Abstract
Helicobacter pylori is the leading risk factor associated with gastric carcinogenesis. H. pylori leads to chronic inflammation because of the failure of the host to eradicate the infection. Chronic inflammation leads to oxidative stress, deriving from immune cells and from within gastric epithelial cells. This is a main contributor to DNA damage, apoptosis and neoplastic transformation. Both pathogen and host factors directly contribute to oxidative stress, including H. pylori virulence factors, and pathways involving DNA damage and repair, polyamine synthesis and metabolism, and oxidative stress response. Our laboratory has recently uncovered a mechanism by which polyamine oxidation by spermine oxidase causes H2O2 release, DNA damage and apoptosis. Our studies indicate novel targets for therapeutic intervention and risk assessment in H. pylori-induced gastric cancer. More studies addressing the many potential contributors to oxidative stress, chronic inflammation, and gastric carcinogenesis are essential for development of therapeutics and identification of gastric cancer biomarkers.
Introduction
Helicobacter pylori is a Gram-negative, microaerophilic bacterium that infects up to 50% of the global human population.Citation1H. pylori selectively colonizes the human gastric mucosa and is directly responsible for gastroduodenal pathologies that include chronic gastritis, peptic ulcers, mucosa-associated lymphoid tissue lymphoma, and gastric adenocarcinoma.Citation2,Citation3 The human immune system mounts a vigorous innate and adaptive response to H. pylori, but it is generally unable to eradicate the pathogen.Citation4,Citation5 This failed immune response leads directly to chronic inflammation, oxidative stress, and DNA damage.Citation5-Citation9 It is well known that oxidative stress has the capacity to cause DNA damage.Citation10 Thus, we hypothesize that these deleterious outcomes are responsible for neoplastic transformation within the gastric mucosa that can ultimately lead to gastric adenocarcinoma. Gastric cancer is the second leading cause of cancer death worldwide, with H. pylori infection serving as the strongest known risk factor for this malignancy.Citation2,Citation11 Moreover, the World Health Organization classifies H. pylori as a Type 1 carcinogen.Citation12 As such, there is high demand for new insights into the roles of oxidative stress and chronic inflammation in the progression from H. pylori infection to gastric cancer. In this review, we dissect the role of H. pylori-derived factors, immune cells, and epithelial signaling events in the generation of oxidative stress, DNA damage, and cancer risk.
H. pylori Factors that Induce Oxidative Stress, DNA Damage and Inflammation
CagA
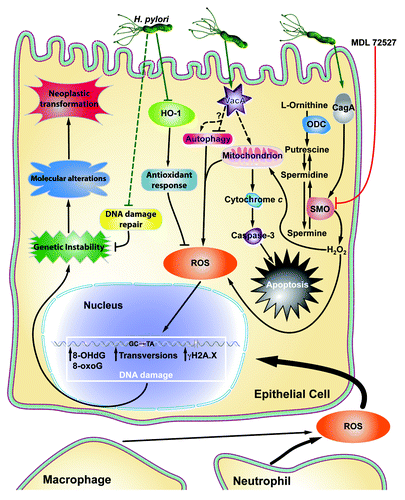
It has long been understood that individuals infected with H. pylori strains possessing cytotoxin associated gene A (cagA), encoding for the protein CagA, are at a substantially increased risk for developing gastric cancer compared with persons infected with cagA-negative strains.Citation11,Citation13 While CagA has been linked to many biological effects, the precise mechanism by which it leads to gastric cancer is poorly understood. A recent report indicates that cagA+ strains induce greater levels of hydrogen peroxide (H2O2) production and oxidative DNA damage ().Citation14 This DNA damage was measured by 8-oxoguanosine levels using flow cytometry in gastric epithelial cells from in vitro and in vivo models, and 8-hydroxy-2′-deoxyguanosine (8-OHdG) levels by immunohistochemistry in tissues.Citation14
Figure 1. Schematic summary of the various pathways by which ROS production and DNA damage are induced in gastric epithelial cells. H. pylori CagA induces SMO expression, leading to H2O2 production, which causes ROS accumulation and apoptosis. MDL 7527 inhibits SMO and attenuates the deleterious effects of its activation. H. pylori VacA targets the mitochondrial membrane, resulting in ROS production and cytochrome c release. Cytochrome c release activates caspase-3, leading to apoptosis. H. pylori can inhibit HO-1 and the antioxidant response. Neutrophils and phagocytes also contribute to ROS accumulation. ROS lead to various forms of DNA damage, including transversions, oxidative DNA damage (8-OHdG, 8-hydroxy-2′-deoxyguanosine; and 8-oxoG, 8-oxoguanosine), and double strand breaks (γH2A.X). This, combined with H. pylori-mediated inhibition of DNA damage repair, leads to genomic instability within gastric epithelial cells. Subsequently, molecular alterations and neoplastic transformations occur, resulting in gastric carcinogenesis.
Moreover, cagA positivity is significantly associated with increased expression of tumor necrosis factor α (TNFα) and interleukin (IL)-8, both of which are markers of inflammation and oxidative stress.Citation6 However, cagA gene status alone is not sufficient to stratify risk of developing gastric cancer. We recently reported that among subjects in Colombia that all harbored cagA+ strains, the severity of preneoplastic lesions and oxidative DNA damage also depended on phylogeographic origin of the strains.Citation15 Thus, other H. pylori-associated factors likely contribute to the oxidative stress and chronic inflammation associated with infection.Citation15
VacA
Vacuolating cytotoxin A (vacA) is another well-studied H. pylori virulence gene. It encodes for the protein VacA, which enhances colonization and correlates with increased inflammation and risk for gastric carcinogenesis.Citation16 Like CagA, the mechanism by which VacA increases disease severity and cancer risk is unknown. However, it has been demonstrated that VacA induces Ca2+ influx and reactive oxygen species (ROS) generation, which can be measured by 7'-dichlorodihydrofluorescein diacetate (DCFDA) staining and flow cytometry,Citation17 in eosinophils and various other cell types by targeting the inner mitochondrial membrane ().Citation18,Citation19 The intracellular Ca2+ and ROS subsequently lead to nuclear factor kappa-light-chain-enhancer of B-cell activation (NFκB) activation and upregulation of chemokine expression, presumably enhancing influx of pro-inflammatory immunocytes to the site of H. pylori infection.Citation18
Moreover, VacA appears to play a role in autophagy (), although there are conflicting reports regarding effect of VacA in this process. It has been reported that VacA disrupts autophagy within human gastric epithelial cells.Citation16 Disruption of autophagy results in an increase in cellular ROS, as this mechanism for clearance of cellular injury is attenuated.Citation16 VacA-induced increases in cellular ROS concentrations can contribute to an environment that favors oxidative DNA damage and subsequent molecular alterations that lead to neoplastic transformations and cancer (). However, it has also been reported that VacA induces autophagy.Citation20 In this second study, VacA was shown to induce autophagy via its binding to low density lipoprotein-receptor related protein 1 (LRP1). LRP1 binding results in the accumulation of ROS, leading to AKT activation, mouse double minute 2 (MDM2) phosphorylation, p53 degradation, and induction of autophagy.Citation20 Regardless of its mechanism of action, VacA leads to increased cellular ROS and contributes to the pro-tumorigenic environment during H. pylori infection.Citation16,Citation20
GGT
γ−glutamyl transferase (GGT) is another H. pylori virulence factor that contributes to disease severity during infection. Purified GGT elicited H2O2 production in gastric epithelial cells, as well as a corresponding increase in NFκB activation and IL-8 production.Citation21 IL-8 is a pro-inflammatory cytokine that signifies the establishment of inflammation at the site of infection; in addition IL-8 has pro-tumorigenic properties.Citation22-Citation26 Moreover, addition of GGT to gastric epithelial cells resulted in increased levels of 8-OHdG, indicative of oxidative DNA damage, as measured by flow cytometry.Citation21 Increased ROS, pro-inflammatory cytokine production and oxidative DNA damage create ideal conditions for the progression from infection to cancer.
Urease and NapA
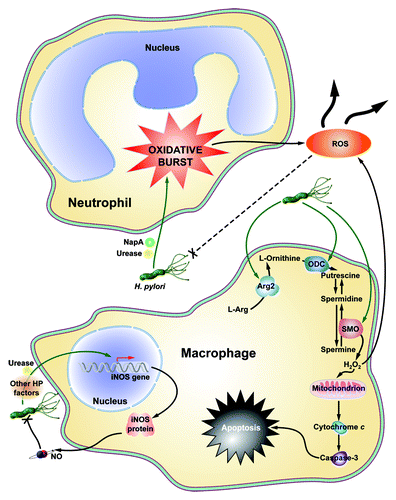
Urease and neutrophil activating protein A (NapA) are two H. pylori virulence factors that contribute to neutrophil infiltration and ROS production (). In a paw edema model in wild-type Swiss mice, recombinant urease induced neutrophil infiltration, high levels of inflammation and ROS production.Citation27 Moreover, the infiltrating neutrophils demonstrated increased resistance to apoptosis and an increased lifespan, potential contributing factors to the chronic inflammation associated with H. pylori infection.Citation4,Citation5,Citation27 NapA also functions to recruit neutrophils to the site of infection.Citation28 NapA is sufficient to induce the oxidative burst from infiltrating neutrophils, which contributes to cellular stress and oxidative DNA damage.Citation28 Hypothetically, neutrophil ROS should non-specifically kill both infected and uninfected cells, and the pathogen. Interestingly, NapA also appears to protect H. pylori from the release of oxyradicals, perpetuating the cycle of neutrophil infiltration and oxidative burst.Citation28
Figure 2. Schematic summary of macrophage and neutrophil contributions to ROS production during H. pylori infection. H. pylori NapA and Urease cause neutrophils to undergo an oxidative burst, leaking substantial amounts of ROS into the infection microenvironment. Moreover, NapA leads to increased survival in neutrophils, compounding their effects on oxidative stress. Macrophage iNOS is induced by urease during infection, leading to the production of NO, a reactive nitrogen species. Also, as yet undetermined H. pylori factors cause Arg2 activation in macrophages, resulting in apoptosis. ODC is also induced in macrophages during infection. This leads to downstream H2O2 production by SMO, and apoptosis.
Cellular Processes that are Dysregulated during H. pylori Infection Leading to Oxidative Stress, DNA Damage and Inflammation
Increased expression of spermine oxidase during H. pylori infection
Spermine oxidase (SMO) is an enzyme in the polyamine metabolic pathway that is responsible for the back-conversion of spermine to spermidine (). In doing so, SMO is directly responsible for generating H2O2, which leads to mitochondrial membrane depolarization and the activation of caspase-mediated apoptosis.Citation14,Citation29 H2O2 can be directly measured by an Amplex® Red enzyme assay, in addition to flow cytometry using DCFDA.Citation10,Citation29 It has been reported that infection with H. pylori leads to increased expression of SMO in gastric epithelial cells, and that this effect is more profound in cagA+ strains than in isogenic mutants lacking cagA.Citation10,Citation14 Additionally, in human subjects, patients harboring strains with functional CagA secretion exhibited higher levels of SMO when assessed by flow cytometry or immunohistochemistry.Citation13
CagA phosphorylation is an integral step in determining CagA function within host cells.Citation30,Citation31 Our laboratory found that levels of both CagA and phospho-CagA (pCagA) in clinical isolates co-cultured with gastric epithelial cells correlated with levels of SMO expression in gastritis tissues from the source patients for these strains.Citation14 While pCagA is likely involved in inducing ROS production, further studies using in vitro and in vivo models are needed to determine the role of pCagA vs. native CagA in the stimulation of ROS generation.
In parallel with increased SMO expression, there is a corresponding increase in oxidative DNA damage and apoptosis in vitro and in vivo.Citation10,Citation14 Intriguingly, we have demonstrated that there is a subpopulation of cells in our in vitro models and in cells isolated from infected mice and gerbils with dysplasia and carcinoma, which have a substantial increase in DNA damage, but are resistant to apoptosis.Citation13 In macrophages, H. pylori also upregulates SMO expression and release of H2O2, which causes apoptosis in these cells ().Citation29 This is another mechanism contributing to the oxidative stress in tissues, as the upstream enzyme, ornithine decarboxylase (ODC), the rate-limiting enzyme for polyamine synthesis ( and ) is upregulated in lamina propria macrophages during H. pylori infection.Citation32
In addition to the survival of a subgroup of cells with DNA damage, there is also the pro-carcinogenic effect of constant stimulation of apoptosis in other gastric epithelial cell populations, which can serve as a pathologic inducer of proliferation.Citation33 Thus, upregulation of SMO expression is a potential mechanism that contributes to the development of gastric carcinoma.
An important question is whether treatment of H. pylori infection has a beneficial effect on SMO expression and DNA damage. We have reported that in gastric tissues from human subjects that were eradicated, levels of SMO mRNA in gastric epithelial cells isolated by laser capture microdissection were significantly decreased compared with their pre-eradications levels.Citation10 We speculate that there is the potential to reverse DNA damage by eradicating the infection, but this may depend on the histological stage of disease. More in vivo studies are needed to address this issue. Chemical inhibition of SMO with MDL 72527 attenuates H2O2 release, DNA damage, and apoptosis, providing further evidence that DNA damage could be reversible or preventable.Citation29
Increased DNA damage and defective DNA damage repair
One of the primary risks of increased ROS and oxidative cellular stress is DNA damage (). Without proper control of oxidative stress and DNA damage repair, neoplastic transformation that leads to cancer can occur. H. pylori is well known to cause enhanced ROS levels from both neutrophil infiltration and de novo synthesis in epithelial cells both in vivo and in vitro.Citation34-Citation36 The increase in ROS has been directly linked to oxidative DNA damage, and DNA deamination and alkylation.Citation37-Citation39 Indeed, studies in Big Blue transgenic mice have directly demonstrated the induction of H. pylori-induced DNA damage in this reporter system.Citation40,Citation41 The oxidative damage frequently consisted of AT to GC and GC to TA transversions () within the genome of gastric epithelial cells.Citation41 In fact, these transversions were 4-fold higher in mice infected with H. pylori after just six months.Citation41 Moreover, as a result of decreased inflammation, Big Blue mice deficient for 8-oxoguanosine DNA glycosylase (OGG1) had significantly decreased numbers of transversions and 8-oxoguanosine lesions.Citation41 This further highlights the deleterious role of chronic inflammation in inducing oxidative DNA damage.
An additional consideration is that H. pylori has been shown to induce DNA strand breaks in vitro and this may directly contribute to genomic instability and neoplastic transformation ().Citation42 Recent studies from our laboratory have demonstrated that in addition to increased levels of oxidative DNA damage, in vivo infection in a rodent model is associated with increased levels of phospho-histone H2AX.2, a marker of DNA double strand breaks, when assessed by flow cytometry in gastric epithelial cells. In addition to our in vitro data showing that siRNA knockdown or chemical inhibition of SMO ameliorates H. pylori induced DNA damage,Citation13 we have found that treatment of gerbils infected with H. pylori with inhibitors of SMO or ODC results in marked attenuation of gastric epithelial cell DNA damage and of development of dysplasia and carcinoma.
Not only does H. pylori infection induce oxidative DNA damage and double strand breaks, but infection also dysregulates DNA repair mechanisms. Both mismatch repair and base excision repair mechanisms are inhibited during H. pylori infection ().Citation43,Citation44 Northern and western blot analyses confirmed decreased expression of mismatch repair proteins, MSH2, MSH6, and MLH1.Citation43 In addtion, mismatch repair genes MSH3, PMS1, and PMS2 were also markedly reduced in expression in further studies.Citation44 Each of these genes have also been implicated in the progression to colon cancer.Citation43
Furthermore, apurinic/apyrimidinic endonuclease 1 (APE-1) mRNA expression is significantly decreased in human AGS cells, indicating a defect in base excision repair, making cells susceptible to both nuclear and mitochondrial genomic instability.Citation44 However, a previous study in human epithelial cells indicates that H. pylori-induced ROS actually increase APE-1 expression.Citation45 This would indicate an increased capacity to correct oxidative DNA damage. Nevertheless, APE-1 becomes acetylated upon H. pylori infection, causing the suppression of Bax expression and p53-mediated apoptosis.Citation46 Given our recent data that there are increased numbers of cells with DNA damage that are resistant to apoptosis, this could be a potential mechanism by which apoptosis is inhibited during infection, leading to increasing numbers of transformed cells.
Within the cancer field, biomarkers are becoming essential for early detection and determination of cancer prognosis. We and others posit that 8-oxoguanosine could be a potential biomarker for gastric cancer.Citation15,Citation47 8-oxoguanosine and 8-OHdG assays measure oxodized nucleotide adducts, which form as a result of oxidative DNA damage.Citation47 They are considered direct markers for DNA damage and surrogate markers for ROS production in vivo.Citation47 Quantitative measurement of ROS in gastric tissues in vivo is difficult, but dihydroethidine staining has been utilized for qualitative studies in infected mice.Citation48 H. pylori causes an increase in ROS, both from immune cell infiltration and de novo synthesis by epithelial cells.Citation6,Citation9,Citation11,Citation14,Citation15,Citation22,Citation34,Citation35,Citation37 Oxidative stress leads to oxidative DNA damage, making 8-oxoguanosine, a marker for oxidative DNA damage, an ideal candidate biomarker for detecting and assessing gastric cancer.Citation15,Citation47
A defective antioxidant response
Heme oxygenase 1 (HO-1), a potent antioxidant and anti-inflammatory enzyme that is responsible for the degradation of heme, has well-studied and wide-ranging protective functions across many disease models.Citation49 HO-1 expression is induced by the presence of nitric oxide (NO), leading to decreased IL-8 expression and inflammation.Citation50 Importantly, HO-1 has the capacity to prevent CagA phosphorylation, significantly attenuating H. pylori virulence and pathogenesis.Citation51 However, the protective role of HO-1 is subverted during H. pylori infection; HO-1 expression is dependent on NFκB activation and signaling, but H. pylori disrupts NO-induced NFκB activation ().Citation50 The loss of NFκB signaling has substantial effects on gastric epithelial cells, leading to increased ROS, apoptosis and necrosis, and DNA damage, and a more rapid development of preneoplasia in vivo.Citation48
H. pylori-mediated HO-1 inhibition results in many of the same consequences, leading to markedly enhanced inflammation.Citation50 Overexpression of HO-1 during H. pylori infection reverses these effects and restores the protective function of HO-1.Citation50 The loss of an effective antioxidant response to H. pylori-induced oxidative stress is another potential contributing factor to the progression to gastric carcinoma.
Furthermore, other components of the oxidative stress response are inhibited in gastric cancer. Expression of glutathione peroxidase 3 (GPX3) is silenced by DNA hypermethylation in gastric carcinomas, and is associated with tumor invasion and lymph node metastases.Citation52 As H. pylori is the strongest known risk factor for gastric cancer, GPX3 inhibition could potentially be caused, either directly or indirectly, by the bacterium. Further studies are needed to determine the role of H. pylori in dysregulating the oxidative stress response.
A role for nitrosative stress?
While no direct causal link has been established between nitrosative stress and H. pylori-induced gastric cancer, reactive nitrogen species (RNS) may also cooperate with ROS to create an environment conducive to DNA damage and neoplastic transformation. In fact, RNS have been implicated as mediators of DNA damage in H. pylori infected mice.Citation53 Moreover, H. pylori is known to upregulate the expression of inducible nitric oxide synthase (iNOS) in macrophages ().Citation54 This leads to a marked increase in nitric oxide (NO) production. NO itself is a powerful signaling molecule that regulates immune functionsCitation50 and can kill H. pylori, but only when released in high concentrations.Citation7,Citation32 In humans, increased expression of iNOS occurs in both immune cells and gastric epithelial cells, indicating that NO may also play a role in H. pylori-induced injury.Citation22 However, its exact mode of action remains unclear; more studies addressing the role of nitrosative stress and carcinogenesis are needed.
Contributions of immune cells
H. pylori infection induces a dynamic innate immune response.Citation4,Citation5 A major component of this response is the oxidative burst from phagocytes like macrophages and neutrophils ().Citation6 As was previously discussed, H. pylori is capable of inducing the oxidative burst, which releases significant amounts of oxyradicals into the infection microenvironment.Citation9 Similarly, several models of colon cancer have established that chronic inflammation leads to more ROS production, greatly increased DNA damage, and more severe disease pathology.Citation39,Citation55
To the disadvantage of the host, H. pylori has the ability to survive the oxidative burst, via catalase and superoxide dismutase (SOD), and continue to persistently colonize the human gastric mucosa.Citation9 Further still, H. pylori can survive oxidative damage by subverting phagolysome-mediated killing in a VacA-dependent mechanism.Citation56 This creates a vicious cycle of immunocyte infiltration, oxidative burst, and DNA damage that contributes to gastric carcinogenesis ().
Additionally, H. pylori stimulates iNOS expression and NO release from macrophages, contributing to nitrosative stress.Citation54,Citation57 H. pylori also activates NFκB-dependent expression of arginase II (Arg2) and induces ODC expression ().Citation58 This leads to macrophage apoptosis, directly contributing to the failed immune response to H. pylori infection.Citation58 By failing to eradicate the bacteria, the innate immune system is likely continually mounting a response to H. pylori, further contributing to the malicious cycle of immune cell ROS production, oxidative stress, and DNA damage.
Contributions of microbiota and environmental factors
As with any disease, the human microbiome and various environmental factors likely play a role. A recent study in wild-type C57BL/6 mice demonstrated that the amount of inflammation associated with H. pylori is directly related to the pre-infection microbiota.Citation59 Specifically, the Clostridium species present in the gastric microbiome, after treatment with antibiotics prior to infection, determine the degree of CD4+ T-cell infiltration and interferon-γ expression.Citation59 Analysis of the gastric microbiota could potentially be used as a tool to determine the risk of developing severe disease.Citation59
Diet may also play a role in disease severity. High salt intake appears to exacerbate disease severity in humans infected with H. pylori, while fruit and vegetable intake decreased disease pathology.Citation60 Moreover, gerbils maintained on an iron-depleted diet also experience increased H. pylori-induced inflammation, dysplasia, and carcinoma.Citation61 While the latter study did not specifically address oxidative stress and DNA damage, we speculate that increased inflammation would lead to increased ROS and oxidative DNA damage under these conditions. This could be a potential mechanism by which lack of iron contributes to carcinogenesis. Other environmental factors, like smoking, alcohol use, and high consumption of red meat also contribute to cancer risk.Citation62
Conclusions
H. pylori infection is clearly linked to chronic inflammation-associated oxidative stress and significant amounts of DNA damage.Citation38,Citation42,Citation44,Citation53,Citation55 We hypothesize that these consequences of infection are the primary mediators of H. pylori-induced carcinogenesis. Ongoing work in our laboratory has implicated the generation of oxidative stress-induced DNA damage in the progression from infection to gastric cancer. Mouse and gerbil models with suppression of these events exhibit reduction in dysplasia and carcinoma.Citation7,Citation14 However, more studies in state-of-the-art model systems, such as 3-dimensional organoid cultures, are needed to address the molecular signaling events that lead to pathologic enhancement of oxidative stress. This will lead to molecular targets for therapeutic intervention in animal models and in humans. Further understanding of these pathways will also lead to improved risk stratification for undertaking such interventions, such as inhibition of polyamine synthesis and oxidation by blocking ODC and SMO, respectively.
| Abbreviations: | ||
| ROS | = | reactive oxygen species |
| CagA | = | cytotoxin associated gene A |
| VacA | = | vacuolating cytotoxin A |
| 8-OHdG | = | 8-hydroxy-2′-deoxyguanosine |
| 8-oxoG | = | 8-oxoguanosine |
| TNFα | = | tumor necrosis factor alpha |
| IL | = | interleukin |
| NFκB | = | nuclear factor kappa-light-chain-enhancer of B-cell activation |
| GGT | = | γ−glutamyl transferase |
| NapA | = | neutrophil activating protein A |
| SMO | = | spermine oxidase |
| H2O2 | = | hydrogen peroxide |
| ODC | = | ornithine decarboxylase |
| OGG1 | = | 8-oxoguanosine DNA glycosylase |
| APE-1 | = | apurinic/apyrimidinic endonuclease 1 |
| HO-1 | = | heme oxygenase 1 |
| NO | = | nitric oxide |
| GPX3 | = | glutathione peroxidase 3 |
| RNS | = | reactive nitrogen species |
| iNOS | = | inducible nitric oxide synthase |
| Arg2 | = | arginase II |
| siRNA | = | small interfering ribonucleic acid |
| MDL 72527 | = | N1,N4-bis(2,3-butandienyl)-1,4-butanediamine |
| DCFDA | = | 7'-dichlorodihydrofluorescein diacetate |
| MDM2 | = | mouse double minute 2 |
| LRP1 | = | low density lipoprotein-receptor related protein 1 |
Acknowledgments
This work was supported by the National Institutes of Health grants R01DK053620, R01AT004821, P01CA028842, and P01CA116087 (to K.T.W.), the Vanderbilt University Digestive Disease Research Center supported by grant P30DK058404, UL1RR024975 (Vanderbilt CTSA, Pilot Project to K.T.W.), K01AT007324 (to R.C), and a Merit Review Grant 1I01BX001453 from the Office of Medical Research, Department of Veterans Affairs (to K.T.W.). D.M.H. was supported by NIH grant 5T32GM008554 and the Thomas F. Frist, Sr. endowment (to K.T.W.).
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Related Research Data
References
- Nomura ASG, Stemmermann GN, Chyou PH, Kato I, Perez-Perez GI, Blaser MJ. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N Engl J Med 1991; 325:1132 - 6; http://dx.doi.org/10.1056/NEJM199110173251604; PMID: 1891021
- Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, et al. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med 1991; 325:1127 - 31; http://dx.doi.org/10.1056/NEJM199110173251603; PMID: 1891020
- Peek RM Jr., Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer 2002; 2:28 - 37; http://dx.doi.org/10.1038/nrc703; PMID: 11902583
- Wilson KT, Crabtree JE. Immunology of Helicobacter pylori: insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology 2007; 133:288 - 308; http://dx.doi.org/10.1053/j.gastro.2007.05.008; PMID: 17631150
- Peek RM Jr., Fiske C, Wilson KT. Role of innate immunity in Helicobacter pylori-induced gastric malignancy. Physiol Rev 2010; 90:831 - 58; http://dx.doi.org/10.1152/physrev.00039.2009; PMID: 20664074
- Augusto AC, Miguel F, Mendonça S, Pedrazzoli J Jr., Gurgueira SA. Oxidative stress expression status associated to Helicobacter pylori virulence in gastric diseases. Clin Biochem 2007; 40:615 - 22; http://dx.doi.org/10.1016/j.clinbiochem.2007.03.014; PMID: 17466292
- Chaturvedi R, de Sablet T, Coburn LA, Gobert AP, Wilson KT. Arginine and polyamines in Helicobacter pylori-induced immune dysregulation and gastric carcinogenesis. Amino Acids 2012; 42:627 - 40; http://dx.doi.org/10.1007/s00726-011-1038-4; PMID: 21874531
- Touati E, Michel V, Thiberge J-M, Wuscher N, Huerre M, Labigne A. Chronic Helicobacter pylori infections induce gastric mutations in mice. Gastroenterology 2003; 124:1408 - 19; http://dx.doi.org/10.1016/S0016-5085(03)00266-X; PMID: 12730880
- Ramarao N, Gray-Owen SD, Meyer TF. Helicobacter pylori induces but survives the extracellular release of oxygen radicals from professional phagocytes using its catalase activity. Mol Microbiol 2000; 38:103 - 13; http://dx.doi.org/10.1046/j.1365-2958.2000.02114.x; PMID: 11029693
- Xu H, Chaturvedi R, Cheng Y, Bussière FI, Asim M, Yao MD, et al. Spermine oxidation induced by Helicobacter pylori results in apoptosis and DNA damage: implications for gastric carcinogenesis. Cancer Res 2004; 64:8521 - 5; http://dx.doi.org/10.1158/0008-5472.CAN-04-3511; PMID: 15574757
- Blaser MJ, Perez-Perez GI, Kleanthous H, Cover TL, Peek RM, Chyou PH, et al. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res 1995; 55:2111 - 5; PMID: 7743510
- IARC. Infection with Helicobacter pylori.. IARC Monogr Eval Carcinog Risks Hum 1994; 61:177 - 240; PMID: 7715070
- Parsonnet J, Friedman GD, Orentreich N, Vogelman H. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut 1997; 40:297 - 301; PMID: 9135515
- Chaturvedi R, Asim M, Romero-Gallo J, Barry DP, Hoge S, de Sablet T, et al. Spermine oxidase mediates the gastric cancer risk associated with Helicobacter pylori CagA. Gastroenterology 2011; 141:1696 - , e1-2; http://dx.doi.org/10.1053/j.gastro.2011.07.045; PMID: 21839041
- de Sablet T, Piazuelo MB, Shaffer CL, Schneider BG, Asim M, Chaturvedi R, et al. Phylogeographic origin of Helicobacter pylori is a determinant of gastric cancer risk. Gut 2011; 60:1189 - 95; http://dx.doi.org/10.1136/gut.2010.234468; PMID: 21357593
- Raju D, Hussey S, Ang M, Terebiznik MR, Sibony M, Galindo-Mata E, et al. Vacuolating cytotoxin and variants in Atg16L1 that disrupt autophagy promote Helicobacter pylori infection in humans. Gastroenterology 2012; 142:1160 - 71; http://dx.doi.org/10.1053/j.gastro.2012.01.043; PMID: 22333951
- Eruslanov E, Kusmartsev S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol Biol 2010; 594:57 - 72; http://dx.doi.org/10.1007/978-1-60761-411-1_4; PMID: 20072909
- Kim JM, Kim JS, Lee JY, Kim Y-J, Youn H-J, Kim IY, et al. Vacuolating cytotoxin in Helicobacter pylori water-soluble proteins upregulates chemokine expression in human eosinophils via Ca2+ influx, mitochondrial reactive oxygen intermediates, and NF-kappaB activation. Infect Immun 2007; 75:3373 - 81; http://dx.doi.org/10.1128/IAI.01940-06; PMID: 17452475
- Willhite DC, Blanke SR. Helicobacter pylori vacuolating cytotoxin enters cells, localizes to the mitochondria, and induces mitochondrial membrane permeability changes correlated to toxin channel activity. Cell Microbiol 2004; 6:143 - 54; http://dx.doi.org/10.1046/j.1462-5822.2003.00347.x; PMID: 14706100
- Tsugawa H, Suzuki H, Saya H, Hatakeyama M, Hirayama T, Hirata K, et al. Reactive oxygen species-induced autophagic degradation of Helicobacter pylori CagA is specifically suppressed in cancer stem-like cells. Cell Host Microbe 2012; 12:764 - 77; http://dx.doi.org/10.1016/j.chom.2012.10.014; PMID: 23245321
- Gong M, Ling SSM, Lui SY, Yeoh KG, Ho B. Helicobacter pylori γ-glutamyl transpeptidase is a pathogenic factor in the development of peptic ulcer disease. Gastroenterology 2010; 139:564 - 73; http://dx.doi.org/10.1053/j.gastro.2010.03.050; PMID: 20347814
- Fu S, Ramanujam KS, Wong A, Fantry GT, Drachenberg CB, James SP, et al. Increased expression and cellular localization of inducible nitric oxide synthase and cyclooxygenase 2 in Helicobacter pylori gastritis. Gastroenterology 1999; 116:1319 - 29; http://dx.doi.org/10.1016/S0016-5085(99)70496-8; PMID: 10348815
- Li M, Zhang Y, Feurino LW, Wang H, Fisher WE, Brunicardi FC, et al. Interleukin-8 increases vascular endothelial growth factor and neuropilin expression and stimulates ERK activation in human pancreatic cancer. Cancer Sci 2008; 99:733 - 7; http://dx.doi.org/10.1111/j.1349-7006.2008.00740.x; PMID: 18307536
- Eftang LL, Esbensen Y, Tannæs TM, Bukholm IR, Bukholm G. Interleukin-8 is the single most up-regulated gene in whole genome profiling of H. pylori exposed gastric epithelial cells. BMC Microbiol 2012; 12:9; http://dx.doi.org/10.1186/1471-2180-12-9; PMID: 22248188
- Wang Y, Xu RC, Zhang XL, Niu XL, Qu Y, Li LZ, et al. Interleukin-8 secretion by ovarian cancer cells increases anchorage-independent growth, proliferation, angiogenic potential, adhesion and invasion. Cytokine 2012; 59:145 - 55; http://dx.doi.org/10.1016/j.cyto.2012.04.013; PMID: 22579115
- Asfaha S, Dubeykovskiy AN, Tomita H, Yang X, Stokes S, Shibata W, et al. Mice that express human interleukin-8 have increased mobilization of immature myeloid cells, which exacerbates inflammation and accelerates colon carcinogenesis. Gastroenterology 2013; 144:155 - 66; http://dx.doi.org/10.1053/j.gastro.2012.09.057; PMID: 23041326
- Uberti AF, Olivera-Severo D, Wassermann GE, Scopel-Guerra A, Moraes JA, Barcellos-de-Souza P, et al. Pro-inflammatory properties and neutrophil activation by Helicobacter pylori urease. Toxicon 2013; 69:240 - 9; http://dx.doi.org/10.1016/j.toxicon.2013.02.009; PMID: 23466444
- Wang G, Hong Y, Olczak A, Maier SE, Maier RJ. Dual Roles of Helicobacter pylori NapA in inducing and combating oxidative stress. Infect Immun 2006; 74:6839 - 46; http://dx.doi.org/10.1128/IAI.00991-06; PMID: 17030577
- Chaturvedi R, Cheng Y, Asim M, Bussière FI, Xu H, Gobert AP, et al. Induction of polyamine oxidase 1 by Helicobacter pylori causes macrophage apoptosis by hydrogen peroxide release and mitochondrial membrane depolarization. J Biol Chem 2004; 279:40161 - 73; http://dx.doi.org/10.1074/jbc.M401370200; PMID: 15247269
- Argent RH, Kidd M, Owen RJ, Thomas RJ, Limb MC, Atherton JC. Determinants and consequences of different levels of CagA phosphorylation for clinical isolates of Helicobacter pylori.. Gastroenterology 2004; 127:514 - 23; http://dx.doi.org/10.1053/j.gastro.2004.06.006; PMID: 15300584
- Lee IO, Kim JH, Choi YJ, Pillinger MH, Kim SY, Blaser MJ, et al. Helicobacter pylori CagA phosphorylation status determines the gp130-activated SHP2/ERK and JAK/STAT signal transduction pathways in gastric epithelial cells. J Biol Chem 2010; 285:16042 - 50; http://dx.doi.org/10.1074/jbc.M110.111054; PMID: 20348091
- Chaturvedi R, Asim M, Hoge S, Lewis ND, Singh K, Barry DP, et al. Polyamines impair immunity to Helicobacter pylori by inhibiting L-arginine uptake required for nitric oxide production. Gastroenterology 2010; 139:1686 - 98, e1-6; http://dx.doi.org/10.1053/j.gastro.2010.06.060; PMID: 20600019
- Valentin-Vega YA, Okano H, Lozano G. The intestinal epithelium compensates for p53-mediated cell death and guarantees organismal survival. Cell Death Differ 2008; 15:1772 - 81; http://dx.doi.org/10.1038/cdd.2008.109; PMID: 18636077
- Davies GR, Banatvala N, Collins CE, Sheaff MT, Abdi Y, Clements L, et al. Relationship between infective load of Helicobacter pylori and reactive oxygen metabolite production in antral mucosa. Scand J Gastroenterol 1994; 29:419 - 24; http://dx.doi.org/10.3109/00365529409096832; PMID: 8036457
- Bagchi D, Bhattacharya G, Stohs SJ. Production of reactive oxygen species by gastric cells in association with Helicobacter pylori.. Free Radic Res 1996; 24:439 - 50; http://dx.doi.org/10.3109/10715769609088043; PMID: 8804987
- Mannick EE, Bravo LE, Zarama G, Realpe JL, Zhang XJ, Ruiz B, et al. Inducible nitric oxide synthase, nitrotyrosine, and apoptosis in Helicobacter pylori gastritis: effect of antibiotics and antioxidants. Cancer Res 1996; 56:3238 - 43; PMID: 8764115
- Baik SC, Youn HS, Chung MH, Lee WK, Cho MJ, Ko GH, et al. Increased oxidative DNA damage in Helicobacter pylori-infected human gastric mucosa. Cancer Res 1996; 56:1279 - 82; PMID: 8640814
- Obst B, Wagner S, Sewing KF, Beil W. Helicobacter pylori causes DNA damage in gastric epithelial cells. Carcinogenesis 2000; 21:1111 - 5; http://dx.doi.org/10.1093/carcin/21.6.1111; PMID: 10836997
- Mangerich A, Knutson CG, Parry NM, Muthupalani S, Ye W, Prestwich E, et al. Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc Natl Acad Sci U S A 2012; 109:E1820 - 9; http://dx.doi.org/10.1073/pnas.1207829109; PMID: 22689960
- Jenks PJ, Jeremy AHT, Robinson PA, Walker MM, Crabtree JE. Long-term infection with Helicobacter felis and inactivation of the tumour suppressor gene p53 cumulatively enhance the gastric mutation frequency in Big Blue transgenic mice. J Pathol 2003; 201:596 - 602; http://dx.doi.org/10.1002/path.1488; PMID: 14648663
- Touati E, Michel V, Thiberge J-M, Avé P, Huerre M, Bourgade F, et al. Deficiency in OGG1 protects against inflammation and mutagenic effects associated with H. pylori infection in mouse. Helicobacter 2006; 11:494 - 505; http://dx.doi.org/10.1111/j.1523-5378.2006.00442.x; PMID: 16961812
- Toller IM, Neelsen KJ, Steger M, Hartung ML, Hottiger MO, Stucki M, et al. Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double-strand breaks and a DNA damage response in its host cells. Proc Natl Acad Sci U S A 2011; 108:14944 - 9; http://dx.doi.org/10.1073/pnas.1100959108; PMID: 21896770
- Kim JJ, Tao H, Carloni E, Leung WK, Graham DY, Sepulveda AR. Helicobacter pylori impairs DNA mismatch repair in gastric epithelial cells. Gastroenterology 2002; 123:542 - 53; http://dx.doi.org/10.1053/gast.2002.34751; PMID: 12145807
- Machado AMD, Figueiredo C, Touati E, Máximo V, Sousa S, Michel V, et al. Helicobacter pylori infection induces genetic instability of nuclear and mitochondrial DNA in gastric cells. Clin Cancer Res 2009; 15:2995 - 3002; http://dx.doi.org/10.1158/1078-0432.CCR-08-2686; PMID: 19383819
- Ding S-Z, O’Hara AM, Denning TL, Dirden-Kramer B, Mifflin RC, Reyes VE, et al. Helicobacter pylori and H2O2 increase AP endonuclease-1/redox factor-1 expression in human gastric epithelial cells. Gastroenterology 2004; 127:845 - 58; http://dx.doi.org/10.1053/j.gastro.2004.06.017; PMID: 15362040
- Bhattacharyya A, Chattopadhyay R, Burnette BR, Cross JV, Mitra S, Ernst PB, et al. Acetylation of apurinic/apyrimidinic endonuclease-1 regulates Helicobacter pylori-mediated gastric epithelial cell apoptosis. Gastroenterology 2009; 136:2258 - 69; http://dx.doi.org/10.1053/j.gastro.2009.02.014; PMID: 19505426
- Borrego S, Vazquez A, Dasí F, Cerdá C, Iradi A, Tormos C, et al. Oxidative stress and DNA damage in human gastric carcinoma: 8-Oxo-7'8-dihydro-2'-deoxyguanosine (8-oxo-dG) as a possible tumor marker. Int J Mol Sci 2013; 14:3467 - 86; http://dx.doi.org/10.3390/ijms14023467; PMID: 23389043
- Shibata W, Takaishi S, Muthupalani S, Pritchard DM, Whary MT, Rogers AB, et al. Conditional deletion of IkappaB-kinase-β accelerates helicobacter-dependent gastric apoptosis, proliferation, and preneoplasia. Gastroenterology 2010; 138:1022 - , e1-10; http://dx.doi.org/10.1053/j.gastro.2009.11.054; PMID: 19962981
- Ryter SW, Alam J, Choi AMK. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 2006; 86:583 - 650; http://dx.doi.org/10.1152/physrev.00011.2005; PMID: 16601269
- Gobert AP, Asim M, Piazuelo MB, Verriere T, Scull BP, de Sablet T, et al. Disruption of nitric oxide signaling by Helicobacter pylori results in enhanced inflammation by inhibition of heme oxygenase-1. J Immunol 2011; 187:5370 - 9; http://dx.doi.org/10.4049/jimmunol.1102111; PMID: 21987660
- Gobert AP, Verriere T, de Sablet T, Peek RM Jr., Chaturvedi R, Wilson KT. Haem oxygenase-1 inhibits phosphorylation of the Helicobacter pylori oncoprotein CagA in gastric epithelial cells. Cell Microbiol 2013; 15:145 - 56; http://dx.doi.org/10.1111/cmi.12039; PMID: 23051580
- Peng D-F, Hu T-L, Schneider BG, Chen Z, Xu Z-K, El-Rifai W. Silencing of glutathione peroxidase 3 through DNA hypermethylation is associated with lymph node metastasis in gastric carcinomas. PLoS One 2012; 7:e46214; http://dx.doi.org/10.1371/journal.pone.0046214; PMID: 23071548
- Katsurahara M, Kobayashi Y, Iwasa M, Ma N, Inoue H, Fujita N, et al. Reactive nitrogen species mediate DNA damage in Helicobacter pylori-infected gastric mucosa. Helicobacter 2009; 14:552 - 8; http://dx.doi.org/10.1111/j.1523-5378.2009.00719.x; PMID: 19889073
- Wilson KT, Ramanujam KS, Mobley HL, Musselman RF, James SP, Meltzer SJ. Helicobacter pylori stimulates inducible nitric oxide synthase expression and activity in a murine macrophage cell line. Gastroenterology 1996; 111:1524 - 33; http://dx.doi.org/10.1016/S0016-5085(96)70014-8; PMID: 8942731
- Meira LB, Bugni JM, Green SL, Lee C-W, Pang B, Borenshtein D, et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest 2008; 118:2516 - 25; PMID: 18521188
- Terebiznik MR, Vazquez CL, Torbicki K, Banks D, Wang T, Hong W, et al. Helicobacter pylori VacA toxin promotes bacterial intracellular survival in gastric epithelial cells. Infect Immun 2006; 74:6599 - 614; http://dx.doi.org/10.1128/IAI.01085-06; PMID: 17000720
- Gobert AP, Mersey BD, Cheng Y, Blumberg DR, Newton JC, Wilson KT. Cutting edge: urease release by Helicobacter pylori stimulates macrophage inducible nitric oxide synthase. J Immunol 2002; 168:6002 - 6; PMID: 12055207
- Gobert AP, Cheng Y, Wang J-Y, Boucher J-L, Iyer RK, Cederbaum SD, et al. Helicobacter pylori induces macrophage apoptosis by activation of arginase II. J Immunol 2002; 168:4692 - 700; PMID: 11971019
- Rolig AS, Cech C, Ahler E, Carter JE, Ottemann KM. The degree of Helicobacter pylori-triggered inflammation is manipulated by preinfection host microbiota. Infect Immun 2013; 81:1382 - 9; http://dx.doi.org/10.1128/IAI.00044-13; PMID: 23429529
- D’Elia L, Rossi G, Ippolito R, Cappuccio FP, Strazzullo P. Habitual salt intake and risk of gastric cancer: a meta-analysis of prospective studies. Clin Nutr 2012; 31:489 - 98; http://dx.doi.org/10.1016/j.clnu.2012.01.003; PMID: 22296873
- Noto JM, Gaddy JA, Lee JY, Piazuelo MB, Friedman DB, Colvin DC, et al. Iron deficiency accelerates Helicobacter pylori-induced carcinogenesis in rodents and humans. J Clin Invest 2013; 123:479 - 92; http://dx.doi.org/10.1172/JCI64373; PMID: 23257361
- Bonequi P, Meneses-González F, Correa P, Rabkin CS, Camargo MC. Risk factors for gastric cancer in Latin America: a meta-analysis. Cancer Causes Control 2013; 24:217 - 31; http://dx.doi.org/10.1007/s10552-012-0110-z; PMID: 23224270