Abstract
Helicobacter pylori is the primary cause of peptic ulcer disease and is estimated to account for about 60% of all cases of gastric cancer, the second most common cause of cancer death worldwide. Among the H. pylori virulence factors associated with disease, in addition to the well-known cag pathogenicity island, is the BabA adhesin, an outer membrane protein that binds with high affinity to fucosylated glycans on the gastric epithelium, such as Lewis B (Leb) and related terminal fucose residues found on the blood group O (H antigen), A and B antigens. BabA-mediated attachment to the gastric mucosa promotes chronic inflammation and gastric pathology, which from the bacterial perspective carries both risks and benefits. We recently described modulation in expression of BabA and related outer membrane proteins that occurs during colonization of experimental animals. Here we put these findings into a broader context, and speculate on their implications for the host-pathogen relationship.
The Ecology of H. pylori
H. pylori inhabits the 100 µm mucus layer that coats the gastric epithelium, particularly the 25 µm closest to the gastric epithelial cells, to which it is sometimes intimately attached near tight junctions.Citation3,Citation4 Colonization of the gastric mucosa is not without obstacles: the high viscosity of the mucus layer, low pH of the stomach lumen, shedding of mucin and epithelial cells, and relative lack of nutrients all pose strong challenges for the bacterium to reach and colonize its niche. Moreover, although ineffective at clearing the infection, the host mounts an immune response that delivers inflammatory mediators across the gastric epithelium. These features of the gastric mucosa are what might be called the “physicochemical facts of life” for H. pylori, and they vary over time, space and even within the microenvironment of the mucus layer. H. pylori must adapt to these challenges in order to achieve long-term colonization in this dynamic environment.
pH.
The low pH of the stomach lumen is generally thought to be inhibitory to bacterial growth. H. pylori has a unique capacity to survive and grow in acid that is largely mediated by large quantities of urease, a urea channel and carbonic anhydrase, resulting in the production of ammonia and bicarbonate. Both urease and carbonic anhydrase are regulated by the acid-responsive ArsRS two-component signal transduction system.Citation5 The elevation in local pH also decreases the viscosity of the mucus and permits H. pylori to swim toward the epithelial layer.Citation6 Surprisingly, the pH at the gastric surface is controversial. Microelectrode studies suggest that there is a pH gradient from around 1.5–2.0 in the gastric lumen to near neutral at the epithelial surface, which is maintained by secretion of bicarbonate ions by gastric surface mucus cells.Citation7 H. pylori itself has also been used as a bioassay to measure pH at the gastric surface, by comparing gene expression profiles under varying pH conditions to that in vivo. These studies suggest that the pH at the gastric surface is acidic, with a pH ≤ 4.0.Citation8 In any case, it is likely that there is a mucus pH gradient, and that H. pylori can alter its local environmental pH by regulation of adhesins and perhaps other molecules that determine its precise position within the mucus layer.
Mucins.
Mucins such as MUC5AC and MUC1 are large glycoproteins that are the major structural component of the gastric mucus gel layer. The glycans that decorate mucins serve as receptors for H. pylori adhesins, such as BabA, which attaches to Lewis B (Leb) and related terminal fucose residues, SabA, which attaches to sialyl- Lewis X (sLex), and no doubt others. At first glance this seems simple enough: H. pylori has evolved adhesins that mediate attachment to carbohydrates that are abundantly expressed on the gastric epithelium and in the mucus layer. However, glycans that decorate mucin proteins are variable from person to person, depending on expression of particular glycosyl and fucosyl transferases. The glycans also vary in different locations in the stomach and change within an individual, from predominantly fucosylation, such as Leb, to inflammation-associated sialylation such as sLex and sLea as a result of H. pylori-induced gastritis.Citation9–Citation11 H. pylori may directly influence this progression by inducing epithelial cells to express β3GnT5, a GlcNAc transferase essential for the biosynthesis of Lewis antigens.Citation12 Mucins may also have complex interactions with H. pylori. For example, mice with deletions of the cellassociated mucin MUC1 have increased H. pylori colonization and increased inflammation compared to wild type mice,Citation13 which would be unexpected if MUC1 serves simply as a receptor. It appears that MUC1 limits H. pylori colonization both by steric hindrance and acting as a decoy that is released when the H. pylori-bound extracellular domain is cleaved,Citation14 and also by blocking NFκB activation that inhibits downstream inflammatory responses.Citation15
Nutrients.
Early theoretical considerations first suggested that adherent H. pylori induces an inflammatory response that provokes tissue damage and nutrient release, which may be required to sustain bacterial growth.Citation16 Experimental evidence has elucidated some of the details. H. pylori targets epithelial tight junctions, where it injects the CagA oncoprotein via a type IV secretion system encoded on the cag pathogenicity island (cagPAI). CagA associates with the epithelial tight-junction scaffolding protein ZO-1 and the transmembrane protein junctional adhesion molecule, causing an ectopic assembly of tight-junction components at sites of bacterial attachment, and altering the composition and function of the apical-junctional complex. 3 Successful colonization of this niche appears to be dependent on the expression of the virulence factor, CagA.Citation17 In the absence of nutrients necessary for H. pylori growth, wild type but not CagA deletion mutants were able to grow on the apical side of polarized epithelial cells. Since the mutant phenotype could not be rescued by the presence of wild type H. pylori cells, the role of CagA in promoting colonization appears highly localized, and does not result simply from disruption of the integrity of the epithelial barrier. However, growth of a CagA knockout was rescued by inhibition of the Par1b pathway, which plays an important role in the establishment and maintenance of cell polarity. Recent structural studies indicate that a subdomain of CagA binds to the substrate binding domain of the PAR1b/MARK2 kinase and inhibits its activity.Citation18 Together these results suggest that attachment and injection of CagA allow H. pylori to obtain nutrients directly from the epithelial cells by mimicking eukaryotic kinase inhibitors and altering cell polarity.
Inflammation.
The hallmark of H. pylori infection is chronic inflammation (gastritis), which consists largely of polymorphonuclear leukocytes (PMNs), plasma cells and Th1/Th17-biased CD4+ T cells that secrete interferon gamma (IFNγ), interleukin-17 and other proinflammatory cytokines. The inflammatory host response is especially robust after infection with H. pylori bearing the cag-PAI encoding the Type IV secretion system,Citation19 which is found more commonly in strains that cause clinical disease rather than simply asymptomatic gastritis.Citation20 The type IV secretion system binds β1 integrinsCitation21,Citation22 and injects CagA into epithelial cells where it disrupts host-cell signaling. The type IV secretion system also mediates translocation of peptidoglycan into host cells, where it activates nucleotide-binding oligomerization domain 1 (NOD1), NFκB and perhaps also type I interferons.Citation23,Citation24 The end result is the release of reactive oxygen and nitric oxide species that can be lethal to bacteria. In response, H. pylori mounts an antiinflammatory response to the host, such as induction of CD4+CD25+ regulatory T cellsCitation25 and inhibition of T cell proliferation by the vacuolating cytotoxin, VacA,Citation26 to name just two examples.
On the other hand, inflammation may actually be necessary for H. pylori colonization, so we should not be surprised to find experiments that appear to give conflicting impressions about the role of inflammation from the bacterial perspective. For example, IL-17-/- mice and mice treated with anti-IL-17 show less inflammation and reduced H. pylori colonization, while administration of recombinant adenovirus encoding mouse IL-17 produces the opposite effect.Citation27 These results suggest that inflammation may be good for the bacterium. On the other hand, infection of neonatal mice produces Treg-mediated immune tolerance that is characterized by less inflammation and higher bacterial load,Citation28 which would seem to suggest that inflammation is bad for the bacterium. Our interpretation of these seemingly conflicting results is that inflammation is neither bad nor good for H. pylori, but rather that its optimization is critical for persistent infection.
The Attachment Dilemma
Thus, H. pylori faces what might be called the attachment dilemma (). On the one hand, planktonic colonization in the outer portion of the mucus layer (near the gastric lumen) risks frequent exposure to unacceptably low pH and poor nutrient availability that cannot sustain long-term colonization. In the inner part of the mucus layer (near the epithelium) H. pylori risks clearance by natural shear forces generated by peristalsis. Bacterial attachment to the epithelium, injection of the effector protein CagA and interference with cell signaling may surmount these challenges. However, this comes at the cost of encountering toxic reactive oxygen species and being shed into the lumen as the gastric epithelium turns over, which is thought to occur, on average, twice weekly, but in the setting of injury can be much more frequent. Thus, attachment to glycans such as membrane bound glycolipids, cell anchored glycoproteins or gastric mucins brings many costs, especially in the mucus gel layer that is renewed several times a day by natural turnover and by release of membrane bound mucins that seem to function as decoys.Citation14 The bacteria must maintain a careful balance between replication in a nutritive and stable niche within the mucus layer and exposure to host defenses. Careful micropipette measurement of explanted gastric mucosa from experimentally infected gerbils suggests that H. pylori is predominantly localized to the 25 µm of the mucus layer that lies closest to the epithelium,Citation4 though we know from other studies that occasional cells attach intimately to pedestals similar to those induced by enteropathogenic Escherichia coli. A small portion of the population may even become internalized in epithelial or immune cells,Citation29,Citation30 which may be a mechanism by which H. pylori can avoid host immunity and recover after antibiotic treatment. Given the importance of the spatial localization of H. pylori, it seems likely that attachment is tightly and carefully regulated by modulation of surface adhesins.
Regulation of Attachment in H. Pylori
Approximately 4% of the H. pylori genome encodes a diverse repertoire of OMPs, the largest of which is the 21-gene Hop (H. pylori outer protein) family.Citation31 Hops are identified by a C-terminal membrane-spanning β-sheet motif, a conserved N-terminal signal peptide sequence and a variable mid-region. BabA is a Hop protein that binds Leb and related terminal fucose residues found on blood group O (H antigen), A and B antigens expressed on mucins and gastric epithelial cells.Citation32–Citation34 Not all H. pylori strains express babA, which can be silent owing to a truncated signal peptide or a single base pair mutation that results in a stop codon.Citation34 Moreover, some strains that express BabA do not bind Leb or the common ABO antigens. These strains might have mutated BabA into a non-binding, albeit expressed protein, or they may bind to some other as yet unknown oligosaccharides that are distinct from the Leb and ABO antigens.Citation35
Expression of BabA is clinically important because strains that carry a functional babA are considered more virulent since they are more commonly found in patients with duodenal ulcer and gastric adenocarcinoma than in patients with asymptomatic gastritis.Citation36 This is consistent with the observation that peptic ulcer is more common among blood group O individuals, in whom H and Leb are the predominant blood group antigens expressed on the gastric epithelium.Citation37 On the other hand, peptic ulcer has also been found to be more common in non-secretors,Citation38 who lack the α1,2-fucose typical for the ABO blood group antigens, and instead express the shorter Lea antigen that H. pylori does not bind. At first glance this seems to contradict the importance of BabA-mediated binding in the development of peptic ulcer. However, nonsecretors do have small amounts of Leb on their gastric cell surface, but they lack α1,2-fucosylated secreted glycans present in the gastric mucus that might otherwise compete with the fucosylated cell surfaces for binding. Thus, lack of fucosylated “decoys” could be a key to understanding the higher prevalence of gastric disease among non-secretors.
Sequence analysis suggests that among the genes encoding BabA and other Hop family proteins there is considerable potential for both antigenic variation, in which combinatorial DNA shuffling creates antigenically distinct proteins and phase variation, in which there is reversible on/off switching of gene expression. For example, there is extensive 5′ and 3′ homology between babA and two other H. pylori genes encoding OMPs of unknown function, called BabB and BabC. The extensive homology between babA and babB can select for recombination events that promote activation of silent babA, and hence reconstituted expression of Leb binding.Citation39 The greater similarity among bab paralogues (within a genome) than among orthologues (across genomes) suggests that there is frequent recombination and concerted evolution among these genes.Citation40,Citation41 In some strains regions of dinucleotide CT repeats in the 5′ coding region of babA, babB and other Hop genes may promote phase variation by slippedstrand mispairing during DNA replication. Finally, polynucleotide (A or T) tracts in babA and other Hop promoters might alter expression by subtle changes in the spacing between-35 and -10 hexamers.
We previously found that H. pylori strains recovered from experimentally challenged rhesus macaques had lost expression of BabA.Citation1 In some cases there was a gene conversion event in which the babA gene was replaced by babB, while in other cases the babA gene was present but was not expressed because of phase variation. Absence of babA and duplication of babB was also seen in about 15% of H. pylori isolates derived from human clinical samples, suggesting that this recombination event is not unique to experimentally infected monkeys.Citation42 More recently we sought to test the generality of this observation by comparison of different H. pylori strains and different animal hosts.Citation2 Again we observed loss of BabA expression and Leb attachment in H. pylori strains recovered 2–3 months after challenge of monkeys, and also in mice and gerbils, which are the two most commonly used animal models of H. pylori infection. In mice, strains lost attachment to Leb and expression of BabA by phase variation. In the gerbil, Leb binding was lost by replacement of babA with a second babA allele that differed at six amino acid residues and encodes a BabA that does not bind Leb. What is the function of this babA paralogue (or for that matter babB) that does not bind Leb? Since the babA paralogue was already present in the human isolate used to infect gerbils, and so had presumably been adapted to its human host, it may confer an alternative binding activity that is sometimes present in the human but not in the gerbil. For example, the six amino acid changes in BabA might reflect adaptation to changes in glycosylation induced by H. pylori-associated gastric inflammation.Citation9,Citation10 Alternatively, the variant babA might serve to inactivate Leb binding by gene conversion when conditions arise under which attachment is disadvantageous, perhaps leaving a small population of “memory” cells that can expand and reconstitute expression of Leb binding BabA when conditions change. Finally, a non-binding BabA may maintain its location and complex partners in the bacterial outer membrane, which may contribute to enhanced membrane stability. Thus, BabA expression in vivo is highly dynamic, and there are robust pressures that in some cases can act early in infection to select for strains with reduced expression or altered Leb binding activity.
What might be the selective pressures that modulate expression of BabA, BabB and perhaps other H. pylori OMPs? Since BabA is an important surface protein that mediates adherence, strains that express it might be selected against by the host adaptive immune response. However, we found that frequency of BabA expression was markedly reduced in both WT and RAG2-/- mice, which do not have functional B or T cells, suggesting that evasion of adaptive immunity is not sufficient to explain loss of BabA expression in mice (unpublished results). These findings are also consistent with lack of immune responses to BabA in rhesus monkeysCitation1 and in humans.Citation43,Citation44 Interestingly, infection of C3H/HeJ mice did not result in loss of BabA expression.Citation2 Since these mice express a defective TLR4, it is possible that innate immune responses in part select against expression of BabA. Modulation in the expression of BabA and other proteins involved in adherence may be one mechanism by which H. pylori regulates exposure to the inflammatory response in the gastric mucosa, a concept that is also supported by mathematical modeling.Citation45 In addition, both nutrient availability and local pH, which are higher near the epithelial surfaces than near the acidic gastric lumen, might also regulate expression of BabA. The presence of gene duplications and dinucleotide repeats in other genes within the Hop family46 suggests that gene conversion and slipped strand mispairing are probably general mechanisms utilized by H. pylori to modulate expression of surface proteins and adapt to changing environmental conditions within the host.
Conclusion
Modulation of bacterial adhesion in H. pylori may be a strategy essential for life at the margins to regulate exposure to physicochemical properties in the gastric niche, particularly the host inflammatory response.
Abbreviations
| BabA | = | blood group antigen binding adhesin A |
| CagA | = | cytotoxin associated gene A |
| cagPAI | = | cytotoxin associated gene pathogenicity island |
| Leb | = | lewis B |
| Lex | = | lewis X |
| sLex | = | sialyl lewis X |
| Lea | = | lewis A |
| sLea | = | sialyl lewis A |
| OMP | = | outer membrane protein |
| MUC5AC | = | mucin 5AC |
| MUC1 | = | mucin 1 |
| Hop | = | Helicobacter outer protein |
Figures and Tables
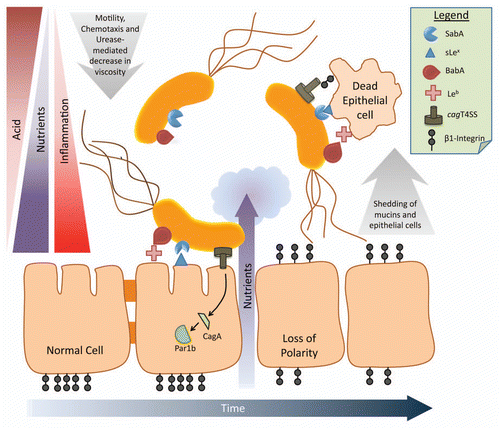
Figure 1 Schematic diagram of the ecology of H. pylori. As H. pylori moves through the mucus layer toward the epithelial surface, it encounters a gradient of decreased acid and increased nutrients, which promote viability and bacterial cell growth. A proportion of bacterial cells attach to the epithelium via BabA, SabA and probably other adhesins that bind their cognate glycan receptors. Attachment to the epithelium permits injection of the CagA oncoprotein via the type IV secretion system (T4SS), disruption of cell signalling and polarity and loss of tight junctions. These inflammatory changes further increase nutrient availability but at the cost of encountering toxic reactive oxygen species. Epithelial cells and the mucus layer are continuously shed into the lumen, carrying attached H. pylori cells, which will be lost into the lumen and expelled unless binding is inhibited. Modulation of attachment proteins gives H. pylori the flexibility to regulate its exposure to these “physicochemical facts of life”, and promote chronic infection.
Acknowledgements
This work was supported by grants from the National Institutes of Health (AI081037, AI070803) to J.V.S., and by grants from the Swedish Research Council (11218) and the Swedish Cancer Foundation to T.B.
Extra View to: Styer CM, Hansen LM, Cooke CL, Gundersen AM, Choi SS, Berg DE, et al. Expression of the BabA adhesin during experimental infection with Helicobacter pylori. Infect Immun 2010; 78:1593 - 1600; PMID: 20123715; http://dx.doi.org/10.1128/IAI.01297-09
References
- Solnick JV, Hansen LM, Salama NR, Boonjakuakul JK, Syvanen M. Modification of Helicobacter pylori outer membrane protein expression during experimental infection of rhesus macaques. Proc Natl Acad Sci USA 2004; 101:2106 - 2111
- Styer CM, Hansen LM, Cooke CL, Gundersen AM, Choi SS, Berg DE, et al. Expression of the BabA adhesin during experimental infection with Helicobacter pylori. Infect Immun 2010; 78:1593 - 1600
- Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 2003; 300:1430 - 1434
- Schreiber S, Konradt M, Groll C, Scheid P, Hanauer G, Werling HO, et al. The spatial orientation of Helicobacter pylori in the gastric mucus. Proc Natl Acad Sci USA 2004; 101:5024 - 5029
- Pflock M, Finsterer N, Joseph B, Mollenkopf H, Meyer TF, Beier D. Characterization of the ArsRS regulon of Helicobacter pylori, involved in acid adaptation. J Bacteriol 2006; 188:3449 - 3462
- Celli JP, Turner BS, Afdhal NH, Keates S, Ghiran I, Kelly CP, et al. Helicobacter pylori moves through mucus by reducing mucin viscoelasticity. Proc Natl Acad Sci USA 2009; 106:14321 - 14326
- Schade C, Flemstrom G, Holm L. Hydrogen ion concentration in the mucus layer on top of acid-stimulated and -inhibited rat gastric mucosa. Gastroenterol 1994; 107:180 - 188
- Scott DR, Marcus EA, Wen Y, Oh J, Sachs G. Gene expression in vivo shows that Helicobacter pylori colonizes an acidic niche on the gastric surface. Proc Natl Acad Sci USA 2007; 104:7235 - 7240
- Mahdavi J, Sonden B, Hurtig M, Olfat FO, Forsberg L, Roche N, et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 2002; 297:573 - 578
- Cooke CL, An HJ, Kim J, Canfield DR, Torres J, Lebrilla CB, et al. Modification of gastric mucin oligosaccharide expression in rhesus macaques after infection with Helicobacter pylori. Gastroenterol 2009; 137:1061 - 1071
- Linden SK, Mahdavi J, Semino-Mora C, Olsen C, Carlstedt I, Boren T, et al. Role of ABO Secretor Status in Mucosal Innate Immunity and H. pylori Infection. PLoS Pathogens 2008; 4:2
- Marcos NT, Magalhaes A, Ferreira B, Oliveira MJ, Carvalho AS, Mendes N, et al. Helicobacter pylori induces beta3GnT5 in human gastric cell lines, modulating expression of the SabA ligand sialyl-Lewis x. J Clin Invest 2008; 118:2325 - 2336
- McGuckin MA, Every AL, Skene CD, Linden SK, Chionh YT, Swierczak A, et al. Muc1 mucin limits both Helicobacter pylori colonization of the murine gastric mucosa and associated gastritis. Gastroenterol 2007; 133:1210 - 1218
- Linden SK, Sheng YH, Every AL, Miles KM, Skoog EC, Florin TH, et al. MUC1 limits Helicobacter pylori infection both by steric hindrance and by acting as a releasable decoy. PLoS Pathogens 2009; 5:1000617
- Guang W, Ding H, Czinn SJ, Kim KC, Blanchard TG, Lillehoj EP. Muc1 cell surface mucin attenuates epithelial inflammation in response to a common mucosal pathogen. J Biol Chem 2010; 285:20547 - 20557
- Kirschner DE, Blaser MJ. The dynamics of Helicobacter pylori infection of the human stomach. J Theor Biol 1995; 176:281 - 290
- Tan S, Tompkins LS, Amieva MR. Helicobacter pylori usurps cell polarity to turn the cell surface into a replicative niche. PLoS Pathogens 2009; 5:1000407
- Nesic D, Miller MC, Quinkert ZT, Stein M, Chait BT, Stebbins CE. Helicobacter pylori CagA inhibits PAR1-MARK family kinases by mimicking host substrates. Nat Struct Mol Biol 2010; 17:130 - 132
- Hornsby MJ, Huff JL, Kays RJ, Canfield DR, Bevins CL, Solnick JV. Helicobacter pylori induces an antimicrobial response in rhesus macaques in a Cag Pathogenicity Island-Dependent Manner. Gastroenterol 2008; 134:1049 - 1057
- Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, et al. Cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci USA 1996; 93:14648 - 14653
- Jimenez-Soto LF, Kutter S, Sewald X, Ertl C, Weiss E, Kapp U, et al. Helicobacter pylori type IV secretion apparatus exploits beta1 integrin in a novel RGD-independent manner. PLoS Pathogens 2009; 5:1000684
- Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, et al. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 2007; 449:862 - 866
- Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol 2004; 5:1166 - 1174
- Watanabe T, Asano N, Fichtner-Feigl S, Gorelick PL, Tsuji Y, Matsumoto Y, et al. NOD1 contributes to mouse host defense against Helicobacter pylori via induction of type I IFN and activation of the ISGF3 signaling pathway. J Clin Invest 2010; 120:1645 - 1662
- Harris PR, Wright SW, Serrano C, Riera F, Duarte I, Torres J, et al. Helicobacter pylori gastritis in children is associated with a regulatory T-cell response. Gastroenterol 2008; 134:491 - 499
- Gebert B, Fischer W, Weiss E, Hoffmann R, Haas R. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 2003; 301:1099 - 1102
- Shi Y, Liu XF, Zhuang Y, Zhang JY, Liu T, Yin Z, et al. Helicobacter pylori-induced Th17 responses modulate Th1 cell responses, benefit bacterial growth and contribute to pathology in mice. J Immunol 2010; 184:5121 - 5129
- Arnold IC, Lee JY, Amieva MR, Roers A, Flavell RA, Sparwasser T, et al. Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterol 2011; 140:199 - 209
- Amieva MR, Salama NR, Tompkins LS, Falkow S. Helicobacter pylori enter and survive within multivesicular vacuoles of epithelial cells. Cell Microbiol 2002; 4:677 - 690
- Dubois A, Boren T. Helicobacter pylori is invasive and it may be a facultative intracellular organism. Cellular Microbiol 2007; 9:1108 - 1116
- Alm RA, Bina J, Andrews BM, Doig P, Hancock RE, Trust TJ. Comparative genomics of Helicobacter pylori: analysis of the outer membrane protein families. Infect Immun 2000; 68:4155 - 4168
- Aspholm-Hurtig M, Dailide G, Lahmann M, Kalia A, Ilver D, Roche N, et al. Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science 2004; 305:519 - 522
- Boren T, Falk P, Roth KA, Larson G, Normark S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science 1993; 262:1892 - 1895
- Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, et al. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science 1998; 279:373 - 377
- Odenbreit S, Swoboda K, Barwig I, Ruhl S, Boren T, Koletzko S, et al. Outer membrane protein expression profile in Helicobacter pylori clinical isolates. Infect Immun 2009; 77:3782 - 3790
- Gerhard M, Lehn N, Neumayer N, Borén T, Rad R, Schepp W, et al. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc Natl Acad Sci USA 1999; 96:12778 - 12783
- Mentis A, Blackwell CC, Weir DM, Spiliadis C, Dailianas A, Skandalis N. ABO blood group, secretor status and detection of Helicobacter pylori among patients with gastric or duodenal ulcers. Epidemiol Infect 1991; 106:221 - 229
- Cederberg A, Varis K, Salmi HA, Sipponen P, Harkonen M, Sarna S. Young onset peptic ulcer disease and non-ulcer dyspepsia are separate entities. Scand J Gastroenterol Suppl 1991; 186:33 - 44
- Backstrom A, Lundberg C, Kersulyte D, Berg DE, Boren T, Arnqvist A. Metastability of Helicobacter pylori bab adhesin genes and dynamics in Lewis b antigen binding. Proc Natl Acad Sci USA 2004; 101:16923 - 16928
- Pride DT, Blaser MJ. Concerted evolution between duplicated genetic elements in Helicobacter pylori. J Mol Biol 2002; 316:629 - 642
- Pride DT, Meinersmann RJ, Blaser MJ. Allelic variation within Helicobacter pylori babA and babB. Infect Immun 2001; 69:1160 - 1171
- Colbeck JC, Hansen LM, Fong JM, Solnick JV. Genotypic profile of the outer membrane proteins BabA and BabB in clinical isolates of Helicobacter pylori. Infect Immun 2006; 74:4375 - 4378
- Haas G, Karaali G, Ebermayer K, Metzger WG, Lamer S, Zimny-Arndt U, et al. Immunoproteomics of Helicobacter pylori infection and relation to gastric disease. Proteomics 2002; 2:313 - 324
- Kimmel B, Bosserhoff A, Frank R, Gross R, Goebel W, Beier D. Identification of immunodominant antigens from Helicobacter pylori and evaluation of their reactivities with sera from patients with different gastroduodenal pathologies. Infect Immun 2000; 68:915 - 920
- Blaser MJ, Kirschner D. Dynamics of Helicobacter pylori colonization in relation to the host response. Proc Natl Acad Sci USA 1999; 96:8359 - 8364
- Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 1999; 397:176 - 180