
Abstract
A multitude of metagenomic studies has brought to light an enormous richness of human gut microbiota compositions. In this space of possible configurations, clinical specialists are trying to mine the markers of healthy microbiota via case-control and longitudinal studies. We have discovered potentially beneficial communities while examining the microbial diversity in rural Russians in comparison with the urban dwellers. In this addendum, we further examine the data by elaborating on some of the less common types and suggesting the possible co-metabolism of their drivers. In the light of the first validated clinically effective bacterial transplantation, we discuss the concept of a reference healthy microbiota, outline the problems encountered on the way to its restoration in the developed world, and speculate if rural communities can serve as a source for its prototype.
Introduction
The research of human gut microbiota was recently marked by effective treatment of C. difficile infection using fecal mass transplantation (FMT).Citation1 The promising results encouraged similar experiments targeting other intestinal and non-intestinal diseases.Citation2 The question arises as to what is the reference healthy microbiota suitable for transfer. A plethora of comparative studies identified the distinctions between the microbiota in health and various diseases.Citation3-Citation6 However, the relativity of “normality” was indicated by the descriptive studies of microbiota in the world populations that highlighted significant diversity of composition.Citation7-Citation10 Such diversification is contributed by multiple sociocultural and geographic factors. In this light, it would be interesting to examine metagenome in a cohort of people living in a wide range of these factors.
One such cohort is the population of Russian Federation, the largest country in the world by area and the ninth most populous nation (143 million people) inhabited by more than 150 ethnic groups with diverse cultural traditions, including diet. The climate of inhabited areas ranges from subtropics to permafrost zone, and the quality of life varies greatly. Considering such wide ranges of existence, the gut microbiota of the Russian population is of significant scientific interest for extending our view of global microbiota diversity.
In our study,Citation11 we expected that a descriptive examination of microbiota composition in a large cohort of healthy people would display certain novel features not observed previously that would contribute to the understanding of human microbiota richness. The cohort included 96 subjects enrolled from metropolitan (n = 50) and rural (n = 46) areas corresponding to the most populated part of Russia including areas in Europe and south of Siberia. Certain distinctions between the urban and rural microbiotas were expected, as the urban diet was characterized by Western-style diets (higher meat and preserved food consumption), while people in the rural areas consumed more natural products, including those obtained from the household plots.
DNA was extracted from the stool samples and subject to whole-genome sequencing on SOLiD 4 platform yielding 2.7 ± 1.1 Gbp of 50 bp fragment reads. The resulting reads were mapped to the reference sets of 444 gut microbe genomes and 3.3 million gene catalogCitation10 to yield taxonomic and functional profiles of microbiota, accordingly. The results were validated using Ion Torrent, 454, and Illumina sequencing that showed high correlation of microbial composition. The Russian metagenomes were then compared with the existing data on urban adult populations from DenmarkCitation10 (n = 85), United StatesCitation8 (n = 137), rural communities from VenezuelaCitation7 (n = 10) and MalawiCitation7 (n = 5), and 70 subjects from ChinaCitation4 obtained using similar methods.
In fact, along with the above-mentioned studies, our study was one of the first to examine human gut microbiota for a large cohort of people using whole-genome sequencing. The general set of the microbial components in the Russian metagenomes was consistent with the other studies and the fraction of the identified reads was similar to the other studies (percentage of the reads mapped to the reference sets was 26.32 ± 8.08% for genomes and 46.66 ± 7.84 for the genes). However, several samples had lower genomes mapping fractions, as low as 12% for one sample from rural Omsk region, indicating the presence of the organisms not included in the catalog. Due to high similarity with the result produced by an alternative method based on unique clade-specific gene markers,Citation12 such cases likely correspond to the abundant microbial taxa that lack representative sequenced genomes that are typically observed in human stool when 16S rRNA sequencing is used (for example, Oscillibacter). Additionally, among the samples with low fraction of genome-wise identification, there are those that are also low in gene-wise identification, reflecting presence of microbes not present in Western European microbiota (as the gene catalog has been constructed from Spanish and Danish metagenomes). De novo assembly did not detect any novel high-abundance taxa in the Russian metagenomes.
Novel Microbial Community Structures
Analysis of similarities (ANOSIM)Citation13 showed significant differences between the Russian and the US, Danish, and Chinese cohorts (R ≥ 0.26; P = 9.999 × 10−5, 10 000 permutations). While the applied DNA extraction and sample preparation methods were similar in all of these studies, other details of study design like subject enrolment criteria might have contributed to the observed variation. Moreover, we have discovered that almost two-thirds of the Russian metagenomes are dominated neither by Bacteroides nor by Prevotella, previously described drivers of the two enterotypes.Citation14 The originality of such novel community structures was roughly assessed by considering the sets of three of the most abundant genera (triplets) for each sample. About 43% of the Russian metagenomes were dominated by triplets not occurring in the non-Russian groups. Most of the genera of these triplets belonged to Firmicutes, with the presence of Bacteroidetes, Verrucomicrobia, Actinobacteria, Proteobacteria, Tenericutes, and Archaea. Interestingly, the novel microbial community structures occurred in the hosts from urban populations 2.6-fold less frequently than in the rural hosts, suggesting that further exploration of rural microbiota will reveal even greater diversity of microbiota configurations.
The Firmicutes drivers included Roseburia, Coprococcus, Faecalibacterium, Eubacterium, Ruminococcus, Blautia, Butyrivibrio, and unclassified Lachnospiraceae. Besides Firmicutes-dominated triples, we identified other original structures not present in the non-Russian metagenomes which were even more phylogenetically distant - from other phyla and kingdoms. Two samples were dominated by Akkermansia muciniphila and Methanobrevibacter smithii. Each of the bacteria alone has its own history of associations from the existing studies; however, as a combination, this pair has only a few recently observed associations—with a healthy gut: as markers of microbiota rich in metabolic potential (high gene count, HGC)Citation6 and as components enriched in lean minipigs microbiota in comparison with their obese counterpartsCitation15; additionally, the two bacteria were found to be considerably overrepresented in the fecal microbiota of guinea pigs in comparison with the humans.Citation16 The dominance of Methanobrevibacter is likely to reflect high levels of hydrogen provided by fermentative bacteria. It looks logical, owing to the lack of competition from sulfate-reducing bacteria (SRB) which are more efficient hydrogenotrophs;Citation17 we observed zero levels of Desulfovibrio and Desulfitobacterium. Hypothetically, introduction of SRB into such system could significantly modify its composition—considering the fact that A. muciniphila genome contains glycosulfatases yielding host-derived sulfate for SRB.
Among the other singular original compositions are Phascolarctobacterium and Lactobacillus, each one as the most abundant genus in a single rural sample (from rural Omsk region and Khakassia, respectively). Intriguingly, in a recent study of gut microbiota in urban and rural Mongolians, these bacteria were found to be dominant in the rural microbiotas, on the contrary to the urban ones.Citation18 As the above-mentioned original drivers of community structure were present in single metagenomes, the insights into their ecology and temporal stability could be revealed within a deeper longitudinal study of respective subjects or on a larger cohort.
Some of the original compositions were dominated by opportunistic pathogens. Several samples had high fractions of Escherichia coli sometimes observed in inflammatory bowel diseases; varying coverage profiles for reference E. coli genomes suggest that it is not due to laboratory contamination. In one sample from Saint-Petersburg, an unusually high level of Streptococcus infantarius was detected; the bacterium is often isolated from human infections and has associations with colorectal cancer.Citation19 One sample contained an abnormal level of human DNA (44%); the fact that among the bacteria, the second most abundant genome was Enterococcus faecium, hints at possible inflammatory processes. The presence of the mentioned bacteria could be linked to certain pathological states which were not detected during subject enrollment.
Compact Rural Subtypes of Microbiota
Initially we expected that Russian metagenomes would cluster by taxonomic composition into two or three distinct clusters similar to enterotypes.Citation14 However, our results of cluster analysis showed that while the optimal number of the clusters varied from two to three, their overall quality metrics were not definitive: average silhouette width, ASW was modest (ASW < 0.389) but the predictive strength was quite high (PS > 0.8 for Jensen-Shannon and Bray-Curtis metrics). Moreover, while commonly the Russian clusters included Prevotella enterotype that has been previously observed in the other studies,Citation14,Citation20 remarkably, the Bacteroides enterotype was not present (in contrast to the U.S. cohort where it absolutely dominates the microbiota diversity).
As the global clustering was not certain, we decided to look for significant tight subgroups using cluster mining with bootstrapping implemented in R package pvclust.Citation21 Surprisingly, three of the resulting subgroups turned out to be dominated by a single rural region: Omsk Region, Tatarstan, and Tyva. These subgroups were driven by novel genus triplets. Moreover, Omsk cluster mostly contains metagenomes from the members of a single family likely reflecting the co-exposure to similar environment and host-to-host bacterial transfer.
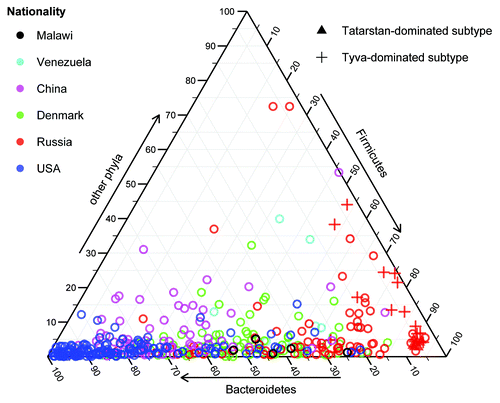
The Tatarstan and Tyva subgroups were distinguished by their composition (): both were formed by similar metagenomes driven by several butyrate-producing Firmicutes (Eubacterium rectale, Coprococcus eutactus, Faecalibacterium prausnitzii, and Ruminococcus bromii), while the latter also had high proportions of Bifidobacterium adolescentis. The discovered rural subtypes were particularly interesting due to the strong associations of these bacteria with the host health that have been shown in the previous studies and keep growing—for example, supported by the latest large-scale metagenomic research of the gut microbiota in metabolic syndromeCitation6,Citation22 and type 2 diabetes.Citation23 Surprisingly, the above-mentioned Firmicutes and Bifidobacterium spp. were recently identified as the main drivers of the Japanese gut microbiota (as reported by Suguru Nishijima at the International Human Microbiome Congress 2013) suggesting that the subtypes we have discovered do in fact represent a significantly novel subspace of microbiota configurations not observed previously in the populations of the world.
Figure 1. Phylum-level taxonomic composition shows the distribution of the Tatarstan- and Tyva-dominated compact subtypes identified among Russian metagenomes. Tyva samples are distinguished by their high abundance of Bifidobacterium genus (Actinobacteria phylum).
Discussion
High modern living standards in the developed countries ensure a comfort zone of human interactions with the complex microbial component of the environment. However, the advent of antibiotics which started in the 1940s with the widespread application of penicillin as well as the prevalence of the industrial food in the diet and excessive hygiene have led to an unprecedented interference in the human holobiont ecology which is yet to be estimated. Particularly, it has been proposed that the decreased exposure to microorganisms as well as the loss of the ancestral indigenous organisms has resulted in the prevalence of modern allergic and metabolic diseases.Citation24
Obviously, the next several years will be dedicated to the identification of “gold standard” healthy microbiota as a prototype transplant for diseases prevention and treatment. It is worth noting that the idea of the bacterial transplantation is not novel: it has been used in the traditional Chinese medicine for at least 17 centuries, in Western medicine its concept was introduced by Russian biologist Elie Metchnikoff, and in wildlife it is manifested by many animals in the form of coprophagy. Recently, the efficiency of FMT was demonstrated for the treatment of Clostridium difficile infection.Citation1 There are preliminary results on ulcerative colitisCitation25 and severe Crohn disease,Citation26 and other socially significant diseases are being targeted. The need for a standardized gnotobiotic composition replacement for donor stool poses several fundamental questions: which composition should it have, where to find the donor for its prototype, and is there actually a universal “magic” donor; is the bacterial transplant transient or stable in the recipient, and, most importantly, what is the healthy microbiota.
While a number of metagenomic studies have identified the gut microbial biomarkers distinguishing the group of patients with particular disease from the control group,Citation4,Citation6,Citation23 the latter are quite abstractly “healthy,” as they have been sampled from the same population and thus their microbiota was subject to a similar complex set of environmental exposures inherent to the urban lifestyle in the developed countries. It suggests that these conditional “healthy” subjects might be at high risk of the disease, too—they just have not developed its symptoms yet. A truly representative control subject should be the one who will never get this disease, owing to the protective effect of his or her lifestyle and microbiota.
As an alternative way to mine the healthy microbiota suitable as a prototype transplant, we suggest searching among the members of the rural communities who sustain natural diets with low consumption of industrial-type food and antimicrobial agents. Living in a more tight connection with an unpolluted environment, in particular, under a higher bacterial exposure, they represent a reservoir of indigenous richness of microbiota.
Previously, rural and urban microbiota were compared in a number of studies.Citation7,Citation9,Citation27 We performed such comparison for Russian cities and villages. Even our limited cohort allowed discovery of the previously unobserved community structures. The existing data support association of their bacterial drivers with the human health, and this evidence keeps growing with the latest independent studies. These communities are promising candidates for the experimental transplantation in animal models and, potentially, in people. Remarkably, the drivers of the Tatarstan and Tyva rural microbiota—Lachnospiraceae and other Firmicutes, as well as Bifidobacterium—formed the majority of the composition of the first semi-artificial stool transplant successfully used for treating C. difficile,Citation28 indicating acceptability, stability and health benefits conferred by these microbial community structures.
One should be wary, however, of transferring these communities into human recipients. On the one hand, a mere fact that a stool donor has been living in a village for life doesn’t make his or her microbiota a beneficial transplant. Besides the potential presence of pathogens, the negative effects contributed by advancing industrialization and associated with low living standards including malnutrition might be present. For instance, the rural Malawian gut metagenomes were enriched in antibiotic resistance genes likely due to excessive application of broad-spectrum antibiotics.Citation29 As the resistance genes are accessible to pathogens with the help of horizontal transfer, enriched resistome is an undesired feature potentially hazardous to human health. A rigorous screening of the stool and its donor is mandatory—from clinical data to metagenomic analysis (including taxonomic and gene-centric profiling).
On the other hand, a long history of coevolution of human and associated microbial communities brought them to a hologenomic equilibrium. Corresponding maxima of resilience might vary among the human populations, and the stability is likely to be disrupted with such rapid procedure as switching to the microbiota from a genetically-distant donor on a different type of diet. Evidence of negative effects caused by evolutionary discordance between the components of holobiont start to appear—for example, in a recent study of Helicobacter pylori where its strain which didn’t co-evolve with a host was associated with an increased risk of gastric cancer.Citation30 Such phenomena indicate the importance of stratifying stool donors by ethnogeographic and social factors.
Even now the quest for the healthy gut microbiota has led the researchers to the communities of African hunter-gatherers.Citation31 However, the genetics, culture, and diet in this pre-agrarian society drastically differ from ones of millions of potential Western recipients suffering from autoimmune and metabolic diseases. Achieving a successful transplantation and beneficial effects might be challenging. The equilibrium of the transplanted rural microbiota can become disrupted after the exposure to antibiotics and a Western diet and switch to a state of dysbiosis. From this perspective, we feel that it is also reasonable to examine the rural communities with a closer cultural and genetic background, like the Russian villages. In particular, considering diet, it is closer to the Western and can even formally consist of the same ingredients, but their origin (natural home-grown instead of the industrial processed) is the point that makes the difference for the health of the host and its microbiota.
Recently the richness of gut microbial gene repertoire was proposed as a potential biomarker of the healthy microbiota: higher gene count was associated with better metabolic indicators.Citation6 The universality of this surrogate marker is to be discussed. While the signal was statistically significant for the populations of the two Western European countries, it might as well change for a more global sample. As an example of such variation, the discriminant metagenomic markers for type 2 diabetes were found to be different between the SwedishCitation23 and ChineseCitation4 populations. In our Russian cohort, the gene count was not found to be bimodal and no link with BMI was detected (data not shown)—noting that the obese and overweight people were excluded from the study. The exact mechanism underlying the association between high gene count and health remains unknown. Moreover, a simple microbiota dominated by a single bacterial genus will have a lower gene count; it then follows that such microbiota is associated with an increased health risk. At the same time, a member of a rural community on a simple diet might likely possess such low-complexity microbiota. It would be wise to separate pure statistics from medicine and take a cautious approach to the translation from the correlations to the diagnostic applications. With such observations in mind, we anticipate that other markers of healthy gut are yet to be defined.
| Abbreviations: | ||
| ANOSIM | = | analysis of similarities |
| ASW | = | average silhouette width |
| FMT | = | fecal mass transplantation |
| HGC | = | high gene count |
| SRB | = | sulfate-reducing bacteria |
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JFWM, Tijssen JGP, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med 2013; 368:407 - 15; http://dx.doi.org/10.1056/NEJMoa1205037; PMID: 23323867
- Smits LP, Bouter KEC, de Vos WM, Borody TJ, Nieuwdorp M. Therapeutic potential of fecal microbiota transplantation. Gastroenterology 2013; 145:946 - 53; http://dx.doi.org/10.1053/j.gastro.2013.08.058; PMID: 24018052
- Karlsson FH, Fåk F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, Bäckhed F, Nielsen J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun 2012; 3:1245; http://dx.doi.org/10.1038/ncomms2266; PMID: 23212374
- Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012; 490:55 - 60; http://dx.doi.org/10.1038/nature11450; PMID: 23023125
- Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 2012; 13:R79; http://dx.doi.org/10.1186/gb-2012-13-9-r79; PMID: 23013615
- Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto J-M, Kennedy S, et al, MetaHIT consortium. Richness of human gut microbiome correlates with metabolic markers. Nature 2013; 500:541 - 6; http://dx.doi.org/10.1038/nature12506; PMID: 23985870
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. Human gut microbiome viewed across age and geography. Nature 2012; 486:222 - 7; PMID: 22699611
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012; 486:207 - 14; http://dx.doi.org/10.1038/nature11234; PMID: 22699609
- De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A 2010; 107:14691 - 6; http://dx.doi.org/10.1073/pnas.1005963107; PMID: 20679230
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al, MetaHIT Consortium. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464:59 - 65; http://dx.doi.org/10.1038/nature08821; PMID: 20203603
- Tyakht AV, Kostryukova ES, Popenko AS, Belenikin MS, Pavlenko AV, Larin AK, Karpova IY, Selezneva OV, Semashko TA, Ospanova EA, et al. Human gut microbiota community structures in urban and rural populations in Russia. Nat Commun 2013; 4:2469; http://dx.doi.org/10.1038/ncomms3469; PMID: 24036685
- Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, Huttenhower C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods 2012; 9:811 - 4; http://dx.doi.org/10.1038/nmeth.2066; PMID: 22688413
- Clarke KR. Non-parametric multivariate analyses of changes in community structure. Austral Ecol 1993; 18:117 - 43; http://dx.doi.org/10.1111/j.1442-9993.1993.tb00438.x
- Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto J-M, et al, MetaHIT Consortium. Enterotypes of the human gut microbiome. Nature 2011; 473:174 - 80; http://dx.doi.org/10.1038/nature09944; PMID: 21508958
- Pedersen R, Ingerslev H-C, Sturek M, Alloosh M, Cirera S, Christoffersen BØ, Moesgaard SG, Larsen N, Boye M. Characterisation of gut microbiota in Ossabaw and Göttingen minipigs as models of obesity and metabolic syndrome. PLoS One 2013; 8:e56612; http://dx.doi.org/10.1371/journal.pone.0056612; PMID: 23437186
- Hildebrand F, Ebersbach T, Nielsen HB, Li X, Sonne SB, Bertalan M, Dimitrov P, Madsen L, Qin J, Wang J, et al. A comparative analysis of the intestinal metagenomes present in guinea pigs (Cavia porcellus) and humans (Homo sapiens). BMC Genomics 2012; 13:514; http://dx.doi.org/10.1186/1471-2164-13-514; PMID: 23020652
- Hansen EE, Lozupone CA, Rey FE, Wu M, Guruge JL, Narra A, Goodfellow J, Zaneveld JR, McDonald DT, Goodrich JA, et al. Pan-genome of the dominant human gut-associated archaeon, Methanobrevibacter smithii, studied in twins. Proc Natl Acad Sci U S A 2011; 108:Suppl 1 4599 - 606; http://dx.doi.org/10.1073/pnas.1000071108; PMID: 21317366
- Zhang J, Zheng Y, Guo Z, Qiao J, Gesudu Q, Sun Z, Huo D, Huang W, Huo Q, Kwok L, et al. The diversity of intestinal microbiota of Mongolians living in Inner Mongolia, China. Benef Microbes 2013; 4:319 - 28; http://dx.doi.org/10.3920/BM2013.0028; PMID: 24311315
- Biarc J, Nguyen IS, Pini A, Gossé F, Richert S, Thiersé D, Van Dorsselaer A, Leize-Wagner E, Raul F, Klein J-P, et al. Carcinogenic properties of proteins with pro-inflammatory activity from Streptococcus infantarius (formerly S.bovis). Carcinogenesis 2004; 25:1477 - 84; http://dx.doi.org/10.1093/carcin/bgh091; PMID: 14742316
- Wu GD, Chen J, Hoffmann C, Bittinger K, Chen Y-Y. Keilbaugh S a., Bewtra M, Knights D, Walters W a., Knight R, et al. Linking Long-Term Dietary Patterns with Gut Microbial Enterotypes. Science 2011; 105.
- Suzuki R, Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2006; 22:1540 - 2; http://dx.doi.org/10.1093/bioinformatics/btl117; PMID: 16595560
- Xiao S, Fei N, Pang X, Shen J, Wang L, Zhang B, Zhang M, Zhang X, Zhang C, Li M, et al. A gut microbiota-targeted dietary intervention for amelioration of chronic inflammation underlying metabolic syndrome. FEMS Microbiol Ecol 2013; PMID: 24117923
- Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, Nielsen J, Bäckhed F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013; 498:99 - 103; http://dx.doi.org/10.1038/nature12198; PMID: 23719380
- Blaser MJ, Falkow S. What are the consequences of the disappearing human microbiota?. Nat Rev Microbiol 2009; 7:887 - 94; http://dx.doi.org/10.1038/nrmicro2245; PMID: 19898491
- Khoruts A. Faecal microbiota transplantation in 2013: developing human gut microbiota as a class of therapeutics. Nat Rev Gastroenterol Hepatol 2014; 11:79 - 80; http://dx.doi.org/10.1038/nrgastro.2013.231; PMID: 24296579
- Zhang F-M, Wang H-G, Wang M, Cui B-T, Fan Z-N, Ji G-Z. Fecal microbiota transplantation for severe enterocolonic fistulizing Crohn’s disease. World J Gastroenterol 2013; 19:7213 - 6; http://dx.doi.org/10.3748/wjg.v19.i41.7213; PMID: 24222969
- Ou J, Carbonero F, Zoetendal EG, DeLany JP, Wang M, Newton K, Gaskins HR, O’Keefe SJD. Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am J Clin Nutr 2013; 98:111 - 20; http://dx.doi.org/10.3945/ajcn.112.056689; PMID: 23719549
- Petrof EO, Gloor GB, Vanner SJ, Weese SJ, Carter D, Daigneault MC, Brown EM, Schroeter K, Allen-Vercoe E. Stool substitute transplant therapy for the eradication of Clostridium difficile infection: ‘RePOOPulating’ the gut. Microbiome 2013; 1:3; http://dx.doi.org/10.1186/2049-2618-1-3; PMID: 24467987
- Forslund K, Sunagawa S, Kultima JR, Mende DR, Arumugam M, Typas A, Bork P. Country-specific antibiotic use practices impact the human gut resistome. Genome Res 2013; 23:1163 - 9; http://dx.doi.org/10.1101/gr.155465.113; PMID: 23568836
- Kodaman N, Pazos A, Schneider BG, Piazuelo MB, Mera R, Sobota RS, Sicinschi LA, Shaffer CL, Romero-Gallo J, de Sablet T, et al. Human and Helicobacter pylori coevolution shapes the risk of gastric disease. Proc Natl Acad Sci U S A 2014; 111:1455 - 60; http://dx.doi.org/10.1073/pnas.1318093111; PMID: 24474772
- de Vrieze J. Gut instinct. Science 2014; 343:241 - 3; http://dx.doi.org/10.1126/science.343.6168.241; PMID: 24436401