
Abstract
Increasing evidence has accumulated to support that the human gut is a reservoir for antibiotic resistance genes. We previously identified more than 1000 genes displaying high similarity with known antibiotic resistance genes in the human gut gene set generated from the Chinese, Danish, and Spanish populations. Here, first, we add our new understanding of antibiotic resistance genes in the US and the Japanese populations; next, we describe the structure of a vancomycin-resistant operon in a Danish sample; and finally, we provide discussions on the correlation of the abundance of resistance genes in human gut with the antibiotic consumption in human medicine and in animal husbandry. These results, combined with those we published previously, provide comprehensive insights into the antibiotic resistance genes in the human gut microbiota at a population level.
Antibiotic resistance has been regarded as a serious worldwide public health problem for many years. Until recently, studies were focused mainly on the antibiotic resistance genes and their dissemination in drug-resistant pathogens. Now, more attention is being paid to the antibiotic resistance genes in the human gut commensal bacteria: the gut microbiota.Citation1-Citation4
The human gut microbiota was considered to be a reservoir for antibiotic resistance genes.Citation5 Resistance genes harbored by the native gut bacteria could be transferred among the residents, which will be a problem when the commensal opportunistic pathogens cause post-surgical infections. The transfer can even occur when the bacteria, e.g., swallowed ones, just pass through the intestine; these passersby might return to the mouth or skin by contamination with excreted bacteria. Evidence has accumulated to support the reservoir hypothesis, and the antibiotic resistance gene transfer events had been observed both in vivo and in vitro models.Citation6,Citation7 In fact, due to the dense microbial population in the gut environment, the potential for gene transfer might be higher than expected.Citation8 This situation represents a high risk with regard to the spread of antibiotic resistance genes and to the increased emergence of antibiotic-resistant human pathogenic bacteria.
Along with the developments of molecular biology and sequencing technologies, the focus of research on antibiotic resistance genes in the human gut is gradually changing from a single isolation to a bacterial community level. A study using microarray analysis targeting healthy volunteers from six European countries indicated that TetM and TetW are the most prevalent tetracycline resistance gene types in their oral and fecal metagenomes, respectively.Citation9 Another study applied metagenomic expression library to functionally characterize antibiotic resistance genes from saliva and fecal samples of two healthy humans and identified a total of 210 antibiotic resistance genes.Citation4 Though these studies have helped us to better understand the antibiotic resistance gene reservoir in human gut, the story of the whole profile of these resistance genes—especially at a human population level—is just beginning.
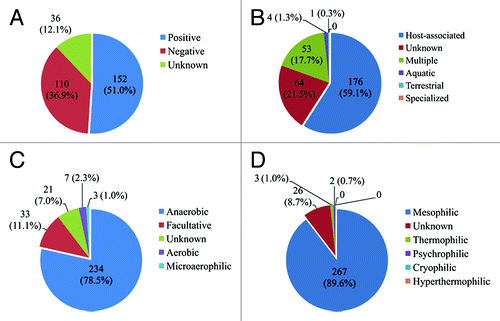
We previously analyzed the diversity and abundance of the antibiotic resistance genes in gut microbiota of 162 individuals belonging to three different countries, China, Denmark, and Spain.Citation10 From about 4 million human gut non-redundant common gene, we identified a total of 1093 antibiotic resistance genes, displaying more than 80% similarity at protein level with those in the Antibiotic Resistance Genes Database (ARDB).Citation11 We found that the microbial origin of the antibiotic resistance genes was not consistent with other gut genes at the phylum level. In fact, among these 1093 antibiotic resistance genes, 298 could be assigned to 106 specific strains at species and subspecies level by using the MEGAN program, and approximately 51%, 59%, 79%, and 90% of these assigned genes were affiliated to gram positive, host-associated, anaerobic, and mesophilic organisms, respectively ().
Figure 1. The distribution of 298 antibiotic resistance genes assigned to specific strains. The classification of the strains was according to the NCBI taxonomy feature: (A) gram positive or gram negative; (B) environment/host/reservoir; (C) anaerobic/aerobic/facultative; and (D) temperature growth characteristics, e.g., mesophilic, psychrophilic, etc. The taxonomic affiliation analysis was based on the use of the MEtaGenome Analyzer (MEGAN, version 3.9) (LCA parameters: mini-score 35, top percentage 10%).
The 1093 antibiotic resistance genes were further classified into 149 gene types according to ARDB. We found both the Chinese and Danish population harbored 133 resistance gene types, and the Spanish population harbored 128 gene types.Citation10 Here, we further analyzed the metagenomic sequencing data from 18 US adultsCitation12,Citation13 and 13 Japanese adults and infants.Citation14 By using the same searching method we used previously, we identified a total of 98 antibiotic resistance gene types in the US samples and 63 types in the Japanese (Table S1). The widely distributed gene type in the US population was TetW, existing in 19 out of 20 samples, followed by MacB (18 samples) and BacA (17 samples), while in the Japanese population were BacA (12 samples), TetO (10 samples), and TetQ and TetW (both in 9 samples). Interestingly, among the 13 Japanese individuals, a 7 month old female child F1-U contained 26 resistance gene types, which was the highest in the cohort; only 11 and 14 gene types were found in her mother and father’s guts, respectively. Further analysis shown that, among the 26 gene types in this child, only 7 were the same as either her mother’s, her father’s, or both. Another child who was 4 months old contained the second highest number of gene types (20 types). As neither subject encountered antibiotics for at least 4 weeks prior to sampling, neither had a history of gastrointestinal disorder or unusual eating behaviors.Citation14 The reason and means by which so many antibiotic resistance genes were acquired in these children’s guts are interesting.
As these data were generated by using Roche/454 and Sanger sequencing methods (Roche/454 data for 18 US samples, Sanger data for two US samples, and 13 Japanese samples) and the sequencing depth was limited (on average, 0.11 Gb and 0.057 Gb data per sample for US and Japan, respectively, while 2.31, 3.32, and 3.19 Gb data for China, Denmark, and Spain, respectively), the results here were not compared with our previous publication. In other words, variance (batch effectCitation15) will be undoubtedly introduced when conducting comparative analysis. We therefore merely give a short glimpse into the profile of the antibiotic resistance genes in the American and Japanese samples, instead of providing a comparative result.
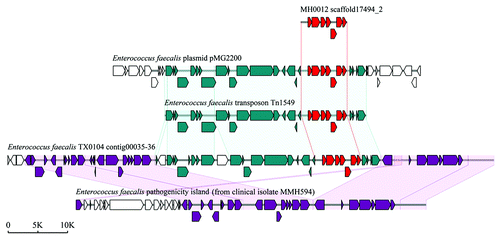
Previously, we used a method similar to RPKM (reads per kilo bases per million reads) to determine the relative abundance of each antibiotic resistance gene identified. Interestingly, the Danish female individuals have higher abundance of resistance genes than male (P = 0.002), but no obvious differences were observed in the Chinese and Spanish populations (). Further, we found the TetQ gene type was the most abundant in all three countries, and the abundance of other gene types differs from country to country. It is noteworthy that several genes belonging to vancomycin-resistant operons, VanRG for example, are also very common in each country. As not all structural gene sequences of individual Van-type operons (at least ðve genes for each Van operon) can be assembled together, we cannot estimate the real abundance of the vancomycin resistance determinants. However, we found a complete VanB operon sequence containing seven structural genes on the scaffold of a Danish sample, MH0012. Detailed analysis indicated that the sequence of the VanB operon in this sample was consistent with that carried on Enterococcus faecalis transposon Tn1549; this transposon was also found to be harbored by E. faecalis plasmid pMG2200 and by E. faecalis TX0104 genome (), suggesting that the VanB operon is frequently horizontally transferred. Interestingly, in E. faecalis TX0104, the VanB operon-carrying transposon was inserted exactly into its pathogenecity island. So, whether the acquisition of a vancomycin resistance at the cost of reducing the virulence in E. faecalis TX0104 is unclear and is deserved to be further investigated.
Figure 2. Comparison of the relative abundance of antibiotic resistance genes between female and male individuals in different populations. China: female, n = 18; male, n = 20. Denmark: female, n = 45; male, n = 40. Spain: female, n = 24; male, n = 15.
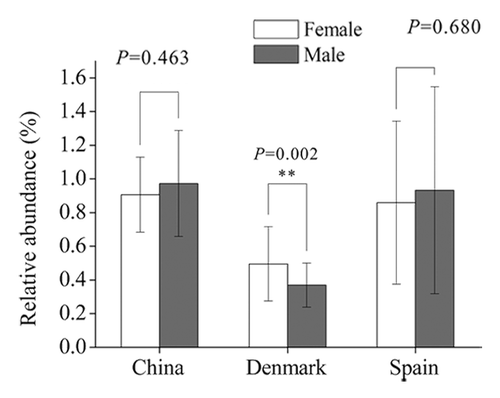
Figure 3. Schematic representations of the vancomycin-resistant VanB operon and comparison of the operon with representative counterparts in NCBI database. The VanB operon (red ORFs) was identified in a scaffold of Danish sample MH0012. The accession numbers of E. faecalis plasmid pMG2200, E. faecalis transposon Tn1549, E. faecalis TX0104 genome, and E. faecalis MMH594 pathogenecity island are AB374546, AF192329, ACGL00000000, and AF454824, respectively.
It is recognized that antibiotic consumption is the main cause of the emerging resistance, and antibiotics used in animals have been shown to contribute to the emergence of antibiotic resistance in humans. Therefore, to correlate the abundance of antibiotic resistance genes in human gut microbiota with antibiotic consumption, we first reviewed the total outpatient antibacterial use in 20 European countries from 1999 to 2008 (). On average, in this ten-year period, the highest antibiotic consumption rate was found in Greece (34.6 Defined Daily Dose [DDD] per 1000 inhabitants daily) and the lowest in the Netherlands (10.3 DDD per 1000 inhabitants daily). Spain exhibited a 1.4 times higher consumption rate than Denmark (19.0 vs. 14.0 DDD per 1000 inhabitants daily). The different consumption rates in these two countries seems to be responsible for the different antibiotic resistance gene abundances in the two populations, for the relative abundance of resistance genes in Spain was nearly 1.8 times higher than in Denmark.Citation10 However, both in Denmark and Spain, β-lactam antibiotics were the most commonly prescribed agents for ambulatory care, but tetracycline resistance genes were the most abundant resistance genes in their gut microbiota, accounting for nearly 50% of all resistance genes in each country.
Figure 4. (A) The total outpatient antibacterial use in 20 European countries from 1999 to 2008 (cumulative annual average). DDD, defined daily dose. Data obtained from the ESAC (European Surveillance of Antimicrobial Consumption) project (now ESAC-Net) (http://www.ecdc.europa.eu/en/activities/surveillance/ESAC-Net/). (B) The estimated total consumption (kg active compound) of prescribed antimicrobials for production animals from 2000 to 2008 in Denmark. Data obtained from DANMAP 2008 (http://www.danmap.org/).
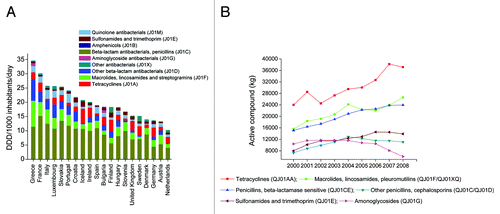
Next, we surveyed the estimated total consumption of prescribed antimicrobials for production animals from 2000 to 2008 (data available only for Denmark). Among six major classes of antimicrobial agents, tetracyclines were the most commonly used in production animals, and their consumption increased from 24 000 kg (in 2000) to 38 200 kg (in 2007) in Denmark (). These results give an indication that the abundance of tetracycline resistance genes in the Danish population could be, to a large extent, related to the consumption of tetracyclines in animals, which may also be the case for China and Spain. Data also shown that, in Demark, from 2000 to 2008, the average veterinary consumption of antimicrobial substances was 30 206 kg per year; in contrast, only 1662 kg of antibacterial agents were used in humans. In addition, previously we found antibiotic resistance genes of tetracyclines, MLSs, and beta-lactams or their analogs accounted for >75% of the total antibiotic resistance genes in each of the three countries. It happened that these antibiotic classes were the most commonly used antimicrobials in animal (). Taken together, we presumed that animal use antibiotics make greater contribution to the enrichment of antibiotic resistance genes in human guts. This view is also supported by another study where the authors use the metagenomic data sets of human gut microbiota from individuals of Spain, Denmark, and the United States, and find that antibiotics used in animal husbandry and the long-term use of antibiotics are the strongest determinants of the antibiotic resistance genes repertoire in the human gut microbiome.Citation16
Summary
The results presented here and in our previously publication provide a comprehensive view of antibiotic resistance genes in human gut microbiota at a population level. The source of the resistance genes in human gut is complex, but antibiotic usage is undoubtedly a major factor.Citation17-Citation19 We suggest that, compared with human medicine use, antibiotics used in animal husbandry exert greater impact on the enrichment of antibiotic resistance genes in human; some evidence of antibiotic-resistant bacteria and antibiotic resistance genes spread from animals, even from soil, to humans have been provided.Citation20,Citation21 However, all our analysis and the suggestions are based on the available data to date; the source of antibiotic resistance genes in human gut microbiota would be more complicated than we now expected. Many other questions are still need to be answered—for example, which genes are intrinsic and which are acquired; to what extent the difference of resistance genes in different population was affected by the structure of the bacterial community; and how other factors, such as diet, compounds and heavy metal ions we encountered daily, and so on contributed to the enrichment of antibiotic resistance genes in our gut. All in all, future efforts on large scale and paralleled sampling of a reasonable selection of subjects (age, race, sex, antibiotic use history, etc.), should be made to fully profile the antibiotic resistance gene reservoir in human gut microbiota.
Additional material
Download Zip (104.5 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was funded by the National Natural Science Foundation of China (NSFC) (31270168) and the Beijing Municipal Science and Technology Development Program (Z131102002813063).
References
- de Vries LE, Vallès Y, Agersø Y, Vaishampayan PA, García-Montaner A, Kuehl JV, Christensen H, Barlow M, Francino MP. The gut as reservoir of antibiotic resistance: microbial diversity of tetracycline resistance in mother and infant. PLoS One 2011; 6:e21644; http://dx.doi.org/10.1371/journal.pone.0021644; PMID: 21738748
- Smith DL, Harris AD, Johnson JA, Silbergeld EK, Morris JG Jr.. Animal antibiotic use has an early but important impact on the emergence of antibiotic resistance in human commensal bacteria. Proc Natl Acad Sci U S A 2002; 99:6434 - 9; http://dx.doi.org/10.1073/pnas.082188899; PMID: 11972035
- Sommer MO, Church GM, Dantas G. The human microbiome harbors a diverse reservoir of antibiotic resistance genes. Virulence 2010; 1:299 - 303; http://dx.doi.org/10.4161/viru.1.4.12010; PMID: 21178459
- Sommer MO, Dantas G, Church GM. Functional characterization of the antibiotic resistance reservoir in the human microflora. Science 2009; 325:1128 - 31; http://dx.doi.org/10.1126/science.1176950; PMID: 19713526
- Salyers AA, Gupta A, Wang Y. Human intestinal bacteria as reservoirs for antibiotic resistance genes. Trends Microbiol 2004; 12:412 - 6; http://dx.doi.org/10.1016/j.tim.2004.07.004; PMID: 15337162
- SchjørringS, KrogfeltKA. Assessment of bacterial antibiotic resistance transfer in the gut. Int J Microbiol 2011; 2011:312956; doi: http://dx.doi.org/10.1155/2011/312956.
- Moubareck C, Bourgeois N, Courvalin P, Doucet-Populaire F. Multiple antibiotic resistance gene transfer from animal to human enterococci in the digestive tract of gnotobiotic mice. Antimicrob Agents Chemother 2003; 47:2993 - 6; http://dx.doi.org/10.1128/AAC.47.9.2993-2996.2003; PMID: 12937011
- Kazimierczak KA, Scott KP. Antibiotics and resistance genes: influencing the microbial ecosystem in the gut. Adv Appl Microbiol 2007; 62:269 - 92; http://dx.doi.org/10.1016/S0065-2164(07)62009-7; PMID: 17869608
- Seville LA, Patterson AJ, Scott KP, Mullany P, Quail MA, Parkhill J, Ready D, Wilson M, Spratt D, Roberts AP. Distribution of tetracycline and erythromycin resistance genes among human oral and fecal metagenomic DNA. Microb Drug Resist 2009; 15:159 - 66; http://dx.doi.org/10.1089/mdr.2009.0916; PMID: 19728772
- Hu Y, Yang X, Qin J, Lu N, Cheng G, Wu N, Pan Y, Li J, Zhu L, Wang X, et al. Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat Commun 2013; 4:2151; http://dx.doi.org/10.1038/ncomms3151; PMID: 23877117
- Liu B, Pop M. ARDB—antibiotic resistance genes database. Nucleic Acids Res 2009; 37:D443 - 7; http://dx.doi.org/10.1093/nar/gkn656; PMID: 18832362
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature 2009; 457:480 - 4; http://dx.doi.org/10.1038/nature07540; PMID: 19043404
- Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE. Metagenomic analysis of the human distal gut microbiome. Science 2006; 312:1355 - 9; http://dx.doi.org/10.1126/science.1124234; PMID: 16741115
- Kurokawa K, Itoh T, Kuwahara T, Oshima K, Toh H, Toyoda A, Takami H, Morita H, Sharma VK, Srivastava TP, et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res 2007; 14:169 - 81; http://dx.doi.org/10.1093/dnares/dsm018; PMID: 17916580
- Leek JT, Scharpf RB, Bravo HC, Simcha D, Langmead B, Johnson WE, Geman D, Baggerly K, Irizarry RA. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet 2010; 11:733 - 9; http://dx.doi.org/10.1038/nrg2825; PMID: 20838408
- Forslund K, Sunagawa S, Kultima JR, Mende DR, Arumugam M, Typas A, Bork P. Country-specific antibiotic use practices impact the human gut resistome. Genome Res 2013; 23:1163 - 9; http://dx.doi.org/10.1101/gr.155465.113; PMID: 23568836
- Goossens H, Ferech M, Vander Stichele R, Elseviers M, ESAC Project Group. Outpatient antibiotic use in Europe and association with resistance: a cross-national database study. Lancet 2005; 365:579 - 87; http://dx.doi.org/10.1016/S0140-6736(05)17907-0; PMID: 15708101
- Austin DJ, Kristinsson KG, Anderson RM. The relationship between the volume of antimicrobial consumption in human communities and the frequency of resistance. Proc Natl Acad Sci U S A 1999; 96:1152 - 6; http://dx.doi.org/10.1073/pnas.96.3.1152; PMID: 9927709
- Enright MC. The evolution of a resistant pathogen--the case of MRSA. Curr Opin Pharmacol 2003; 3:474 - 9; http://dx.doi.org/10.1016/S1471-4892(03)00109-7; PMID: 14559091
- Forsberg KJ, Reyes A, Wang B, Selleck EM, Sommer MO, Dantas G. The shared antibiotic resistome of soil bacteria and human pathogens. Science 2012; 337:1107 - 11; http://dx.doi.org/10.1126/science.1220761; PMID: 22936781
- Smillie CS, Smith MB, Friedman J, Cordero OX, David LA, Alm EJ. Ecology drives a global network of gene exchange connecting the human microbiome. Nature 2011; 480:241 - 4; http://dx.doi.org/10.1038/nature10571; PMID: 22037308