Abstract
The composition of the gastrointestinal microbiome is increasingly recognized as a crucial contributor to immune and metabolic homeostasis—deficiencies in which are characteristic of cystic fibrosis (CF) patients. The murine model (CFTR−/−, CF), has, in previous studies, demonstrated characteristic CF gastrointestinal (GI) manifestations including slowed transit and significant upregulation of genes associated with inflammation. To determine if characteristics of the microbiome are associated with these phenotypes we used a phylogenetic microarray to compare small intestine bacterial communities of wild type and congenic CF mice. Loss of functional CFTR is associated with significant decreases in GI bacterial community richness, evenness and diversity and reduced relative abundance of putative protective species such as Acinetobacter lwoffii and a multitude of Lactobacilliales members. CF mice exhibited significant enrichment of Mycobacteria species and Bacteroides fragilis, previously associated with GI infection and immunomodulation. Antibiotic administration to WT and CF animals resulted in convergence of their microbiome composition and significant increases in community diversity in CF mice. These communities were characterized by enrichment of members of the Lactobacillaceae and Bifidobacteriaceae and reduced abundance of Enterobacteriaceae and Clostridiaceae. These data suggest that Enterobacteria and Clostridia species, long associated with small intestinal overgrowth and inflammatory bowel disease, may suppress both ileal bacterial diversity and the particular species which maintain motility and immune homeostasis in this niche. Thus, these data provide the first indications that GI bacterial colonization is strongly impacted by the loss of functional CFTR and opens up avenues for alternative therapeutic approaches to improve CF disease management.
Introduction
Cystic fibrosis (CF), an autosomal recessive genetic disorder is characterized by mutation(s) in the cystic fibrosis transmembrane conductance regulator (CFTR;Citation1-Citation3) gene which encodes a large trans-membrane protein, critical to cAMP-regulated anion transport across epithelial cells. Thus CFTR mutation impacts mucosal physiology of the respiratory, gastrointestinal, and reproductive tracts, leading to multi-organ defects and system-wide disease. CFTR knockout mice (CFTR−/−; CF) exhibit gastrointestinal manifestations similar to those observed in human CF patients, including accumulation of gastrointestinal mucus, obstruction of the distal intestine, inflammation,Citation4 small intestine bacterial overgrowth (SIBO) and slower transit times through the GI tract,Citation5 which collectively contribute to a failure of these animals, like their human counterparts, to thrive. More recently functional gastrointestinal disorders have been linked to perturbations in the GI microbiome, e.g., inflammatory bowel disease (reviewed inCitation6,Citation7), leading to speculation that the composition and function of these assemblages may play a key role in such diseases and offer a novel therapeutic avenue for management or prevention of CF-related and other GI conditions.
We hypothesized that because of its key role in defining conditions at the epithelial surface that influence bacterial community colonization, lack of functional CFTR (characteristic of severe CF phenotypes), is associated with an aberrant GI microbiome composition which contributes to intestinal disease phenotypes associated with this patient population. If true, initial studies demonstrating this phenomenon would lay the foundation for future investigations to determine the functional contribution of the intestinal microbiome to CF-associated GI disorders. CF mice represent a good model for gastrointestinal aberrations associated with this disease in humans. For example, a previous study of CF mice demonstrated significantly reduced motility (as measured by gastric emptying and transit through the ileum;Citation5) and upregulation of genes associated with inflammation when compared with wild type (WT) controls.Citation8 Using this model and a high-resolution phylogenetic microarray, we investigated two factors fundamental to CF patient health, loss of functional CFTR and the impact of antibiotic administration on the composition of the GI microbiome of these model animals.
Results
Comparison of WT and CF small intestine microbiota
To determine if gross structural aspects of ileal bacterial communities differed between CF and WT mice, 16S rRNA PhyloChip (G2 version) reported community richness (number of taxa detected), evenness (relative distribution of taxa) and diversity were calculated for each sample and relative changes in these metrics compared across treatment groups. CF GI communities were significantly less rich (p < 0.03), less even (p < 0.04) and less diverse (p < 0.03) compared with WT (), suggesting that the loss of community diversity in CF mice is driven both by a relative loss of bacterial types and establishment of communities with a more skewed distribution – a phenomenon typically associated with species overgrowth.Citation9 Though animals were congenic, fed the same diet and housed under identical conditions, CF mice exhibited substantially less bacterial community composition variability compared with that observed among WT animals (). Collectively these data suggest that a lack of functional CFTR exerts a substantial selective pressure on ileal microbiota resulting in a distinct, dysbiotic communities which lack diversity and are associated with the well described GI phenotypes observed in this animal model.
Figure 1. Comparison of GI microbiome community (A) Richness; (B) Evenness and (C) Diversity in WT and CF animals.
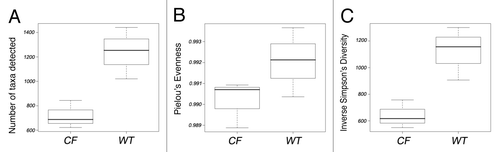
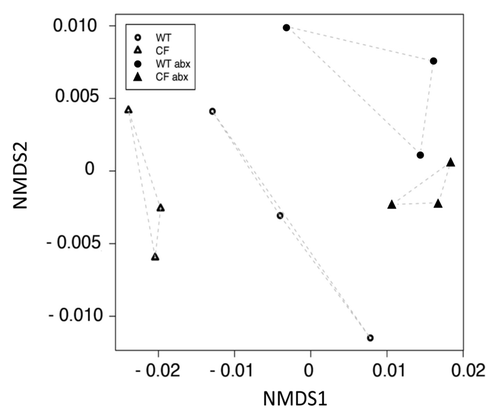
Figure 2. NMDS illustrates substantial differences in CF and WT bacterial community profiles and that antibiotic administration results in relatively similar GI community composition in these genotypically distinct animal groups.
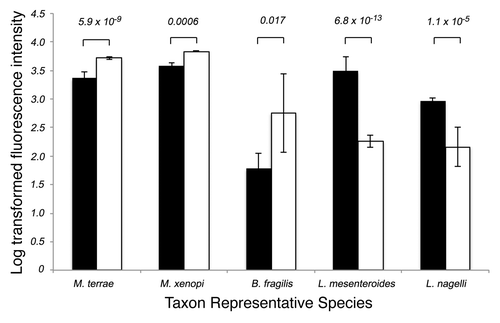
To more specifically identify the taxa that discriminate WT from CF animals, we performed a two-way ANOVA. Following correction for false discovery, and imposing a rank order on the findings (based on magnitude of change in relative fluorescence intensity) a total of 157 taxa belonging to distinct bacterial families including the Bacteroidaceae, Mycobacteriaceae and Pseudonocardiaceae were most increased in relative abundance in CF compared with WT mice (Bonferroni-corrected p < 0.05). For the purpose of this study, taxa are defined as groups of organisms sharing at least 97% 16S rRNA sequence identity. Though multiple species or strains may exist in a given taxon, each taxon is represented by a single species. As such, while the representative species is provided for reference, that species or indeed any phylogenetically related member of a given taxon may trigger the probe set for that taxon and result in a positive “hit” on the array. Taxa exhibiting the greatest increase in relative abundance in CF compared with WT animals, included two Mycobacteriaceae represented by Mycobacterium terrae (p < 5.9 x 10−9) and Mycobacterium xenopi (p < 1 x 10−9). Other taxa of note included one represented by Clostridium botulinum (p < 0.04), a known GI pathogen (). However, the taxon exhibiting the greatest enrichment in CF animals was a member of the Bacteroidaceae, represented by the species B. fragilis, an immunogenic obligate anaerobe, enterotoxigenic strains of which are commonly associated with gastrointestinal infections.Citation10 WT animals, in comparison, exhibited 305 taxa that were significantly increased in relative abundance (compared with CF animals; p < 0.05). These taxa largely belonged to the phylum Proteobacteria which included members of the Moraxellaceae (represented by the species Acinetobacter lwoffii) and Firmicutes which included a number of Lactobaciliales (represented by Leuconostoc mesenteroides, Lactobacillus murinus, Lactobacillus nagelii and Lactobacillus plantarum; ; Table S1).
Figure 3. Specific taxa exhibiting Benjamini-Hochberg-corrected significant differences in relative abundance between CF (white bars) and wild type (black bars) mice.
Impact of antibiotics on WT and CF GI microbiota
Since antibiotic administration represents a significant perturbation to GI microbiotaCitation11 and a frequent feature of CF patient management, we examined the impact of the combination of ciprofloxacin and metronidazole administration on the GI microbiome of both WT and CF animals. Non-metric multi-dimensional scaling (NMDS) analysis demonstrated that antibiotic administration elicited the most significant change in community composition, eliminating the observed baseline variation between CF and WT mice and resulting in compositional convergence of the GI microbiota of these animals (). In fact, antibiotic exposure accounted for 43.4% of the variance in community composition of all samples profiled, underscoring the profound impact of such treatment on the GI microbiota. This is substantially greater than the influence of CFTR knockout alone which, in the context of all samples examined in this study, only accounted for 7.5% of the variance in bacterial community structure (p = 0.15).
Though a change in community composition was evident following the three week course of antibiotics administered to WT mice, this effect was not as pronounced as that observed upon treatment of CF animals (). This observation is supported by the fact that WT animals, when treated with antibiotics did not exhibit significant changes in gross metrics of community composition, e.g., richness, evenness and diversity. In contrast, CF animals exhibited significant gains in all of these metrics following treatment with antibiotics. These data indicate that while WT and CF animals exhibit quite distinct GI microbiota, administration of a three week course of ciprofloxacin and metronidazole to these animals results in establishment of a relatively similar community of organisms in this niche.
As expected from the lack of significant change in gross metrics of community composition, relatively few taxa discriminated WT and antibiotic treated WT animals. A total of 63 taxa were significantly increased in WT animals (compared with antibiotic treated WT) and included members of the Pasteurellaceae, Prevotellaceae, Bacteroidaceae and Clostridaceae. In comparison, 156 taxa were significantly increased in abundance in antibiotic treated WT mice, largely belonging to the phylum Firmicutes, including members of the Bacillales and Lactobacillales (Table S2).
CF animals characteristically present with reduced transit time and small intestinal bowel overgrowth (SIBO), which is releived upon antibiotic treatment of these animals,Citation8 implicating a microbial component in this phenomenon. Given the significant gain in diversity observed following antibiotic administration, we hypothesized that comparison of CF and antibiotic treated CF animals could identify ileal microbiome members associated with SIBO. Therefore, we compared the relative abundance of each taxon detected in these two groups. Taxa exhibiting significantly increased relative abundance in CF animals compared with antibiotic treated CF mice were again ranked based on the magnitude of change in relative abundance. Among the taxa most significantly enriched in CF mice were several members of the Clostridiaceae (represented by C. paraputrificum, C. perfringens and C. collagenovorans) and a Bacteroidaceae member (B. fragilis; Table S3). In addition, we noted that a large majority of these highest ranked taxa were members of the Enterobacteriaceae, suggesting that these microbiome members may individually, or collectively contribute to the SIBO phenotype in CF animals.
As expected based on the significant gains in diversity observed in CF mice administered antibiotics, these animals exhibited significantly increased relative abundance of 545 taxa (Table S3). Taxa enriched in antibiotic-treated CF mice included a Bifidobacteriaceae (B. pseudolongum), a Paenibacillaceae (P. macquariensis) and a Propionibacteriaceae member (P. acnes; Table S3). In addition, L. murinus and L. mesenteroides, which were enriched in WT animals (compared with CF), were also significantly increased in relative abundance in antibiotic-treated CF animals.
Discussion
This study demonstrates using a murine model, that CFTR knockout, critical to trans-epthelial anion transport, exerts a substantial influence on GI bacterial community composition. In the absence of CFTR, mucus accumulates in the intestines and provides a niche for abnormal microbial colonization and development of SIBO.Citation8 At a very gross level, bacterial communities in the knockout animals are characterized by the presence of dysbiotic assemblages significantly depleted in community membership. More specifically, the CFTR knockout animals exhibited significant enrichment for Mycobacteria species which have previously been reported in antibiotic resistant infections of the human skin, lung, and GI tract,Citation12,Citation13 though defining whether such organisms play a role in CF-associated pathology in the GI tract, obviously requires further study.
Of particular note was our finding that the taxon most enriched in the CF animals was represented by B. fragilis. This species is a prominent gut commensal that has recently been shown to signal (via polysaccharide A) through TLR2 directly on Foxp3(+) regulatory T cells to promote host immunologic tolerance, permitting this species to colonize the mucosal surface of the gastrointestinal tract.Citation14 However B. fragilis is also well recognized as an opportunistic pathogen responsible for a range of gastrointestinal infections and diarrheal disease.Citation10 Though further analyses are necessary to confirm such hypotheses, it is plausible that in the context of a dysbiotic microbiome, an over-abundance of B. fragilis or phylogenetically related species plays a role in the gastrointestinal inflammation which characterizes these CF animals.Citation4 Indeed induction of inflammation is likely a primary factor in reducing both GI motility and community diversity in this niche. Recent studies have demonstrated that other GI pathogens such as Salmonella enterica serotype Typhimurium, induce inflammation to afford themselves a growth advantage over microbial competitors in the microbiome,Citation15 a strategy that is likely exercised by multiple species in this and other mucosal-associated niches.
In comparison with the CFTR knockout animals, WT mice exhibited enrichment for A. lwoffii, a species previously isolated from cowsheds, which has been shown to afford protection against airway disease (asthma) via TLR-signalingCitation16 and, more recently, via epigenetic mechanisms.Citation17 These mice also exhibited enrichment for a variety of Lactobacillales, many of which are synonymous with promotion of host health by protecting against enteropathogen infection via competitive colonization of mucosal surfaces and manipulation of host immune responses.Citation18 In particular, WT community members detected in higher relative abundance included Lactobacillus murinis (p < 0.0095), which exhibits anti-inflammatory responses in mouse models of chronic inflammation (e.g., colitis;Citation19), and Lactobacillus plantarum (p < 1.13 × 10−5) which protects against gastrointestinal epithelial villous atrophy.Citation20 More recently, mouse supplementation studies using multiple Lactobacillus species together with Bifidobacterium bifidium and Streptococcus thermophilus demonstrated induction of CD4+ Foxp3+ T-reg populations and reduced inflammatory responses in multiple mouse models of chronic inflammatory disease.Citation21 This suggests a potential mechanism by which members of the WT and antibiotic-treated CF animal GI microbiota may contribute to reduced inflammation (which has previously been reported following antibiotic administration to CF animals;Citation8). It is also notable that known pathogenic species such as Pasteurella pneumotropica and Propionibacterium acnes were detected in significantly higher relative abundance in WT animals, though these animals are healthy and are known to exhibit normal GI histology.Citation22 This suggests that the composition of the microbiome in a given niche may well define the behavior of the pathogenic species present in this niche, a thesis supported by several recent studies of both mouseCitation9 and insect GI microbiota.Citation23
The other key factor that strongly influences GI bacterial community composition is antibiotic administration which occurs with frequency in the CF patient population for disease management and has been shown to increase the life expectancy.Citation24 Antimicrobial administration is known to exert a dramatic impact on the enteric microbiota resulting in loss of discrete microbiome members which fail to return even after protracted periods of time post-treatment.Citation11 The clinical relevance of such losses is currently unknown but given that the GI microbiome plays such a key role in regulating host immune responseCitation25-Citation29 and performing key metabolic functions,Citation30-Citation32 it has been suggested that repeated perturbations and membership losses result in a cumulative detrimental effect on immune and metabolic function over time in this patient population. In our murine based model, it should be noted that the animals examined only received a single course of antimicrobial treatment. This treatment resulted in increased bacterial community richness, evenness and diversity in the CF compared with all other groups examined, indicating antibiotic-mediated relief of community diversity suppression, putatively by those species who exhibit the greatest depletion following antibiotic treatment e.g., Enterobacteriaceae or Clostridaceae members. Importantly, in a previous study, antibiotic administration also significantly increased weight gain in CF mice,Citation8 indicating that gains in microbiome diversity following antibiotic treatment may restore microbial-based metabolic capacity and improve nutrition in these animals. However, given recent studies demonstrating relatively rapid reassembly of the GI microbiome following antibiotic administration,Citation11 it is likely that these effects are short lived and that the selective pressure of both host genetics and diet contrive to reassemble an aberrant community.
Our study suggests that the presence of members of the Enterobacteriaceae, Clostridaceae or B. fragilis which are enriched in CF mice contribute to a persistent state of inflammation that maintains low community diversity. Such strategies are not unprecedented and have recently been described as a tactic employed by Porphyromonas gingivalis to promote its presence in the oral microbiome.Citation33 Indeed a recent study of 26 SIBO patients identified overgrowth of Enterobacteriaceae in 77% and Bacteroides in 27% of the patients examined,Citation34 implicating the same organisms identified in our study, in human cases of SIBO. Moreover, that study also demonstrated that features of SIBO such as elevated lamina propria IgA levels were ameliorated in subjects administered antimicrobials.Citation34 Given the concordance of data in both murine models and human with SIBO, and the availability of a readily manipulable mouse model, further investigation is necessary to define the mechanistic basis of how these species contribute to physiological features of CF-associated GI manifestations that typify the disease.
Materials and Methods
CF murine model
Cftr(+/−) (CF) mice congenic on the C57BL/6 background, originally obtained from Jackson Labs, were bred to obtain wild type (WT, Cftr+/+) and CF (Cftr−/−) genotypes. Mice used in this study originated from 6 different litters. Because of the severity of the CF phenotype, typically only 1 or at most 2 CF mice are obtained per litter, so it is not possible that all animals for such studies are littermates. Both males and females (6 each) all born within one month of each other, were used for this study and housed by gender on corncob bedding (Bed-o’Cobs; Andersons Lab Bedding Products) throughout the study period. At weaning, mice were maintained on a liquid diet (Peptamen, ad libitum; Nestle) to prevent lethal intestinal obstructionCitation35 Where indicated, at weaning, antibiotics (ABX; ciprofloxacin, 50 mg kg−1 day−1; and metronidazole, 100 mg kg−1 day−1) were supplemented to the liquid diet for a period of three weeks. Small intestinal bacterial community composition was compared in four groups of age-matched (6 week old) animals: wild type (WT; n = 3), wild type administered antibiotics (WT/ABX; n = 3), CF (CF; n = 3), and CF administered antibiotics (CF/ABX; n = 3). All animal protocols were approved by the Committee on Animal Research at the University of Kansas. As previously described,Citation8 the entire small intestine was flushed with saline containing 10 mM dithiothreitol, and DNA was extracted from the particulate material using the Qiagen Stool DNA kit (Qiagen).
PCR amplification of 16S rRNA genes
Genomic DNA from the 12 samples was quantified by OD260, and diluted in nuclease-free water to achieve a standard concentration of 500 ng μl−1. Polymerase chain reactions were prepared for each sample containing final concentrations of 10 ng μl-1 DNA template, 0.02 U/μL ExTaq (Takara Bio Inc.), 1X ExTaq buffer, 0.2 mM dNTP mixture, 1 μg μl−1 Bovine Serum Albumin (BSA), and 300 pM each of universal bacterial primers: 27F (5′-AGAGTTTGATCCTGGC TCAG-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′). To minimize PCR bias due to variable template annealing efficiencies and random effects, reactions were performed on a BioRad iCycler with an eight temperature annealing gradient (48–58°C) and the following conditions: 95°C (3 min), followed by 30 cycles of 95°C (30 sec), annealing (30 sec), 72°C (2 min), and a final extension at 72°C (10 min). Reactions were combined for each sample and concentrated with 0.8 volumes isopropanol, washed twice with ice cold 70% ethanol and resuspended in 50 μl nuclease-free water.
PhyloChip microarray profiling
Phylogenetic microarray profiling of the bacterial communities present in each sample was performed as described in Ivanov et al.Citation25 Briefly, 500 ng of pooled PCR product from each sample were spiked with known concentrations of amplicons derived from yeast and bacterial metabolic genes. This mix was fragmented to 50–200 bp using DNase I (0.02 U μg−1 DNA, Invitrogen) and One-Phor-All buffer (GE Healthcare) following the manufacturer's protocols. The mixture was then incubated at 25°C for 20 min and 98°C for 10 min before biotin labeling with a GeneChip DNA labeling reagent kit (Affymetrix) following the manufacturer's instructions. Labeled DNA was denatured at 99°C for 5 min and hybridized to G2 PhyloChips (Affymetrix) at 48°C and 60 rpm for 16 h. PhyloChip washing and staining were performed according to the standard Affymetrix protocols described previously.Citation36
Each array was scanned and recorded as a pixel image, and initial data acquisition and intensity determination were performed using standard Affymetrix software (GeneChip microarray analysis suite, version 5.1). Background subtraction, data normalization and probe pair scoring were performed as reported previously.Citation37-Citation39 The positive fraction (PosFrac) was calculated for each probe set as the number of positive probe pairs divided by the total number of probe pairs in a probe set. Taxa were deemed present when the PosFrac value exceeded 0.90. Intensities were summarized for each taxon/probe-set using a trimmed average (highest and lowest values removed before averaging) of the intensities of the perfect match probes (PM) minus their corresponding mismatch probes (PM).
Statistical analyses
All statistical analyses were performed in the R software environment (http://www.r-project.org/). To correct for technical variation associated with array processing, a two-step normalization procedure previously reported by Ivanov et al.Citation25 was employed. First, for each PhyloChip experiment, a scaling factor best explaining the intensities of the spiked control probes under a multiplicative error model was estimated using a maximum-likelihood procedure. The intensities in each experiment are multiplied with its corresponding optimal scaling factor. In addition, the intensities for each experiment are corrected for the variation in total array intensity by dividing the intensities by its corresponding total array intensity for bacterial probe sets.
Following normalization and scaling, microarray intensity data was log transformed. For comparison of bacterial community structure between samples, only those bacterial taxa detected in at least 2 out of 3 replicates from either treatment group were considered. A distance matrix was calculated using the trimmed average fluorescence intensity data using the Bray-Curtis distance metric within the function ‘vegdist’ in the R package ‘vegan’Citation40. The distance matrix was represented as an ordination plot by NMDS using the metaMDS function in the ‘vegan’ package. Richness was expressed as the number of taxa with PosFrac (pf) values ≥ 0.90 per sample, while evenness was calculated using Pielou's evenness index and diversity, using inverse Simpson’s index (in the ‘vegan’ package). To determine organisms that varied significantly in relative abundance between the treatment groups we applied a two-sample ANOVA. The raw p-values were corrected for multiple testing using Benjamini-Hochberg procedure. To determine the partitioning of variance in bacterial composition across factors (e.g., strain, antibiotics) and their interactions, we performed permutational multivariate ANOVA, using the Bray-Curtis distance matrix to represent the variance in bacterial community structure. This was performed in R using the function ‘adonis’ also in the ‘vegan’ package.
Conclusion
Collectively, this pilot study indicates that loss of functional CFTR is associated with enrichment of known pathogenic bacteria and the concomitant loss of organisms known to afford protection at mucosal surfaces. A single course of antimicrobial administration to these animals results in dramatic restructuring of the GI microbiome involving a significant increase in community diversity and depletion of species previously associated with GI disease manifestations, e.g., SIBO. These studies have identified target organisms for future, more focused investigations to determine relationships between GI community composition and disease state in this animal model.
| Abbreviations: | ||
| CF | = | cystic fibrosis |
| WT | = | wild type |
| Abx | = | antibiotics, NMDS, non-metric dimensional scaling |
Additional material
Download Zip (557 KB)Disclosure of Potential Conflicts of Interest
The authors declare no conflicts of Interest.
Acknowledgments
S.V.L. is supported in part by the Rainin Foundation. W.D.K. is supported by a fellowship from the CFRI. R.C.D. was supported by NIH grant AI-083479. Part of this work was performed at Lawrence Berkeley National Laboratory under the Department of Energy Contract no. de-AC02–05CH11231.
Supplemental Material
Supplemental material may be found here:
http://www.landesbioscience.com/journals/gutmicrobes/article/22430/
References
- Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, et al. Identification of the cystic fibrosis gene: genetic analysis. Science 1989; 245:1073 - 80; http://dx.doi.org/10.1126/science.2570460; PMID: 2570460
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989; 245:1066 - 73; http://dx.doi.org/10.1126/science.2475911; PMID: 2475911
- Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 1989; 245:1059 - 65; http://dx.doi.org/10.1126/science.2772657; PMID: 2772657
- Norkina O, Kaur S, Ziemer D, De Lisle RC. Inflammation of the cystic fibrosis mouse small intestine. Am J Physiol Gastrointest Liver Physiol 2004; 286:G1032 - 41; http://dx.doi.org/10.1152/ajpgi.00473.2003; PMID: 14739145
- De Lisle RC. Altered transit and bacterial overgrowth in the cystic fibrosis mouse small intestine. Am J Physiol Gastrointest Liver Physiol 2007; 293:G104 - 11; http://dx.doi.org/10.1152/ajpgi.00548.2006; PMID: 17363465
- Dahlqvist G, Piessevaux H. Irritable bowel syndrome: the role of the intestinal microbiota, pathogenesis and therapeutic targets. Acta Gastroenterol Belg 2011; 74:375 - 80; PMID: 22103040
- Goulet O, Joly F. [Intestinal microbiota in short bowel syndrome]. Gastroenterol Clin Biol 2010; 34:Suppl 1 S37 - 43; http://dx.doi.org/10.1016/S0399-8320(10)70019-1; PMID: 20889003
- Norkina O, Burnett TG, De Lisle RC. Bacterial overgrowth in the cystic fibrosis transmembrane conductance regulator null mouse small intestine. Infect Immun 2004; 72:6040 - 9; http://dx.doi.org/10.1128/IAI.72.10.6040-6049.2004; PMID: 15385508
- Lawley TD, Clare S, Walker AW, Goulding D, Stabler RA, Croucher N, et al. Antibiotic treatment of clostridium difficile carrier mice triggers a supershedder state, spore-mediated transmission, and severe disease in immunocompromised hosts. Infect Immun 2009; 77:3661 - 9; http://dx.doi.org/10.1128/IAI.00558-09; PMID: 19564382
- Durmaz B, Dalgalar M, Durmaz R. Prevalence of enterotoxigenic Bacteroides fragilis in patients with diarrhea: a controlled study. Anaerobe 2005; 11:318 - 21; http://dx.doi.org/10.1016/j.anaerobe.2005.06.001; PMID: 16701593
- Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 2008; 6:e280; http://dx.doi.org/10.1371/journal.pbio.0060280; PMID: 19018661
- Smith DS, Lindholm-Levy P, Huitt GA, Heifets LB, Cook JL. Mycobacterium terrae: case reports, literature review, and in vitro antibiotic susceptibility testing. Clin Infect Dis 2000; 30:444 - 53; http://dx.doi.org/10.1086/313693; PMID: 10722426
- Andréjak C, Lescure FX, Pukenyte E, Douadi Y, Yazdanpanah Y, Laurans G, et al, Xenopi Group. Mycobacterium xenopi pulmonary infections: a multicentric retrospective study of 136 cases in north-east France. Thorax 2009; 64:291 - 6; http://dx.doi.org/10.1136/thx.2008.096842; PMID: 19052044
- Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, et al. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 2011; 332:974 - 7; http://dx.doi.org/10.1126/science.1206095; PMID: 21512004
- Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 2010; 467:426 - 9; http://dx.doi.org/10.1038/nature09415; PMID: 20864996
- Conrad ML, Ferstl R, Teich R, Brand S, Blümer N, Yildirim AO, et al. Maternal TLR signaling is required for prenatal asthma protection by the nonpathogenic microbe Acinetobacter lwoffii F78. J Exp Med 2009; 206:2869 - 77; http://dx.doi.org/10.1084/jem.20090845; PMID: 19995952
- Brand S, Teich R, Dicke T, Harb H, Yildirim AÖ, Tost J, et al. Epigenetic regulation in murine offspring as a novel mechanism for transmaternal asthma protection induced by microbes. J Allergy Clin Immunol 2011; 128:618 - 25, e1-7; http://dx.doi.org/10.1016/j.jaci.2011.04.035; PMID: 21680015
- Candela M, Perna F, Carnevali P, Vitali B, Ciati R, Gionchetti P, et al. Interaction of probiotic Lactobacillus and Bifidobacterium strains with human intestinal epithelial cells: adhesion properties, competition against enteropathogens and modulation of IL-8 production. Int J Food Microbiol 2008; 125:286 - 92; http://dx.doi.org/10.1016/j.ijfoodmicro.2008.04.012; PMID: 18524406
- Marcinkiewicz J, Ciszek M, Bobek M, Strus M, Heczko PB, Kurnyta M, et al. Differential inflammatory mediator response in vitro from murine macrophages to lactobacilli and pathogenic intestinal bacteria. Int J Exp Pathol 2007; 88:155 - 64; http://dx.doi.org/10.1111/j.1365-2613.2007.00530.x; PMID: 17504445
- Yoshida Y, Tsukahara T, Ushida K. Oral administration of Lactobacillus plantarum Lq80 and Megasphaera elsdenii iNP-001 induces efficient recovery from mucosal atrophy in the small and the large intestines of weaning piglets. Anim Sci J 2009; 80:709 - 15; http://dx.doi.org/10.1111/j.1740-0929.2009.00692.x; PMID: 20163663
- Kwon HK, Lee CG, So JS, Chae CS, Hwang JS, Sahoo A, et al. Generation of regulatory dendritic cells and CD4+Foxp3+ T cells by probiotics administration suppresses immune disorders. Proc Natl Acad Sci U S A 2010; 107:2159 - 64; http://dx.doi.org/10.1073/pnas.0904055107; PMID: 20080669
- De Lisle RC, Roach E, Jansson K. Effects of laxative and N-acetylcysteine on mucus accumulation, bacterial load, transit, and inflammation in the cystic fibrosis mouse small intestine. Am J Physiol Gastrointest Liver Physiol 2007; 293:G577 - 84; http://dx.doi.org/10.1152/ajpgi.00195.2007; PMID: 17615175
- Dong Y, Manfredini F, Dimopoulos G. Implication of the mosquito midgut microbiota in the defense against malaria parasites. PLoS Pathog 2009; 5:e1000423; http://dx.doi.org/10.1371/journal.ppat.1000423; PMID: 19424427
- Ratjen F. Changes in strategies for optimal antibacterial therapy in cystic fibrosis. Int J Antimicrob Agents 2001; 17:93 - 6; http://dx.doi.org/10.1016/S0924-8579(00)00333-2; PMID: 11165111
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009; 139:485 - 98; http://dx.doi.org/10.1016/j.cell.2009.09.033; PMID: 19836068
- Penders J, Thijs C, van den Brandt PA, Kummeling I, Snijders B, Stelma F, et al. Gut microbiota composition and development of atopic manifestations in infancy: the KOALA Birth Cohort Study. Gut 2007; 56:661 - 7; http://dx.doi.org/10.1136/gut.2006.100164; PMID: 17047098
- Penders J, Thijs C, van den Brandt PA, Kummeling I, Snijders B, Stelma F, et al. Gut microbiota composition and development of atopic manifestations in infancy: the KOALA Birth Cohort Study. Gut 2007; 56:661 - 7; http://dx.doi.org/10.1136/gut.2006.100164; PMID: 17047098
- Penders J, Thijs C, Vink C, Stelma FF, Snijders B, Kummeling I, et al. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 2006; 118:511 - 21; http://dx.doi.org/10.1542/peds.2005-2824; PMID: 16882802
- Penders J, Vink C, Driessen C, London N, Thijs C, Stobberingh EE. Quantification of Bifidobacterium spp., Escherichia coli and Clostridium difficile in faecal samples of breast-fed and formula-fed infants by real-time PCR. FEMS Microbiol Lett 2005; 243:141 - 7; http://dx.doi.org/10.1016/j.femsle.2004.11.052; PMID: 15668012
- Muegge BD, Kuczynski J, Knights D, Clemente JC, González A, Fontana L, et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 2011; 332:970 - 4; http://dx.doi.org/10.1126/science.1198719; PMID: 21596990
- Sonnenburg JL. Microbiology: Genetic pot luck. Nature 2010; 464:837 - 8; http://dx.doi.org/10.1038/464837a; PMID: 20376136
- Sonnenburg ED, Zheng H, Joglekar P, Higginbottom SK, Firbank SJ, Bolam DN, et al. Specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations. Cell 2010; 141:1241 - 52; http://dx.doi.org/10.1016/j.cell.2010.05.005; PMID: 20603004
- Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 2011; 10:497 - 506; http://dx.doi.org/10.1016/j.chom.2011.10.006; PMID: 22036469
- Riordan SM, McIver CJ, Wakefield D, Duncombe VM, Thomas MC, Bolin TD. Small intestinal mucosal immunity and morphometry in luminal overgrowth of indigenous gut flora. Am J Gastroenterol 2001; 96:494 - 500; http://dx.doi.org/10.1111/j.1572-0241.2001.03533.x; PMID: 11232696
- Eckman EA, Cotton CU, Kube DM, Davis PB. Dietary changes improve survival of CFTR S489X homozygous mutant mouse. Am J Physiol 1995; 269:L625 - 30; PMID: 7491981
- Masuda N, Church GM. Escherichia coli gene expression responsive to levels of the response regulator EvgA. J Bacteriol 2002; 184:6225 - 34; http://dx.doi.org/10.1128/JB.184.22.6225-6234.2002; PMID: 12399493
- Brodie EL, Desantis TZ, Joyner DC, Baek SM, Larsen JT, Andersen GL, et al. Application of a high-density oligonucleotide microarray approach to study bacterial population dynamics during uranium reduction and reoxidation. Appl Environ Microbiol 2006; 72:6288 - 98; http://dx.doi.org/10.1128/AEM.00246-06; PMID: 16957256
- Brodie EL, DeSantis TZ, Parker JP, Zubietta IX, Piceno YM, Andersen GL. Urban aerosols harbor diverse and dynamic bacterial populations. Proc Natl Acad Sci U S A 2007; 104:299 - 304; http://dx.doi.org/10.1073/pnas.0608255104; PMID: 17182744
- DeSantis TZ, Brodie EL, Moberg JP, Zubieta IX, Piceno YM, Andersen GL. High-density universal 16S rRNA microarray analysis reveals broader diversity than typical clone library when sampling the environment. Microb Ecol 2007; 53:371 - 83; http://dx.doi.org/10.1007/s00248-006-9134-9; PMID: 17334858
- Oksanen, J., et al., vegan: Community Ecology Package. R package version 1.17-2., 2010.