
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Fecal sampling is widely utilized to define small intestinal tissue-level microbial communities in healthy and diseased newborns. However, this approach may lead to inaccurate assessments of disease or therapeutics in newborns because of the assumption that the taxa in the fecal microbiota are representative of the taxa present throughout the gastrointestinal tract. To assess the stratification of microbes in the newborn gut and to evaluate the probable shortcoming of fecal sampling in place of tissue sampling, we simultaneously compared intestinal mucosa and fecal microbial communities in 15 neonates undergoing intestinal resections. We report three key results. First, when the site of fecal and mucosal samples are further apart, their microbial communities are more distinct, as indicated by low mean Sørensen similarity indices for each patient's fecal and tissue microbiota. Second, two distinct niches (intestinal mucosa and fecal microbiota) are evident by principal component analyses, demonstrating the critical role of sample source in defining microbial composition. Finally, in contrast to adult studies, intestinal bacterial diversity was higher in tissue than in fecal samples. This study represents an unprecedented map of the infant microbiota from intestinal mucosa and establishes discernable biogeography throughout the neonatal gastrointestinal tract. Our results question the reliance on fecal microbiota as a proxy for the developing intestinal microbiota. Additionally, the robust intestinal tissue-level bacterial diversity we detected at these early ages may contribute to the maturation of mucosal immunity.
Introduction
The rapid microbial colonization of the human neonatal gastrointestinal tract after birth is an essential driver of intestinal immune programming and the subsequent development of mature innate and adaptive immune responses.Citation1,Citation2,Citation3 By contrast, the absence of bacterial colonization results in significant underdevelopment of key intestinal immune mediators in germ free animal models, including hypoplastic Peyer’s patches and dramatically reduced numbers of IgA-producing plasma cells and lamina propria CD4+ T cells.Citation4,Citation5 These early derangements in microbial colonization produce altered signal transduction between the microbial inhabitants and the developing immune system. This altered interaction may be a key initiator of disease processes during infancy, such as necrotizing enterocolitis (NEC),Citation6,Citation7 and in auto-immune phenomena occurring in childhood, such as type I diabetesCitation8 and inflammatory bowel disease (IBD).Citation9
Despite recognition of the significant dependence of host immune development on commensal bacteria locally, the neonatal intestinal microbiome has been solely defined through analysis of bacterial communities in fecal samples. This sampling approach may not accurately represent the microbial composition and diversity in contact with the intestinal mucosa.Citation10,Citation11 In addition, different regions of the adult intestine have unique microbial flora that may impact disease processes.Citation12 Two studiesCitation13,Citation14 describe tissue-level bacterial communities in neonates; however, neither used culture-independent techniques or modern sequencing-based approaches to comprehensively define intestinal microbiota. Moreover, we are aware of no study that has analyzed simultaneously sampled feces and tissue from the same newborn patient to determine the relationship between microbial communities from these two different body habitats.
To understand how closely tissue-level and fecal bacterial communities are related in neonates, we obtained fresh intestinal tissue collected during surgery and simultaneous fecal samples from patients admitted to the newborn intensive care unit at the Monroe Carell Jr Children’s Hospital at Vanderbilt. We hypothesized that neonates would have a unique bacterial colonization pattern at the tissue-level compared with fecal samples. However, because high inter-individual variation in both bacterial taxonomy and gene composition has been observed among infants of different ages,Citation15 we predicted that the greatest disparities in microbial composition would be between individuals and not between tissue and fecal samples for a single individual. The most notable finding in our study of the newborn microbiota is that sample location has more impact on the presence of microbial diversity than the individual infant from whom the sample is taken. Understanding the distinct and early differences in the microbiota detected in tissue vs. fecal samples in a cohort of newborn infants has significant implications. In particular, it affects how we interpret studies conducted with fecal samples and how we ultimately diagnose and treat childhood gastrointestinal disorders that originate from abnormal intestinal immune development.
Results
Patient demographics
Fifteen neonates (8 females, 7 males) undergoing intestinal resections were recruited for this study (). The median gestational age and birth weight for study participants was 31 4/7 wk (range 24 to 39) and 1660 g (range 700 to 3454), respectively. The mean age at the time of surgery was 31 d (range 0 to 120). Eight patients were exposed to antibiotics at least 24 h prior to surgery. The majority of tissue samples were obtained from the small intestine (10 ileal and 3 jejunal samples); two colonic samples were also collected. Fecal and tissue samples from a single patient (Patient 6) were excluded from all diversity calculations and principal component analyses (PCoA) due to inadequate depth of coverage in the fecal sample (approximately 73 reads per sample).
Table 1. Characteristics of patients requiring bowel surgery (n = 15)
Increasing anatomical distance between tissue sample site and fecal sample site decreases their microbiome similarity
To compare each individual’s fecal and tissue microbiota, we first used the Sørensen similarity index (SI). SI is a statistic that compares the similarity of two samples, with exact similarity of the feces and tissue microbiota set at 1 and no similarity set at 0. Based on bacterial genera, the mean SI across all 15 samples was 0.40 (range, 0.19–0.52) (). Notably, similarity was highest for tissue and fecal samples adherent to the intestinal mucosa (mean SI 0.51, n = 2), followed by the first post-operative stoma output (mean SI 0.43, n = 7) and then the patient feces retrieved from a diaper (mean SI = 0.32, n = 6). Indeed, mean tissue-adherent fecal material (median difference = 0.19, 95% Bayesian credible interval 0.03–0.034) and stoma output SI values (median difference = 0.19, 95% Bayesian credible interval 0.03–0.34) were significantly higher (posterior probability = 0.99 and >0.99, respectively) than for feces collected from the diaper, confirming that physical distance between the tissue site and location of fecal sample retrieval drives microbial dissimilarity between tissue and feces. The mean SI value for ostomy effluent was only nominally smaller than for tissue adherent feces (median difference = 0.07, 95% Bayesian credible interval -0.22–0.08, posterior probability = 0.18).
Table 2. Sørensen’s Index (SI) comparing the bacterial composition of each patient’s fecal and tissue samples
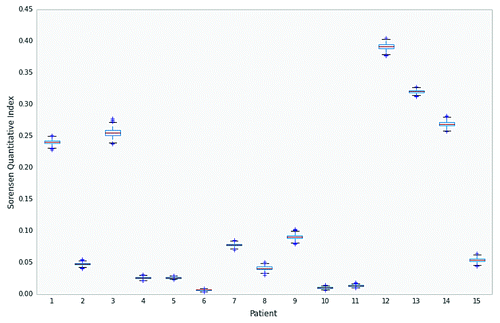
We observed the same trend when we used the Sorensen Quantitative index (CN) with 95% Bootstrap confidence intervals, a similarity metric that compares the similarity of two samples on the basis of taxon presence or absence (). Higher CN values correspond with greater shared similarity between tissue and fecal samples. The boxplots highlight that the five highest CN are for Patients 1, 3, 12, 13, and 14, consistent with each of these five individuals having the greatest similarity between their respective fecal and tissue samples. Two of these fecal samples were tissue adherent and another two were proximal stoma effluents; only one sample (from Patient 13) was collected from a diaper.
Figure 1. Greater overlap between fecal and tissue microbial communities with fecal samples that are in closer proximity to tissue sources. Patients 1–15 and boxplots of Quantitative Sorensen indices (CN) with 95% bootstrap confidence intervals along the x- and y-axis, respectively. Higher indices for Patients 1, 3, 12, 13, and 14, reflective of greater similarity between fecal and tissue microbiota, correspond with fecal samples collected more proximal to the tissue source.
Tissue samples harbor a distinct microbial community
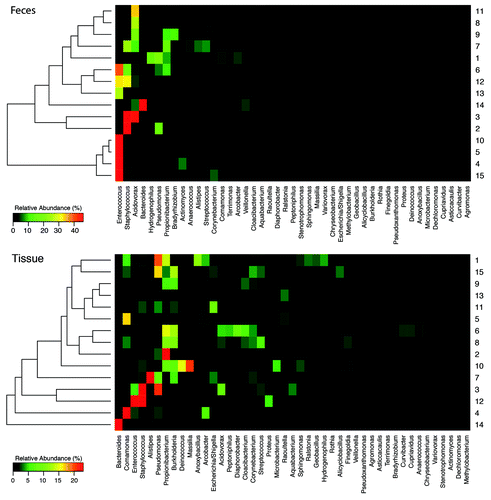
We classified the microbiota of individual patients into genera (). The heat maps delineate the enriched operational taxonomic unit (OTU) diversity and abundance shifts of the microbiota across patients in tissue vs. fecal samples. Fecal samples appear to cluster into two groups: the bottom group, including patients 4, 5, 10, and 15 with the greatest abundance of Enterococcus, and the top group, which includes all remaining patients. This grouping is not a function of tissue type, gestational age, age, fecal sampling, or patient’s diet. However, the surgical indication for three out of four of the clustered patients was ostomy takedown. The fourth patient received bowel resection for a mid-gut volvulus. On the other hand, tissue samples did not display similar hierarchical clustering.
Figure 2. Distinct hierarchical clustering for tissue vs. fecal samples. Patients’ samples are sorted along the y-axis based on the hierarchical clustering of OTU-based UniFrac distances. Heat maps constructed with in house scripts reflect the relative abundance of the 40 most dominant genera in feces (top) and intestinal tissue samples (bottom).
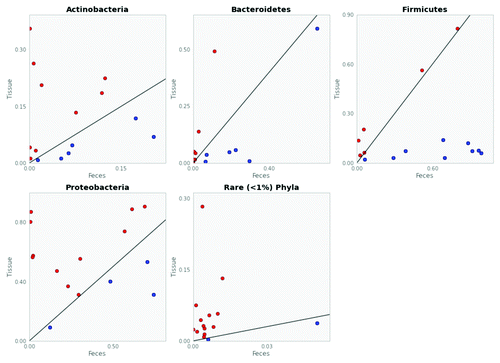
Next, we averaged the patient data for the four major gut phyla (Firmicutes, Proteobacteria, Actinobacteria, and Bacteroidetes)Citation16,Citation17 and other “rare phyla” (<1% abundance) (). We constructed extended error bar plots and associated confidence intervals with bootstrapping in order to indicate the mean difference for each phyla between fecal and tissue samples (). The pie charts () illustrate the marked changes in microbial biogeography of the two dominant phyla. Firmicutes shift from 16% of the reads in tissue to 49% of the reads in feces (P = 0.015, paired t test), while Proteobacteria inversely shift from 55% of the tissue reads to 33% of the fecal reads (P = 0.009, paired t test). In addition, there was a reduction in the detection of rare phyla in tissue (6%) vs. fecal samples (1%) (P = 0.016, paired t test). As illustrated in , differences in the mean proportion of Firmicutes, Proteobacteria, and rare phyla are still statistically significant when variation across fecal or tissue samples is considered as well. Logarithmic plots correlating tissue and fecal samples illustrate an increased abundance of Actinobacteria, Proteobacteria, and rare phyla and decreased abundance of Firmicutes in tissue samples ().
Figure 3. Increased abundance of Proteobacteria and rare phyla, decreased abundance of Firmicutes in tissue samples. (A) Pie charts reflect the proportions of the four major gut phyla as well as rare phyla (<1% abundance) in the 15 tissue and fecal samples. (B) Extended error bar plot, illustrating the difference in the mean proportion of the four major phyla between tissue and fecal samples with bootstrapped confidence intervals.
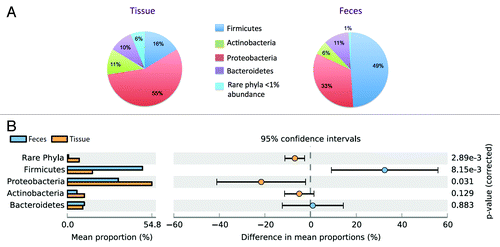
Figure 4. Rare phyla are consistently under-detected in fecal vs. tissue samples. Logarithmic plots of fecal vs. tissue sample microbial abundance for each of the four major gut phyla (Firmicutes, Proteobacteria, Actinobacteria, and Bacteroidetes) and rare phyla <1% compare tissue with fecal samples for individual patients. Each circle represents a single patient. For any given patient, if phyla abundance (OTUs per samples) of a fecal sample (x-axis) is greater than that of tissue (y-axis), it is depicted as a blue circle. Conversely, phyla abundance that is greater in a given tissue vs. fecal sample is represented as a red circle. Exact correlation between a fecal and tissue sample for any given patient would plot along the diagonal line present in each graph. In the “Rare Phyla” plot, clustering of the majority of points above the diagonal line reflects greater detection of rare phyla (<1% abundance) in tissue vs. fecal samples.
Microbial diversity is highly related to sample location
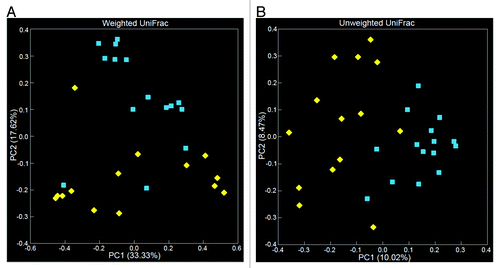
We applied PCoA to categorize the clusters of taxa in each sampling site.Citation18 We detected two distinct clusters of phylogenetic relatedness for tissue and fecal sources in both weighted and unweighted Unifrac measures (). For any individual patient, in both analyses, the microbial composition of a fecal sample was closer to other patient’s fecal communities than to the microbiota of its own matched tissue.
Figure 5. Principal component analyses (PCoA), based on both weighted (A) and unweighted (B), Unifrac distances generated from OTU data, demonstrate grouping by the source of the sample. Blue squares and yellow diamonds represent tissue and fecal samples (Patients 1–15), respectively. Variance explained by PC1 or PC2 is reflected by the values in parentheses listed along the axes.
Ecological diversity is highest in tissue samples, independent of bacterial load
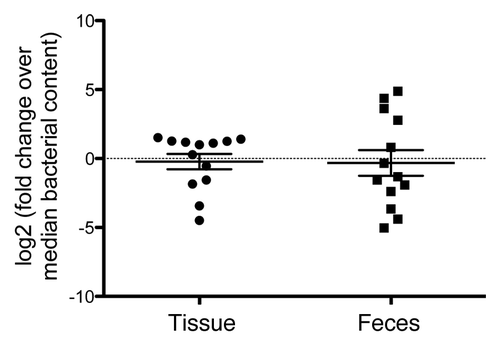
Three different diversity metrics (Simpson’s inverse index, Shannon-Wiener, and Chao 1 estimator) were calculated with genus-level data in order to compare the microbial diversity of tissue vs. fecal samples ().Citation19 The mean Simpson’s inverse diversity index, which measures the probability that two random samples from the same community will belong to the same taxon, was higher in tissue (8.26) vs. fecal samples (4.67) (P = 0.048, paired t test). The mean Shannon index, which measures the uncertainty of predicting the taxon from a random sample, was also higher for tissue (2.70) compared with fecal samples (2.12) (P = 0.009, paired t test). Finally, the Chao 1 species richness estimator was also higher in tissue (234) vs. fecal samples (140) (P = 0.003, paired t test). Since we used different extraction methods for tissue vs. fecal samples that may yield different amounts of DNA and thus different diversity results, we considered that different bacterial yield from these two approaches may confound results. To address this possibility, we performed quantitative PCR on 12 fecal and tissue samples for which we had available bacterial DNA. While the total bacterial load for feces was more variable across samples, the median bacterial content was the same between tissue and feces (). Additionally, when we connected median bacterial content with sample source and diversity metrics (Simpson’s inverse index, Shannon-Wiener, and Chao 1 estimator), there was no correlation between increased diversity, bacterial load, and sample source (Table S1). Therefore, the two extraction methods did not yield different amounts of bacteria and, even in samples with the lowest bacterial yield, there was no bias toward less diversity.
Figure 6. Microbial diversity is greater in tissue vs. fecal samples based on three different diversity metrics: Simpson’s inverse index (A), Shannon-Wiener (B), and Chao 1 (C). Sample source (tissue vs. stool) is listed along the x-axis, with tissue on the left and fecal samples on the right, for each individual plot. Diversity indices are recorded on the y-axis.
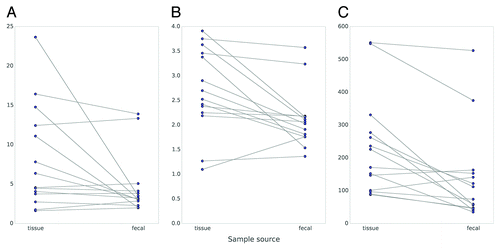
Figure 7. Median bacterial load across tissue and fecal samples is equivalent. For each sample, the log (2) of the calculated ratio of the sample median to the overall population median is determined. A value of “0” correlates with no difference between the median sample and that calculated for each patient sample. Of note, the median bacterial DNA load is more variable across all fecal vs. tissue samples.
Discussion
Tissue-level microbial communities form inseparable relationships with the mucosal immune system that, when altered, can trigger exaggerated inflammatory responses known to characterize a variety of pediatric intestinal disorders, such as NEC and IBD.Citation20,Citation21 While defining the microbiota of the small intestine in neonates is currently not feasible, surrogates employed in other studies such as nasogastric aspirates and ileostomy effluents may not fully represent the tissue-adherent communities that directly impact mucosal immune function.Citation11 Here we show for the first time that the differences between tissue and fecal samples delineate two clear microbial niches. Discrete clusters of fecal and tissue samples on the PCoA plots illustrate how sample source is a critical determinant for bacterial composition, more so than the inherent microbial diversity that is driven by the individual. Site-specific differences in microbial community structure are amplified with respect to physical distance such as when the communities from small intestine are compared with those of feces collected from a diaper (mostly a product of the large intestine). However, amidst the rapid shifts in bacterial species known to characterize this initial gut colonization period,Citation22 the intestinal mucosa quickly emerges with its own microbial niche, even during the immediate postnatal period when newborns’ bacterial load is still relatively low.
We found that rare phyla were consistently under-detected in fecal samples compared with tissue. While missed in conventional microbiome studies, these rare organisms could become abundant as the mucosal immune system develops during infancy and childhood and may be critical for maintenance of an individual’s health and wellbeing. While detection of the presence or absence of rare phyla may be affected by the presence of chimera and host contamination as well as inequalities in sequence depth, we were intrigued by the complex microbial colonization of intestinal mucosa within just a few days of age. In fact, based on three different diversity measures (Simpson’s, Shannon, and Chao 1), the mucosal microbial environment was found to be significantly more diverse than that of simultaneously collected fecal samples. This diversity was independent of the total bacterial load in tissue vs. fecal samples and the opposite of previous reports in adults where feces have a greater microbial diversity than intestinal mucosaCitation10,Citation12 or ileostomy effluent.Citation23 Samples of the neonatal microbiota collected anywhere from seconds to within 24 h after delivery across different body habitats (skin, oral, and rectal mucosa) were relatively homogeneous between body sites at this early age.Citation24 However, our study population, whose age range was anywhere from several hours to several months, had distinct microbiota in two body habitats (feces, intestinal tissue) suggesting that the time frame for establishment of site-specific microbiota as found in adults can be as rapidly and as early as 24 h after birth.
Within fecal samples, hierarchical relationships constructed by Unifrac distances displayed similar microbial community compositions for the three patients undergoing ostomy takedowns. Although not mirrored in tissue samples, this finding could reflect the adaptation of gut microbial communities after bowel resections and intestinal diversions and may warrant further investigation.
Our study has several limitations. One caveat is the deployment of different bacterial DNA extraction methods for tissue (manual) and fecal (automated) samples, including the use of lysozyme for digestion of the more dense tissue samples. We used different extraction methods because of the inherent challenges of processing samples with extremely low bacterial loads, such as meconium, the first feces of newborns. While we cannot rule out a methodological bias, both DNA extraction methods yielded equivalent amounts of DNA. Previous studies performing side-by-side comparisons demonstrated that microbial diversity for fecal samples was higher with the automated easyMAG method than manual DNA extractions.Citation25,Citation26 In contrast, we unexpectedly found less diversity in fecal samples compared with tissue, suggesting a “top to bottom” microbial colonization pattern in the newborn.
Although most patients had received antibiotics based on standard of care practice, this exposure does not confound our results since we compared tissue and feces samples from the same individual. This rationale would also apply to other exposures known to affect the microbiota in this population, such as mode of delivery and introduction of enteral feeds, as well as to the different types of tissue and fecal samples included in this study.
Finally, our method is restricted to newborns undergoing intestinal resections and therefore is not directly transferable to fecal studies in healthy newborns. In addition, extensive replication of mucosa-adherent bacteria has been detected in inflammatory bowel diseases,Citation27,Citation28 which may also be true in this cohort of surgical patients and could be the etiology for the increased tissue-level bacterial abundance. However, because endoscopic biopsies cannot be obtained in neonates, our approach currently provides the only mechanism for examining the microbiota of inaccessible regions of the developing gut. Large-scale inter-institution collaborations, comparable to initiatives already developed in adult cohorts,Citation29 will be necessary to generate a biorepository of fresh tissue samples from neonates with enough breadth to answer complex questions about host-microbiota interactions during this critical developmental window.
In ongoing efforts to define the microbiome as it establishes itself in the neonatal gastrointestinal tract, it will be important to determine how variation in microbial community distribution between anatomic sites translates into distinct functional microbiomes. Adult studies suggest that while divergence in microbial assemblages correlates with a well-preserved core microbiome at a functional level,Citation17 distinct metabolic profiles still exist when samples from different regions of the gastrointestinal tract are examined.Citation30 These findings have not been confirmed in newborns, whose first several months and years are devoted to the simultaneous maturation of the intestinal microbiota and the mucosal immune system, within the context of dynamic and nascent dietary stimuli. We expect that continued development of safe methodologies for studies of the neonatal intestinal tissue-level microbiota and its relationship with mucosal immune function will advance our understanding of the pathogenesis of pediatric gastrointestinal disorders such as NEC and IBD. Furthermore, by continuing to study the relationship between fecal and tissue-level bacteria, we will be able to develop predictive models that infer the tissue microbiome from the fecal microbiome and serve as the basis for practical, bed-side, fecal-based microbial screens aimed at early disease detection.
Patients, Methods, and Materials
Recruitment of subjects and sample collection
This study was approved by the Vanderbilt University Institutional Review Board (protocol no. 090161). All infants born at any gestational age were eligible for the study if they underwent intestinal resection at <180 d of age. Informed consent was obtained from parents or legal guardians to permit collection of metadata from the medical records including gestational age, birth weight, race, sex, mode of delivery, maternal or fetal indications for delivery, antibiotic exposure, and enteral feeding regimen. Tissue was collected at the time of surgery, gently rinsed with sterile saline solution, snap frozen, and stored at -80 °C. Fecal material adjacent to surgical tissue samples was obtained by gently scraping the surface of the tissue. When feces from the tissue surface could not be obtained, the patient’s first post-operative stool was collected, snap frozen, and stored at -80 °C.
DNA extraction and amplification of 16SrRNA gene
DNA was extracted from 15 to 25 mg of intestinal tissue using a modified Qiagen protocol that included pretreatment for lysis of Gram-positive bacteria with 20 mg/ml lysozyme in TRIS-HCl and EDTA buffer (Qiagen, DNeasy Blood & Tissue Kit, catalog no. 69504). The remainder of the tissue DNA extraction protocol proceeded per the manufacturer’s instructions. All DNA samples were evaluated for quality using spectrophotometry and agarose gel electrophoresis. Samples with low yield (<10 ng/µl) or poor quality metrics (O.D. 260/280 ratio < 1.8; overt degradation) were discarded and the corresponding subject excluded unless a repeat extraction yielded high quality DNA. All fecal DNA extractions were conducted with 180–200 mg of feces and were automated with NucliSENS easyMAG (BioMérieux).
PCR amplification of the V1- V3 hypervariable region of the bacterial 16S rRNA was conducted with universal primers 5F (5′-TGGAGAGTTT GATCCTGGCT CAG-3′) and 532R (TACCGCGGCT GCTGGCAC) that had been previously validated.Citation31 For each sample, 50 µl PCR reactions were set up consisting of AmpliTaq Gold® 360 Master Mix and GC Enhancer (Applied Biosystems®, catalog no. 4393901), 50 pmol/µl of each bacterial primer, and up to 1 µg of template DNA (DNA concentration range of 4.3‒254 ng/µl). PCR was conducted on a Bio-Rad Thermal Cycler under the following conditions: 10 min at 95 °C, followed by 30 cycles of 30 s at 95 °C, 30 s at 60 °C, and 45 s at 72 °C, with a final extension period of 7 min at 72 °C. Barcoded 527 bp amplicons were gel purified (Gel Qiaquick Gel Extraction Kit, Qiagen, catalog no. 28704), quantified, and then pooled prior to bi-directional sequencing on a 454 FLX Titanium sequencer.Citation32
Quantification of total fecal and tissue bacterial load
Real-time PCR amplification was performed in triplicates for 13 patients with 20 ng of fecal or tissue DNA on an ABI 7900 TaqMan Real Time PCR System (Applied Biosystems). We used the conserved eubacterial (EUB) 1114 forward (CGGCAACGAG CGCAAGCC), and 1221 reverse (CCATTGTAGC ACGTGTGTGT AGCC) 16S ribosomal primers to detect total bacteria.Citation33 Reaction mixtures consisted of the DNA template, 100 nM concentration of each primer, 1.25 μL Omni Klentaq (DNA Polymerase technology, cat no. 350), and 0.2 mM dNTP (Enzymatics, cat no. N2050L) in a final volume of 50 μL. Cycling conditions were as follows: initial incubation of 95 °C for 3 min, denaturing at 95 °C for 10 s, then 58 for 30 s, then 72 for 30 s, for 40 cycles. We used the Real-time PCR MinerCitation34 to analyze qPCR curves, obtain reaction efficiency, and relative threshold values.
454 pyrosequencing and data analysis
Sequencing of the barcoded 16S rRNA amplicons on a 454 FLX Titanium sequencer generated 129 149 sequences with an average of 4305 sequences per sample and a mean length of 436 base pairs. Bidirectional sequences generated from the pyrosequencing of barcoded 16S rRNA gene PCR amplicons were analyzed using default settings in the open source software package Quantitative Insights Into Microbial Ecology (QIIME; http://qiime.sourceforge.net).Citation35 16S rRNA gene sequences were assigned to OTUs using the QIIME implementation of uclust using a threshold of 97% pairwise identity.Citation36 OTUs were classified taxonomically with the Ribosomal Database Project (RDP) classifier 2.2.Citation37 Potential chimeric sequences were detected via ChimeraSlayer and removed prior to downstream analyses.Citation38 A single representative from each OTU was aligned using PyNast to build the phylogenetic tree used for measuring unweighted UniFrac.Citation39,Citation40 Statistical Analysis of Metagenomic Profiles (STAMP)Citation41 was used to generate extended error bar plots for the comparison of phyla composition between fecal and tissue samples and associated confidence intervals were calculated via bootstrapping. Table S2 is a summary of the total number of OTUs assigned to each phyla for each patient’s tissue and fecal samples. The total and average number of assigned OTUs were similar between feces and tissue samples (P = 0.5, paired t test).
Overlap between bacterial genera within an individual’s fecal vs. tissue sample was determined by Sørensen similarity index (SI) as calculated by: SI = 2C/A + B, where A and B are the number of genera per sample, respectively, and C is the number of shared genera between A and B.Citation42 Sorensen’s Quantitative Index was calculated as follows: CN = 2jN/Na + Nb, where Na is the total number of genera in site A (tissue); Nb is the total number of genera in site B (feces); and 2jN is the sum of the lower of the two abundances for genera found in both sites. Bootstrapping was used to generate confidence intervals around CN calculations. Bacterial diversity in tissue vs. fecal samples was estimated by Shannon-Wiener (H') and Simpson’s inverse (1/D) diversity indices using the following equations:
where pi is the proportion of bacteria in the ith species in the community, s is the total number of species in the community, N is the total number of sequences and n (or ns) is the number of sequences of species s.Citation43–Citation45 Chao 1 (SChao1) estimates of species diversity were calculated as SChao1 = Sobs + n12 / 2n2, where Sobs is the number of observed species, n1 is the number of singletons (species captured once), and n2 is the number of doubletons (species captured twice).Citation46 All sequences reported in this paper have been deposited into the NCBI sequence short read archive (accession number SRA081700).
| Abbreviations: | ||
| NEC | = | necrotizing enterocolitis |
| IBD | = | inflammatory bowel disease |
| SI | = | Sørensen similarity index |
| OTU | = | operational taxonomic unit |
| PCoA | = | principal component analysis |
| QIIME | = | Quantitative Insights Into Microbial Ecology |
| RDP | = | Ribosomal Database Project |
Additional material
Download Zip (160.9 KB)Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
We express our appreciation for the patients and their families who were willing to participate in this study. We thank the Vanderbilt Departments of Pediatric Surgery and Pathology for their efforts with patient recruitment and sample collection. We are grateful to J Aschner and A Stark for discussion and critical review of this manuscript. This project was supported by the American Academy of Pediatrics Marshall Klaus Perinatal Research Award (to J.R.K.) and the Eunice Kennedy Shriver National Institute Of Child Health and Human Development (NICHD) grants T32HD068256 (to J.R.K.) and K08HD061607 (to J.H.W.), the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grant K08DK090146 (to D.J.M.), and the National Science Foundation grant DEB-1046149 (to S.R.B.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NICHD, NIDDK, or the National Institutes of Health (NIH). This project was also funded by the Vanderbilt Digestive Disease Research Center Grant P30DK058404, Vanderbilt Diabetes Center Grant P30DK20593 (both NIDDK and NIH), and the Vanderbilt CTSA Grant UL1 RR024975-01 from the National Center for Research Resources (NCRR and NIH).
References
- Hooper LV. Bacterial contributions to mammalian gut development. Trends Microbiol 2004; 12:129 - 34; http://dx.doi.org/10.1016/j.tim.2004.01.001; PMID: 15001189
- Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 2005; 122:107 - 18; http://dx.doi.org/10.1016/j.cell.2005.05.007; PMID: 16009137
- Hooper LV, Gordon JI. Commensal host-bacterial relationships in the gut. Science 2001; 292:1115 - 8; http://dx.doi.org/10.1126/science.1058709; PMID: 11352068
- Macpherson AJ, Hunziker L, McCoy K, Lamarre A. IgA responses in the intestinal mucosa against pathogenic and non-pathogenic microorganisms. Microbes Infect 2001; 3:1021 - 35; http://dx.doi.org/10.1016/S1286-4579(01)01460-5; PMID: 11580989
- Macpherson AJ, Martinic MM, Harris N. The functions of mucosal T cells in containing the indigenous commensal flora of the intestine. Cell Mol Life Sci 2002; 59:2088 - 96; http://dx.doi.org/10.1007/s000180200009; PMID: 12568335
- Claud EC, Walker WA. Hypothesis: inappropriate colonization of the premature intestine can cause neonatal necrotizing enterocolitis. FASEB J 2001; 15:1398 - 403; http://dx.doi.org/10.1096/fj.00-0833hyp; PMID: 11387237
- Mai V, Young CM, Ukhanova M, Wang X, Sun Y, Casella G, Theriaque D, Li N, Sharma R, Hudak M, et al. Fecal microbiota in premature infants prior to necrotizing enterocolitis. PLoS One 2011; 6:e20647; http://dx.doi.org/10.1371/journal.pone.0020647; PMID: 21674011
- Roesch LF, Lorca GL, Casella G, Giongo A, Naranjo A, Pionzio AM, Li N, Mai V, Wasserfall CH, Schatz D, et al. Culture-independent identification of gut bacteria correlated with the onset of diabetes in a rat model. ISME J 2009; 3:536 - 48; http://dx.doi.org/10.1038/ismej.2009.5; PMID: 19225551
- Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A 2007; 104:13780 - 5; http://dx.doi.org/10.1073/pnas.0706625104; PMID: 17699621
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science 2005; 308:1635 - 8; http://dx.doi.org/10.1126/science.1110591; PMID: 15831718
- Zoetendal EG, von Wright A, Vilpponen-Salmela T, Ben-Amor K, Akkermans AD, de Vos WM. Mucosa-associated bacteria in the human gastrointestinal tract are uniformly distributed along the colon and differ from the community recovered from feces. Appl Environ Microbiol 2002; 68:3401 - 7; http://dx.doi.org/10.1128/AEM.68.7.3401-3407.2002; PMID: 12089021
- Stearns JC, Lynch MD, Senadheera DB, Tenenbaum HC, Goldberg MB, Cvitkovitch DG, Croitoru K, Moreno-Hagelsieb G, Neufeld JD. Bacterial biogeography of the human digestive tract. Sci Rep 2011; 1:170; http://dx.doi.org/10.1038/srep00170; PMID: 22355685
- Bucher BT, McDuffie LA, Shaikh N, Tarr PI, Warner BB, Hamvas A, White FV, Erwin CR, Warner BW. Bacterial DNA content in the intestinal wall from infants with necrotizing enterocolitis. J Pediatr Surg 2011; 46:1029 - 33; http://dx.doi.org/10.1016/j.jpedsurg.2011.03.026; PMID: 21683193
- Smith B, Bodé S, Petersen BL, Jensen TK, Pipper C, Kloppenborg J, Boyé M, Krogfelt KA, Mølbak L. Community analysis of bacteria colonizing intestinal tissue of neonates with necrotizing enterocolitis. BMC Microbiol 2011; 11:73; http://dx.doi.org/10.1186/1471-2180-11-73; PMID: 21486476
- Kurokawa K, Itoh T, Kuwahara T, Oshima K, Toh H, Toyoda A, Takami H, Morita H, Sharma VK, Srivastava TP, et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res 2007; 14:169 - 81; http://dx.doi.org/10.1093/dnares/dsm018; PMID: 17916580
- Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, et al. Evolution of mammals and their gut microbes. Science 2008; 320:1647 - 51; http://dx.doi.org/10.1126/science.1155725; PMID: 18497261
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature 2009; 457:480 - 4; http://dx.doi.org/10.1038/nature07540; PMID: 19043404
- Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 2005; 71:8228 - 35; http://dx.doi.org/10.1128/AEM.71.12.8228-8235.2005; PMID: 16332807
- Hill TC, Walsh KA, Harris JA, Moffett BF. Using ecological diversity measures with bacterial communities. FEMS Microbiol Ecol 2003; 43:1 - 11; http://dx.doi.org/10.1111/j.1574-6941.2003.tb01040.x; PMID: 19719691
- Weitkamp JH, Koyama T, Rock MT, Correa H, Goettel JA, Matta P, Oswald-Richter K, Rosen MJ, Engelhardt BG, Moore DJ, et al. Necrotising enterocolitis is characterised by disrupted immune regulation and diminished mucosal regulatory (FOXP3)/effector (CD4, CD8) T cell ratios. Gut 2013; 62:73 - 82; http://dx.doi.org/10.1136/gutjnl-2011-301551; PMID: 22267598
- Rosen MJ, Chaturvedi R, Washington MK, Kuhnhein LA, Moore PD, Coggeshall SS, McDonough EM, Weitkamp JH, Singh AB, Coburn LA, et al. STAT6 deficiency ameliorates severity of oxazolone colitis by decreasing expression of claudin-2 and Th2-inducing cytokines. J Immunol 2013; 190:1849 - 58; http://dx.doi.org/10.4049/jimmunol.1201373; PMID: 23303670
- Sharon I, Morowitz MJ, Thomas BC, Costello EK, Relman DA, Banfield JF. Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Res 2013; 23:111 - 20; http://dx.doi.org/10.1101/gr.142315.112; PMID: 22936250
- Booijink CC, El-Aidy S, Rajilić-Stojanović M, Heilig HG, Troost FJ, Smidt H, Kleerebezem M, De Vos WM, Zoetendal EG. High temporal and inter-individual variation detected in the human ileal microbiota. Environ Microbiol 2010; 12:3213 - 27; http://dx.doi.org/10.1111/j.1462-2920.2010.02294.x; PMID: 20626454
- Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, Knight R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A 2010; 107:11971 - 5; http://dx.doi.org/10.1073/pnas.1002601107; PMID: 20566857
- Loens K, Bergs K, Ursi D, Goossens H, Ieven M. Evaluation of NucliSens easyMAG for automated nucleic acid extraction from various clinical specimens. J Clin Microbiol 2007; 45:421 - 5; http://dx.doi.org/10.1128/JCM.00894-06; PMID: 17166966
- Nylund L, Heilig HG, Salminen S, de Vos WM, Satokari R. Semi-automated extraction of microbial DNA from feces for qPCR and phylogenetic microarray analysis. J Microbiol Methods 2010; 83:231 - 5; http://dx.doi.org/10.1016/j.mimet.2010.09.003; PMID: 20849891
- Thomazini CM, Samegima DA, Rodrigues MA, Victoria CR, Rodrigues J. High prevalence of aggregative adherent Escherichia coli strains in the mucosa-associated microbiota of patients with inflammatory bowel diseases. Int J Med Microbiol 2011; 301:475 - 9; http://dx.doi.org/10.1016/j.ijmm.2011.04.015; PMID: 21616711
- Bringer MA, Glasser AL, Tung CH, Méresse S, Darfeuille-Michaud A. The Crohn’s disease-associated adherent-invasive Escherichia coli strain LF82 replicates in mature phagolysosomes within J774 macrophages. Cell Microbiol 2006; 8:471 - 84; http://dx.doi.org/10.1111/j.1462-5822.2005.00639.x; PMID: 16469058
- Human Microbiome Project Consortium. A framework for human microbiome research. Nature 2012; 486:215 - 21; http://dx.doi.org/10.1038/nature11209; PMID: 22699610
- Zoetendal EG, Raes J, van den Bogert B, Arumugam M, Booijink CC, Troost FJ, Bork P, Wels M, de Vos WM, Kleerebezem M. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J 2012; 6:1415 - 26; http://dx.doi.org/10.1038/ismej.2011.212; PMID: 22258098
- Tang YW, Ellis NM, Hopkins MK, Smith DH, Dodge DE, Persing DH. Comparison of phenotypic and genotypic techniques for identification of unusual aerobic pathogenic gram-negative bacilli. J Clin Microbiol 1998; 36:3674 - 9; PMID: 9817894
- Nossa CW, Oberdorf WE, Yang L, Aas JA, Paster BJ, Desantis TZ, Brodie EL, Malamud D, Poles MA, Pei Z. Design of 16S rRNA gene primers for 454 pyrosequencing of the human foregut microbiome. World J Gastroenterol 2010; 16:4135 - 44; http://dx.doi.org/10.3748/wjg.v16.i33.4135; PMID: 20806429
- Kriegel MA, Sefik E, Hill JA, Wu HJ, Benoist C, Mathis D. Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc Natl Acad Sci U S A 2011; 108:11548 - 53; http://dx.doi.org/10.1073/pnas.1108924108; PMID: 21709219
- Zhao S, Fernald RD. Comprehensive algorithm for quantitative real-time polymerase chain reaction. J Comput Biol 2005; 12:1047 - 64; http://dx.doi.org/10.1089/cmb.2005.12.1047; PMID: 16241897
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010; 7:335 - 6; http://dx.doi.org/10.1038/nmeth.f.303; PMID: 20383131
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010; 26:2460 - 1; http://dx.doi.org/10.1093/bioinformatics/btq461; PMID: 20709691
- Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 2007; 73:5261 - 7; http://dx.doi.org/10.1128/AEM.00062-07; PMID: 17586664
- Hamady M, Lozupone C, Knight R. Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J 2010; 4:17 - 27; http://dx.doi.org/10.1038/ismej.2009.97; PMID: 19710709
- Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E, et al, Human Microbiome Consortium. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res 2011; 21:494 - 504; http://dx.doi.org/10.1101/gr.112730.110; PMID: 21212162
- Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 2010; 26:266 - 7; http://dx.doi.org/10.1093/bioinformatics/btp636; PMID: 19914921
- Parks DH, Beiko RG. Identifying biologically relevant differences between metagenomic communities. Bioinformatics 2010; 26:715 - 21; http://dx.doi.org/10.1093/bioinformatics/btq041; PMID: 20130030
- Looman J, Campbell JB. Adaptation of Sorensen’s K (1948) for estimating unit affinities in prairie vegetation. Ecology 1960; 41:409 - 16; http://dx.doi.org/10.2307/1933315
- Shannon CE. A mathematical theory of communication. Bell Syst Tech J 1948; 27:379 - 423, 623-656; http://dx.doi.org/10.1002/j.1538-7305.1948.tb01338.x
- Wiener N. Cybernetics: or Control and Communication in the Animal and Machine. 1st ed. New York: John Wiley and Sons; 1948. p. 117.
- Simpson EH. Measurement of diversity. Nature 1949; 163:688; http://dx.doi.org/10.1038/163688a0
- Chao A. Non-parametric estimation of the number of classes in a population 1984. Scand J Stat 1984; 11:265 - 70