-well plates with the QuantiTect SYBR Green PCR kit (Qiagen). Each reaction was performed in duplicate in a final volume of 20 ∝l containing 2.5 ∝l of each primer (10 pmol), 10 ∝l QuantiTect SYBR Green PCR Master Mix and 5 ∝L of template DNA (diluted at 2.5 ng∝l-1), using annealing temperatures specified in Table S8. For SYBR-Green amplifications, a melting step was added to confirm amplification specificity. For generation of standard curves, 16S rDNA amplicons were produced from cecal samples using each of the specific primer-sets. Amplicons were cloned using the pGEM-T Easy PCR Cloning Kit (Promega) and transformed into E. coli JM109. The plasmid DNA containing the insert was extracted, linearized, purified and sequenced, confirming that plasmid DNA pertained to members representing Blautia wexlerae, Parabacteroides distasonis, Faecalibacterium prausnitzii and Coprococcus eutactus and was used as template for the standard curves in reactions targeting Clostridia cluster XIVa, Bacteroides group, C. leptum subgroup and eubacteria, respectively (Table S9). Standard curves were generated from serial dilutions of known concentrations of plasmid DNA.
454-pyrosequencing and phylogenetic analysis.
16S rRNA gene amplicons generated from the cecal contents of representatives from each of the four rodent groups in the first experiment (animals 3, 8, 12 and 17) and the fecal inoculum of the human-donor given on the first day of inoculation (sample II) were evaluated by pyrosequencing using the 454 platform (Roche Diagnostics Corporation) as previously described in reference Citation67 (see Table S8 for primers used).
Initial sequence processing, alignment, clustering and phylotype classification were performed using RDP’s pyrosequencing pipeline (http://pyro.cme.msu.edu).Citation33,Citation68,Citation69 In brief, all 454-pyrosequencing reads were sorted according to their respective sample, the barcode and primers trimmed from the sequence and any sequences of low quality or of less than 150 nucleotides removed. A total of 27,524 sequence reads with an average length of 235 nucleotides (3,018 assigned to the human-donor, 4,841 to the GF WKY recipient rat, 5,297 to the GF C57BL/6 recipient mouse, 4,986 to the antibiotic-treated SPF C57BL/6 recipient mouse and 4,113 to the SPF C57BL/6 non-recipient mouse) were aligned and clustered as previously described in reference Citation67, using the RDP complete linkage clustering tool at a distance cut-off of 0.02. Those clusters that comprised only one sequence (detected across all samples) were excluded from further analysis, leaving a total of 290 sequences representing 290 phylotypes (see Tables S2, S4 and S6). Phylotype sequences were deposited under the GenBank accession numbers HM368767–HM369056.
The sequences of the most closely related validly described type strains and environmental isolates were subsequently aligned with 128 phylotypes assigned to the human-donor sample (Table S2) using MUSCLE.Citation70 A neighbor-joining tree of the human-donor phylotypes and nearest neighbors was constructed using MEGA4,Citation71 with evolutionary distances determined from pairwise dissimilarities using the Jukes-Cantor correction model.Citation72
Statistical analysis.
Non-parametric multivariate statistical analysis was performed using PRIMER (v.6.1.6, PRIMER-E, Plymouth Marine Laboratory).Citation73,Citation74 All multivariate routines were computed on either standardized abundance data or presence/absence data. That is, in all three profiling methods (T-RFLP, RT-qPCR and 454-pyrosequencing) the abundance of bacterial species or bacterial groups was calculated as a percent of their contribution to the total bacterial abundance. From the multivariate data matrix of either T-RFLP, RT-qPCR or pyrosequencing data, a sample-similarity matrix was generated using the Bray-Curtis similarity coefficient by comparing either the abundances of: each T-RF generated from T-RFLP; each specific bacterial group as determined by RT-qPCR; or each bacterial phylotype using pyrosequencing, in regards to every pairwise combination of all samples. Community structures were explored by ordination using nonmetric multidimensional scaling (nMDS) (50 random restarts). Differences in phylotype dominance and diversity of five representative samples used for pyrosequencing were further explored using k-dominance plots (cumulative ranked abundances plotted against species rank). Analysis of similarity (ANOSIM) was used to test for statistically significant differences in bacterial communities between predefined groups using 999 permutations. The accompanying R statistic measures the degree of separation between groups and ranges from -1 to 1, in which the higher its value (closer to 1) the more distinct the groups. Also, no formal mathematical correction was made for multiple comparisons, as only a few scientific sensible planned comparisons were made and reported (rather than every possible pairwise comparison).
| Abbreviations: ANOSIM | = | analysis of similarity |
| C57BL/6 | = | black 6 mouse (C57BL/6JZtm) |
| F/B | = | Firmicutes/Bacteriodetes |
| GF | = | germ-free |
| GI | = | gastrointestinal |
| HZI | = | Helmholtz Centre for Infection Research |
| IBD | = | inflammatory bowel disease |
| IVC | = | individually ventilated cages |
| MHH | = | Hannover Medical School |
| nMDS | = | non-metric multidimensional scaling |
| NMRI | = | a Swiss mouse named after the US Naval Medical Research Institute (NMRI/MaxZtm) |
| PT | = | phylotype |
| RDP | = | Ribosomal Database Project |
| RT-qPCR | = | real-time quantitative polymerase chain reaction |
| SEM | = | standard error of the mean |
| SPF | = | specific pathogen-free |
| SPRD | = | Sprague-Dawley rat (Ztm:SPRD) |
| T-RFLP | = | terminal restriction fragment length polymorphism |
| T-RF | = | terminal restriction fragment |
| WKY | = | Wistar Kyoto rat (WKY/Ztm) |
Additional material
Download Zip (8.8 MB)Acknowledgments
We kindly thank Dr H. Riedesel, K. Kränzler and N. Schukowski for support relating to animal work performed at the HZI. We acknowledge I. Plumeier, A. Krüger, M. Blank, C. Luge, S. Edelmann and M. Schulte for technical support. This work was partly funded by DFG SFB621 (A.B. and H.J.H.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental materials can be found here: www.landesbioscience.com/journals/gutmicrobes/article/19934/
References
- Li P, Hotamisligil GS. Metabolism: Host and microbes in a pickle. Nature 2010; 464:1287 - 8; http://dx.doi.org/10.1038/4641287a; PMID: 20428156
- Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J, et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci U S A 2010; 107:18933 - 8; http://dx.doi.org/10.1073/pnas.1007028107; PMID: 20937875
- Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 2010; 328:228 - 31; http://dx.doi.org/10.1126/science.1179721; PMID: 20203013
- Tamboli CP, Neut C, Desreumaux P, Colombel JF. Dysbiosis in inflammatory bowel disease. Gut 2004; 53:1 - 4; http://dx.doi.org/10.1136/gut.53.1.1; PMID: 14684564
- Matsuda H, Fujiyama Y, Andoh A, Ushijima T, Kajinami T, Bamba T. Characterization of antibody responses against rectal mucosa-associated bacterial flora in patients with ulcerative colitis. J Gastroenterol Hepatol 2000; 15:61 - 8; http://dx.doi.org/10.1046/j.1440-1746.2000.02045.x; PMID: 10719749
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature 2006; 444:1022 - 3; http://dx.doi.org/10.1038/4441022a; PMID: 17183309
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006; 444:1027 - 31; http://dx.doi.org/10.1038/nature05414; PMID: 17183312
- Wang M, Karlsson C, Olsson C, Adlerberth I, Wold AE, Strachan DP, et al. Reduced diversity in the early fecal microbiota of infants with atopic eczema. J Allergy Clin Immunol 2008; 121:129 - 34; http://dx.doi.org/10.1016/j.jaci.2007.09.011; PMID: 18028995
- Verhulst SL, Vael C, Beunckens C, Nelen V, Goossens H, Desager K. A longitudinal analysis on the association between antibiotic use, intestinal microflora, and wheezing during the first year of life. J Asthma 2008; 45:828 - 32; http://dx.doi.org/10.1080/02770900802339734; PMID: 18972304
- Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 2008; 455:1109 - 13; http://dx.doi.org/10.1038/nature07336; PMID: 18806780
- Brugman S, Klatter FA, Visser JT, Wildeboer-Veloo AC, Harmsen HJ, Rozing J, et al. Antibiotic treatment partially protects against type 1 diabetes in the Bio-Breeding diabetes-prone rat. Is the gut flora involved in the development of type 1 diabetes?. Diabetologia 2006; 49:2105 - 8; http://dx.doi.org/10.1007/s00125-006-0334-0; PMID: 16816951
- Marteau P, Pochart P, Doré J, Béra-Maillet C, Bernalier A, Corthier G. Comparative study of bacterial groups within the human cecal and fecal microbiota. Appl Environ Microbiol 2001; 67:4939 - 42; http://dx.doi.org/10.1128/AEM.67.10.4939-4942.2001; PMID: 11571208
- Pang X, Hua X, Yang Q, Ding D, Che C, Cui L, et al. Inter-species transplantation of gut microbiota from human to pigs. ISME J 2007; 1:156 - 62; http://dx.doi.org/10.1038/ismej.2007.23; PMID: 18043625
- Becker N, Kunath J, Loh G, Blaut M. Human intestinal microbiota: characterization of a simplified and stable gnotobiotic rat model. Gut Microbes 2011; 2:25 - 33; http://dx.doi.org/10.4161/gmic.2.1.14651; PMID: 21637015
- Van den Abbeele P, Grootaert C, Marzorati M, Possemiers S, Verstraete W, Gérard P, et al. Microbial community development in a dynamic gut model is reproducible, colon region specific, and selective for Bacteroidetes and Clostridium cluster IX. Appl Environ Microbiol 2010; 76:5237 - 46; http://dx.doi.org/10.1128/AEM.00759-10; PMID: 20562281
- Rawls JF, Mahowald MA, Ley RE, Gordon JI. Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell 2006; 127:423 - 33; http://dx.doi.org/10.1016/j.cell.2006.08.043; PMID: 17055441
- Hirayama K, Itoh K. Human flora-associated (HFA) animals as a model for studying the role of intestinal flora in human health and disease. Curr Issues Intest Microbiol 2005; 6:69 - 75; PMID: 16107039
- Barc MC, Charrin-Sarnel C, Rochet V, Bourlioux F, Sandré C, Boureau H, et al. Molecular analysis of the digestive microbiota in a gnotobiotic mouse model during antibiotic treatment: Influence of Saccharomyces boulardii.. Anaerobe 2008; 14:229 - 33; http://dx.doi.org/10.1016/j.anaerobe.2008.04.003; PMID: 18511310
- Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 2009; 1:ra14; http://dx.doi.org/10.1126/scitranslmed.3000322; PMID: 20368178
- Alpert C, Sczesny S, Gruhl B, Blaut M. Long-term stability of the human gut microbiota in two different rat strains. Curr Issues Mol Biol 2008; 10:17 - 24; PMID: 18525103
- Licht TR, Madsen B, Wilcks A. Selection of bacteria originating from a human intestinal microbiota in the gut of previously germ-free rats. FEMS Microbiol Lett 2007; 277:205 - 9; http://dx.doi.org/10.1111/j.1574-6968.2007.00962.x; PMID: 18031341
- Rehman A, Sina C, Gavrilova O, Häsler R, Ott S, Baines JF, et al. Nod2 is essential for temporal development of intestinal microbial communities. Gut 2011; 60:1354 - 62; http://dx.doi.org/10.1136/gut.2010.216259; PMID: 21421666
- Tlaskalová-Hogenová H, Stěpánková R, Kozáková H, Hudcovic T, Vannucci L, Tučková L, et al. The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: contribution of germ-free and gnotobiotic animal models of human diseases. Cell Mol Immunol 2011; 8:110 - 20; http://dx.doi.org/10.1038/cmi.2010.67; PMID: 21278760
- Manichanh C, Reeder J, Gibert P, Varela E, Llopis M, Antolin M, et al. Reshaping the gut microbiome with bacterial transplantation and antibiotic intake. Genome Res 2010; 20:1411 - 9; http://dx.doi.org/10.1101/gr.107987.110; PMID: 20736229
- Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, Wollam A, et al. Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc Natl Acad Sci U S A 2009; 106:5859 - 64; http://dx.doi.org/10.1073/pnas.0901529106; PMID: 19321416
- Gootenberg DB, Turnbaugh PJ. Companion animals symposium: humanized animal models of the microbiome. J Anim Sci 2011; 89:1531 - 7; http://dx.doi.org/10.2527/jas.2010-3371; PMID: 20833767
- Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A 2005; 102:11070 - 5; http://dx.doi.org/10.1073/pnas.0504978102; PMID: 16033867
- Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A 2004; 101:15718 - 23; http://dx.doi.org/10.1073/pnas.0407076101; PMID: 15505215
- Silley P. Human flora-associated rodents--does the data support the assumptions?. Microb Biotechnol 2009; 2:6 - 14; http://dx.doi.org/10.1111/j.1751-7915.2008.00069.x; PMID: 21261878
- Kibe R, Sakamoto M, Yokota H, Ishikawa H, Aiba Y, Koga Y, et al. Movement and fixation of intestinal microbiota after administration of human feces to germfree mice. Appl Environ Microbiol 2005; 71:3171 - 8; http://dx.doi.org/10.1128/AEM.71.6.3171-3178.2005; PMID: 15933018
- Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, et al. Evolution of mammals and their gut microbes. Science 2008; 320:1647 - 51; http://dx.doi.org/10.1126/science.1155725; PMID: 18497261
- Mariat D, Firmesse O, Levenez F, Guimarăes VD, Sokol H, Doré J, et al. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol 2009; 9:123 - 9; http://dx.doi.org/10.1186/1471-2180-9-123; PMID: 19508720
- Cole JR, Chai B, Farris RJ, Wang Q, Kulam SA, McGarrell DM, et al. The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res 2005; 33:Database issue D294 - 6; http://dx.doi.org/10.1093/nar/gki038; PMID: 15608200
- Song Y, Könönen E, Rautio M, Liu C, Bryk A, Eerola E, et al. Alistipes onderdonkii sp. nov. and Alistipes shahii sp. nov., of human origin. Int J Syst Evol Microbiol 2006; 56:1985 - 90; http://dx.doi.org/10.1099/ijs.0.64318-0; PMID: 16902041
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science 2005; 308:1635 - 8; http://dx.doi.org/10.1126/science.1110591; PMID: 15831718
- Zoetendal EG, Rajilic-Stojanovic M, de Vos WM. High-throughput diversity and functionality analysis of the gastrointestinal tract microbiota. Gut 2008; 57:1605 - 15; http://dx.doi.org/10.1136/gut.2007.133603; PMID: 18941009
- Flint HJ. The significance of prokaryote diversity in the human gastrointestinal tract. In: Logan N, Lappin-Scott H, Oyston P, Eds. Prokaryotic diversity: Mechanisms and significance SGM symposium. Cambridge, UK: Cambridge University Press 2006; 65-90.
- Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al, MetaHIT Consortium. Enterotypes of the human gut microbiome. Nature 2011; 473:174 - 80; http://dx.doi.org/10.1038/nature09944; PMID: 21508958
- Sokol H, Seksik P, Furet JP, Firmesse O, Nion-Larmurier I, Beaugerie L, et al. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis 2009; 15:1183 - 9; http://dx.doi.org/10.1002/ibd.20903; PMID: 19235886
- Zoetendal EG, Vaughan EE, de Vos WM. A microbial world within us. Mol Microbiol 2006; 59:1639 - 50; http://dx.doi.org/10.1111/j.1365-2958.2006.05056.x; PMID: 16553872
- Louis P, Young P, Holtrop G, Flint HJ. Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA:acetate CoA-transferase gene. Environ Microbiol 2010; 12:304 - 14; http://dx.doi.org/10.1111/j.1462-2920.2009.02066.x; PMID: 19807780
- Stackebrandt E, Kramer I, Swiderski J, Hippe H. Phylogenetic basis for a taxonomic dissection of the genus Clostridium. FEMS Immunol Med Microbiol 1999; 24:253 - 8; http://dx.doi.org/10.1111/j.1574-695X.1999.tb01291.x; PMID: 10397308
- Maukonen J, Mättö J, Satokari R, Söderlund H, Mattila-Sandholm T, Saarela M. PCR DGGE and RT-PCR DGGE show diversity and short-term temporal stability in the Clostridium coccoides-Eubacterium rectale group in the human intestinal microbiota. FEMS Microbiol Ecol 2006; 58:517 - 28; http://dx.doi.org/10.1111/j.1574-6941.2006.00179.x; PMID: 17117993
- Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 2006; 124:837 - 48; http://dx.doi.org/10.1016/j.cell.2006.02.017; PMID: 16497592
- Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006; 55:205 - 11; http://dx.doi.org/10.1136/gut.2005.073817; PMID: 16188921
- Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A 2011; 108:Suppl 1 4554 - 61; http://dx.doi.org/10.1073/pnas.1000087107; PMID: 20847294
- Tlaskalová-Hogenová H, Stepánková R, Hudcovic T, Tucková L, Cukrowska B, Lodinová-Zádníková R, et al. Commensal bacteria (normal microflora), mucosal immunity and chronic inflammatory and autoimmune diseases. Immunol Lett 2004; 93:97 - 108; http://dx.doi.org/10.1016/j.imlet.2004.02.005; PMID: 15158604
- Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol 2009; 9:799 - 809; http://dx.doi.org/10.1038/nri2653; PMID: 19855405
- Khachatryan ZA, Ktsoyan ZA, Manukyan GP, Kelly D, Ghazaryan KA, Aminov RI. Predominant role of host genetics in controlling the composition of gut microbiota. PLoS One 2008; 3:e3064; http://dx.doi.org/10.1371/journal.pone.0003064; PMID: 18725973
- Blum S, Schiffrin EJ. Intestinal microflora and homeostasis of the mucosal immune response: implications for probiotic bacteria?. Curr Issues Intest Microbiol 2003; 4:53 - 60; PMID: 14503689
- Hecht G. Innate mechanisms of epithelial host defense: spotlight on intestine. Am J Physiol 1999; 277:C351 - 8; PMID: 10484321
- Peppard JV, Kaetzel CS, Russell MW. Phylogeny and comparative physiology of IgA. In: Mestecky J, Lamm ME, Strober W, Bienenstock J, McGhee JR, Mayer L, Eds. Mucosal Immunology. London, UK: Elsevier Academic Press 2005.
- Ganz T. Defensins in the urinary tract and other tissues. J Infect Dis 2001; 183:Suppl 1 S41 - 2; http://dx.doi.org/10.1086/318838; PMID: 11171012
- Weiss A. The Genus Bordetella. In: Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E, Eds. The Prokaryotes—A handbook on the biology of bacteria. Singapore: Springer Science+Business Media 2006; 648-74.
- Dethlefsen L, McFall-Ngai M, Relman DA. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 2007; 449:811 - 8; http://dx.doi.org/10.1038/nature06245; PMID: 17943117
- Johnson SA, Nicolson SW, Jackson S. The effect of different oral antibiotics on the gastrointestinal microflora of a wild rodent (Aethomys namaquensis). Comp Biochem Physiol A Mol Integr Physiol 2004; 138:475 - 83; http://dx.doi.org/10.1016/j.cbpb.2004.06.010; PMID: 15369837
- Wang RF, Cao WW, Cerniglia CE. PCR detection and quantitation of predominant anaerobic bacteria in human and animal fecal samples. Appl Environ Microbiol 1996; 62:1242 - 7; PMID: 8919784
- Kepner RLJ Jr., Pratt JR. Use of fluorochromes for direct enumeration of total bacteria in environmental samples: past and present. Microbiol Rev 1994; 58:603 - 15; PMID: 7854248
- Kleiber M. The fire of life. New York, USA: John Wiley and Sons Inc. 1961.
- Snipes R. Intestinal absorptive surface in mammals of different sizes. Heidelberg: Springer-Verlag Berlin 1997.
- McConnell EL, Basit AW, Murdan S. Measurements of rat and mouse gastrointestinal pH, fluid and lymphoid tissue, and implications for in-vivo experiments. J Pharm Pharmacol 2008; 60:63 - 70; http://dx.doi.org/10.1211/jpp.60.1.0008; PMID: 18088506
- Smith CJ, Danilowicz BS, Clear AK, Costello FJ, Wilson B, Meijer WG. T-Align, a web-based tool for comparison of multiple terminal restriction fragment length polymorphism profiles. FEMS Microbiol Ecol 2005; 54:375 - 80; http://dx.doi.org/10.1016/j.femsec.2005.05.002; PMID: 16332335
- Suzuki MT, Taylor LT, DeLong EF. Quantitative analysis of small-subunit rRNA genes in mixed microbial populations via 5′-nuclease assays. Appl Environ Microbiol 2000; 66:4605 - 14; http://dx.doi.org/10.1128/AEM.66.11.4605-4614.2000; PMID: 11055900
- Matsuki T, Watanabe K, Fujimoto J, Takada T, Tanaka R. Use of 16S rRNA gene-targeted group-specific primers for real-time PCR analysis of predominant bacteria in human feces. Appl Environ Microbiol 2004; 70:7220 - 8; http://dx.doi.org/10.1128/AEM.70.12.7220-7228.2004; PMID: 15574920
- Barman M, Unold D, Shifley K, Amir E, Hung K, Bos N, et al. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect Immun 2008; 76:907 - 15; http://dx.doi.org/10.1128/IAI.01432-07; PMID: 18160481
- Franks AH, Harmsen HJ, Raangs GC, Jansen GJ, Schut F, Welling GW. Variations of bacterial populations in human feces measured by fluorescent in situ hybridization with group-specific 16S rRNA-targeted oligonucleotide probes. Appl Environ Microbiol 1998; 64:3336 - 45; PMID: 9726880
- Wos-Oxley ML, Plumeier I, von Eiff C, Taudien S, Platzer M, Vilchez-Vargas R, et al. A poke into the diversity and associations within human anterior nare microbial communities. ISME J 2010; 4:839 - 51; http://dx.doi.org/10.1038/ismej.2010.15; PMID: 20182526
- Cole JR, Chai B, Farris RJ, Wang Q, Kulam-Syed-Mohideen AS, McGarrell DM, et al. The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res 2007; 35:Database issue D169 - 72; http://dx.doi.org/10.1093/nar/gkl889; PMID: 17090583
- Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 2009; 37:Database issue D141 - 5; http://dx.doi.org/10.1093/nar/gkn879; PMID: 19004872
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004; 32:1792 - 7; http://dx.doi.org/10.1093/nar/gkh340; PMID: 15034147
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 2007; 24:1596 - 9; http://dx.doi.org/10.1093/molbev/msm092; PMID: 17488738
- Jukes TH, Cantor CR. Evolution of protein molecules. In: Munro H, ed. Mammalian Protein Metabolism. New York, USA: Academic Press 1969; 21-132.
- Clarke KR. Non-parametric multivariate analyses of changes in community structure. Aust J Ecol 1993; 18:117 - 43; http://dx.doi.org/10.1111/j.1442-9993.1993.tb00438.x
- Clarke K, Warwick R. Change in marine communities: An approach to statistical analysis and interpretation. Plymouth, UK: PRIMER-E 2001.