Abstract
Cancer immunotherapy has seen a tremendous number of failures and only few recent regulatory successes. This is a review dedicated to determine major regulatory and developmental issues around cancer immunotherapeutics. A three pillar approach should be used in setting a development path: discovery platforms and sufficient pool of validated tumor antigens, product development strategy enabling to bring the product closer to the patient and clinical development strategy accounting for competitive landscape, treatment paradigm, technical and commercial risks. Regulatory framework existing around cancer vaccines in the EU, US, Japan and some developing countries is outlined. In addition, the review covers some specific issues on the design and conduct of clinical trials with cancer vaccines.
Path for Development of Cancer Vaccines
The technologies for cancer vaccination have been extensively reviewed elsewhere.Citation1-Citation6 In brief, therapeutic cancer vaccines can be off-shelf available (recombinant antigen cocktails, recombinant microorganisms, whole tumor cell derived (allogeneic), oncolytic viruses, anti-idiotypic antibodies, DNA and genetherapy based products) which could be manufactured and distributed worldwide and personalised cancer vaccines (autologous cells and antigens, adoptive cell transfer) which are heavily dependent on specialized centers of expertise and manufacturing. Despite extensive prior efforts and trials, only one vaccine: Dendreon’s Provenge (sipuleucel-T), a dendritic cell for metastatic castration-resistant prostate cancer, in so far has achieved an approval with FDA in 2010. In clinical development, sipuleucel-T was manufactured from autologous APC-containing peripheral blood mononuclear cells (PBMCs) of prostate cancer patients. PBMCs were obtained from a leukapheresis procedure. These cells were co-cultured with PA2024, the recombinant fusion protein of human PAP-GM-CSF, prior to reinfusion. Of note, sipuleucel-T comprises multiple types of mononuclear cells including APCs, CD4 and CD8 T cells, NK cells, and B cells. Provenge provides with approximately 4.1 mo in median OS improvement and has been introduced on US market at hefty $93,000 per treatment course for a regimen (three infusions given over one month). Significant company investment was required to overcome not only considerable development costs but also to solve various logistical hurdles with launching numerous FDA-certified centers across the US for production of Provenge.Citation7-Citation10 Crowded space of prostate cancer treatments along with pricing and logistical issues have created significant barriers for Provenge access across the US. Furthermore, high pricing and logistical barriers in expanding the supply chain makes significantly reduces probability of commercial success for the product in the EU, Japan and emerging markets.
Based on these lessons, current and future cancer vaccine developers will have to take product attributes and product development as one of the key goal-posts in commercial and clinical development. These prerequisites include development of off-shelf available product that will be amenable to distribution network in a reasonable distance from production sites thereby minimizing number of manufacturing sites. In addition, product should be brought closer to potential patients taking into consideration cold-chain issues, and patients living in rural and remotely based communities. Ideally, the bulk of the maintenance vaccination regimen should be administered via subcutaneous route allowing for greater market penetration and benefiting from higher insurance co-payments in some emerging markets. Finally, the manufacturing costs should be kept to a minimum and leveraging reimbursement and pricing across both developed and emerging markets. Reassuringly, Datamonitor (2009) reported that among 142 therapeutic cancer vaccines in phase II-III clinical development across seven major markets, the majority (96 vaccines, 68%) are standardized (off-shelf) products.Citation11
A summary of strategic directions that should be considered during cancer vaccine development is outlined in .
Figure 1. Three pillar approach in cancer vaccine development path.
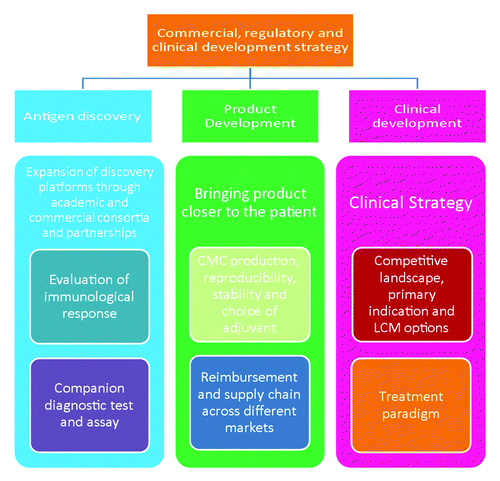
Antigen Discovery
Clinical studies of immunotherapy in cancer have focused on three classes of tumor antigens: tumor-specific and shared antigens (MAGE-3, NY-ESO-1, TRAG-3); mutated unique oncogenes (p53, α-actinin-4 and malic enzymes); oncofetal antigens (carcinoembryonic antigen); self-antigens overexpressed in tumors (HER2, MUC1, survivin); antigens expressed in some normal tissues (WT-1, PRAME, Survivin-2b).Citation12,Citation13 Despite that approximately 20 antigens have reached phase III clinical development, cancer immunotherapy continues to be limited by a paucity of antigens that are universally associated with disease, strongly immunogenic, continuously expressed and cancer specific. It is reported that approximately 50 antigens are currently in different stages of development and they encompass not only membrane-bound but also intracellular targets. Applying an attrition factor to this number in keeping with well recognized high failure rate of potential candidates to progress into late stage development and regulatory submission, it is strikingly clear, that the number of currently studied tumor antigens is highly inadequate to ensure that there will a reasonable number of successful products in years to come. Therefore there is an urgent need for further tumor antigen discovery and their comprehensive immunological target validation. A combination of efforts driven by academic institutions and industry could assist in achieving these goals. Networking and alliance creation between cancer vaccine developers and university groups across the US, EU, Japan, Singapore, South Korea and China could become especially affluent source of future new cancer vaccine candidates.
Potential sources of new tumor antigens may come from the following interesting associations between cancer progression and immunity: (1) molecular mechanisms of tumor evasion are driven by both accumulation of genetic abnormalities and epigenetic changes. Regulatory molecules (such as histone modifying enzymes) might serve an interesting target in maintaining a high level of immunological response in cancerCitation14; (2) increasingly, cancer stem cells are considered to play a significant role in tumor evasion. Although in variable ratios, cancer stem cells have been identified in leukemia, prostate, breast, lung, melanoma, sarcomas and other tumors.Citation15 Most of current anti-cancer therapeutics (including highly targeted therapies) primarily target either semi-differentiated or proliferating cancer cells, and, conceivably, will not be effective against undifferentiated and mostly quiescent cancer stem cells. Encouragingly, recent data suggest that some commonly used drugs such as docetaxel and metformin may actually root out cancer stem cells thereby making them an attractive combination with immunotherapeutic regimens.Citation15 Three molecular pathways are especially prominent in regulation of cancer stem cells: the Notch pathway, the Hedgehog pathway, and the Wnt/β-catenin pathway.Citation14 Recently, a novel tumor antigen DNAJB8 highly-overexpressed in cancer stem cells and cancer progentior cells has been identified and proposed for immunotherapy of several tumors, including renal carcinoma.Citation16 (3) In addition, cancer stem cells may harbor unique antigens and regulatory factors that could constitute a target for cancer immunotherapy. (4) Similarly to MAGE antigens in melanoma, other biological entities might be responsible for interaction between tumor cells and stromal microenvironment thereby increasing a chance of identifying new candidates.Citation17 (5) Extended focus is now given to intimate relationships between cancer, viral and non-virally driven inflammation. The success of complete resolution of inflammation is inherently linked to carcinogenesis, tumor responsiveness to radio- and chemotherapy and ultimately tumor expansion. The role of intra-tumor and peri-tumor (stromal) inflammation can be different in terms of the influence on tumor. A recent study by Haabeth et al. (2011) demonstrated that inflammation driven by tumor-specific Th1 cells protects against B-cell cancer.Citation18 On the contrary, Colotta et al. (2009) reported that intratumoral inflammatory microenvironment acts as a favorable ground for premalignant cells to attain malignant properties via evasion, epigenetic changes, and epithelial to mesenchymal transition.Citation19 Discovery of tumor stromal molecules and pathways which could delineate these conflicting roles of tumor microenvironment and play causative and contributory role in induction of tumor evasion may help to design new effective cancer immunotherapy. (6) Alpizar et al. (2011) indicated that despite a large number of potential cancer antigens were discovered, clinical trials with immunisations were disappointing mainly due to loss of MHC class I expression in tumors. Therefore an identification of specific regulatory factors and elements which are responsible for downregulation of MHC class I and combining them in cocktails with other antigens could emerge as an attractive approach in cancer immunotherapy.Citation14 (7) It is hypothesized that some virally-induced tumors could evade immunity via secondary tolerance toward MHC class I and viral peptide complexes.Citation5 Development of oncolytic viruses conferring immunogenic sequences of viral peptides associated with the life cycle of virus and immune tolerance, could abrogate the tolerance and enhance the anti-tumor response.
Therefore further efforts are required in discovery and validation of novel tumor antigens in order to enrich existing clinical pipeline and account for attrition rate of vaccine candidates.
Clinical Positioning and Treatment Paradigm
The ultimate outcome of the clinical development for a novel product is positioning in a subset of patients in whom the benefit-risk ratio is most favorable and most of target product profile features are well linked with anticipated clinical values and benefits. Any developers should be thinking about a specific market niche in context of crowded and competitive treatment paradigm, efficacy and safety attributes emerging from ongoing clinical studies and biomarkers or companion diagnostic tests predictive of the optimal clinical response.
The market for prostate, breast and kidney cancer drugs has grown increasingly crowded in recent years with multiple agents in clinical development and several products approved across US, EU and Japan markets. For example, the old paradigm of renal cancer treatment was based on use of immunomodulatory therapy which provided a modest survival benefit, at the expense of considerable toxicity. Since 2005, seven targeted agents, bevacizumab, sorafenib, sunitinib, pazopanib, temsirolimus, everolimus and axitinib have been approved by the US FDA for the treatment of different lines of metastatic or locally invasive disease. In general, these agents have higher efficacy against clear cell than non-clear cell histologies.Citation20 Similarly, the treatment paradigm for prostate and breast cancer has become incredibly competitive providing with only limited remaining opportunities and creating a fierce rivalry for immunotherapy products.
By the time Provenge came into US market in 2010, the landscape for treating castrate-resistant prostate cancer has become very crowded. Two new agents: Sanofi's new chemotherapy Jevtana (cabazitaxel) and J&J’s hormone therapy Zytiga (abiraterone) were approved by EMA and FDA in 2011 for patients following chemotherapy and is being positioned for earlier-stage patients after failure of primary androgen deprivation therapy and prior to chemotherapy, which is the same population targeted by Provenge.Citation7,Citation21 In addition, generic oral ketoconazole has a similar mechanism of action as abiraterone and, among other options, has been used for many years as a as a second-line hormonal treatment prior to chemotherapy. US National Comprehensive Cancer Network guidelines include adrenal/paracrine androgen synthesis inhibitors abiraterone and ketoconazole as options for patients after initial castration therapy has failed. The guidelines specify that abiraterone may be used for metastatic CRPC patients who have not received prior chemotherapy, though it isn’t the standard of care.Citation22 Many other treatments are in development for prostate cancer and have shown promise leaving a scope for differentiation either for further improvements in safety/tolerability profile or provide with cost-effectiveness benefits for payers and health care.
There are some oncology indications which are highly desirable for cancer vaccines (). A disproportionately high number (> 50%) of new therapeutic cancer vaccines are in development in most crowded therapeutic settings, such as melanoma, prostate cancer, breast cancer and NSCLC.Citation11 Such an imbalance is driven by success story of Provenge in securing FDA approval and also by especially high number of tumor antigens previously discovered and studied in these tumors. For example, non-small cell lung carcinoma (NSCLC) represents a highly desirable (unmet medical need) but risky area for cancer immunotherapies. GSK’s MAGE-3 is targeting post-resection disease free NSCLC patients as despite surgery, nearly 50% of patients are likely to develop disease recurrence ultimately die.Citation23 In contrast, Stimuvax (Oncothyreon) is currently tested in the ongoing phase III study in a different patient population: locoregional, unresectable stage III NSCLC patients. This is an example how two different products are confined to slightly different patient subpopulations allowing protecting the intended market and mark boundaries for the optimal product use in real life scenario.
Figure 2. The attractiveness of new opportunities for cancer vaccine development is driven not only but level of competition, but is also determined by probability of development success, development costs, past attrition rate and level of other residual commercial and non-commercial risks.
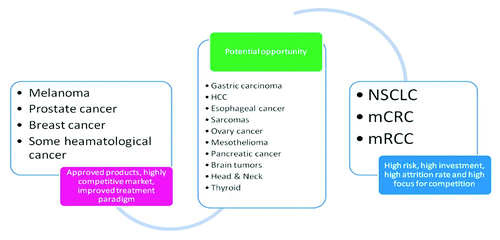
The group of indications representing potential commercial and clinical opportunity (see ) is determined by a relative scarcity of current clinical pipeline, high unmet medical need and a scope for discovery and validation of novel tumor antigens and technologies aiming to tackle these underserved tumor types.
Another example of uncontested oncology opportunity is ovarian cancer. For nearly two decades, frontline therapies for newly diagnosed patients haven’t budged beyond paclitaxel and platinum-based drugs. And neither these nor additional options for second-line treatment, namely, gemcitabine, bevasizumab (Roche) and olaparib (AstraZeneca) and several other chemotherapies have produced appreciable advances in OS. In addition, recent failure of anti-idiotypic CA-125 monoclonal antibody abogovomab (Menarini) to demonstrate statistically significant difference in PFS and OS in phase III MIMOSA study conducted in women with advanced ovarian cancer, was a serious blow to immunotherapy efforts in ovarian cancer field.Citation24
Median life expectancy for ovarian cancer patients remains stuck at barely 5 years, and roughly 80% of those diagnosed with the illness will eventually succumb to recurrent disease despite initial debulking surgery, platinum and taxane therapies.Citation25 With a relatively long life-span in malignancies such as ovarian, thyroid cancer and some tumors masquerading as a chronic disease due to succession line of treatments with considerable impact on OS (breast, prostate and renal cancer) new developers face a real challenge in running studies sufficiently long to capture OS benefits. PFS as a primary endpoint might be more appropriate in these settings. FDA has specifically stipulated that if PFS is used as a primary endpoint, the magnitude of PFS improvement must be substantial and it must outweigh the risk associated with the treatment.Citation25 Based on time-to-progression (TTP) estimates from Provenge studies and other clinical experience with cancer vaccines, it is expected that cancer vaccines are unlikely to provide with TTP/PFS advantages because of the immunologically driven tumor swelling and infiltration observed in initial phases of treatment.Citation10 It is expected that a delayed benefit may arise throughout the treatment, but the experience with PFS interpretation in different truncated phases of treatment is limited and did not go through sufficient regulatory scrutiny. As a consequence, development of immunotherapies may require more sensitive imaging tools and novel validated clinical endpoints in measuring the disease and predicting clinical benefits derived from different domains (survival related, quality of life and symptom/symptom cluster related).
Therefore new immunotherapy studies in competitive clinical settings will grow in required sample size, duration, complexity, costs and level of development risk. Since cancer vaccines are largely developed by small and mid-size biotech companies, these developments will not be feasible for biotech companies unless there is a considerable support from investors or large pharma partners. Unavoidably, it is predicted that competitive and risky oncology indications will be primarily a battleground for large pharma companies. In addition, the evolving and improving treatment paradigm needs to be integrated into the scheme of background therapies for cancer immunotherapy agents which are either in development or about to enter the clinical phase. Developers should now consider that by the time their studies will be completed, regulatory and payer bodies will be expecting that background therapies should represent up-to-date best standards of care in a specific region providing them with appropriate benchmark comparisons in order to appreciate the benefit of a new therapy.
Combination Therapies
The traditional strategy to design combination therapy in clinical trials is to empirically combine agents with proven clinical efficacy. This strategy has been irregularly successful in deriving effective combination treatments. With the wealth of data available from molecular studies on tumor angiogenesis, we can now rationalize novel combinations which can readily be tested in preclinical models and subsequent clinical trials.
Empirical combinations of different products which are individually were able to show significant OS survival has shown inconsistent results. Bevacizumab has been successful in a number of approved oncology indications, including metastatic renal carcinoma (mRCC). However, to date, the great majority of Bevasizumab-containing combination regimens have failed to show a demonstrable clinical benefit. Namely, combinations of bevasizumab with erlotinib and sunitinib were unfruitful in clinical studies.Citation20 In addition, despite being deemed “targeted therapies,” most if not all of the novel small molecule and biological agents currently approved for the treatment of cancer can have significant side effects. Much of what we know about the safety profile and toxicities of these drugs has been elucidated from trials in which these agents were used as a monotherapy. Combination regimens may increase the rate of adverse effects. For example, the incidence of autoimmune manifestations in clinical studies with ipilimumab was a cause for concern and resulted in creation of risk-evaluation and management strategy plan (REMS).Citation26 It is also uncertain whether an anticipated benefit of targeting several signaling pathways will be successful, given a multitude of different tumor escape mechanisms. There are also IP and regulatory considerations that create barriers around cooperation of companies holding individual rights for combination components.Citation27 In particularly, one of the companies may not be so keen on development of combination regimen if their product is near the end of life cycle and novel combination will require considerable investment and may create new unexpected safety risks demanding re-evaluation of the benefit-risk balance. If developer is working on combination regimen composed of two non-approved IND products, the challenge arises that FDA cannot divulge or disclose any information on the product from another file. Exchange of manufacturing files or disclosure of compliance issues between developers is not possible. Combination therapies are not only tricky from IP, commercial and regulatory perspectives, but also problematic from labeling (cross-reference to another product with potentially changing benefit-risk balance), pharmacovigilance (determination of causality for new safety risks and risk management planning), post-marketing (requirements for safety studies, potential impact from the data emerging with components) and reimbursement (potentially prohibitive pricing due to high cost of individual components).
FDA issued draft guidance for co-development of drugs for use in combination in December 2010. The guidance specifically pertains to combination regimens in life-threatening diseases, such as cancer. According to FDA requirement, each component in the combination will require clinical and pre-clinical argumentation for their selection and inclusion.Citation28 Studies comparing monotherapy and combination regimens might be larger in size and longer in duration than studies designed for monotherapy agents. Four arm phase II–III studies comparing dual combination with each individual component and placebo might be required. Adaptive design could be utilized in order to continue with selected arms and reduce the overall sample size. Hence, considerable investment, development, regulatory, commercial and pharmacovigilance resources might be needed to achieve a successful combination therapy. Finally, consideration should be given to the regulatory status of components employed in the combination regimen. For example, granulocyte-macrophage colony stimulating factor (GM-CSF) is often employed in combination with cancer vaccines during priming phase of treatment in order to stimulate dendritic cell growth and activity. However while GM-CSF is licensed in the US, it has never been approved in Europe and therefore the recommendation for its inclusion in EU label for cancer vaccine would effectively mean a recommendation for use of previously unlicensed/unapproved product. Hence, each component of the chosen combination or recommended schedule should be approved in intended regulatory territory.
From cancer vaccine perspective, more rational and risk-based evaluation of potential combination regimens is required. Since cancer vaccines are expected to provide with delayed response and impact on OS, a successful combination could trigger an earlier separation of OS curves resulting in an improved efficacy profile and potentially reducing the duration and size of required clinical studies. Rational selection of immunotherapy combination could be based on some new drug-screening platforms that quantitatively measure the effect of stromal cells on anticancer drug activity.Citation17 It was reported that the anti-tumor effect of agents such as imatinib and doxorubicin against myeloma and certain leukemia cell lines can be significantly reduced in the presence of stromal cells. In contrast, the activity of bortezomib was not affected.Citation17 By using specific combinations of tumor and accessory cells, a screening assay can predict the relationship between targeted therapies and tumor microenvironment and isolate only those small molecule and/or biological agents which could be favorably combined with cancer immunotherapies. Some classes of agents such as proteasome inhibitors or histone deacetylase inhibitors (HDAC) are believed to exert a suppressive effect on tumor stromal cells resulting in enhanced T- and NK-cell driven immunological response against tumor.Citation29 Similarly, combinations of cancer vaccines, monoclonal antibodies, tyrosine kinase inhibitors and other targeted therapies might be rationalised on the basis of their impact on T-regulatory (Tregs), myeloid-derived suppressor cells (MDSCs), natural killer (NK) cells and kinetics of anti-tumor immune response (see overview by Vanneman and Dranoff ().Citation30
Figure 3. Sequential use of specific targeted therapies in order to capture a synergy with key stages of antitumor immune response (adapted with permission from Vanneman and DranoffCitation30).
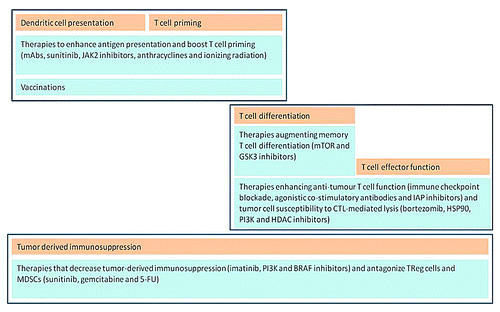
Therapies boosting DC antigen presentation and initial T cell priming should be delivered before vaccines and continued through initial T cell priming. Agents promoting T cell memory formation or enhancing T cell function and tumor cell lysis should be given during specific stages of immune response and discontinued upon completion of immune response phase in order to avoid deleterious effect. Tumor-derived immunosuppression constantly antagonizes anti-tumor immune responses; and therapies that are designed to mitigate this should be given before vaccination and continued throughout treatment. 5-FU, 5-fluorouracil; CTL, cytotoxic T lymphocyte; GSK3β, glycogen synthase kinase 3β; HDAC, histone deacetylase; HSP90, heat shock protein 90; IAP, inhibitor of apoptosis protein; JAK2, janus kinase 2; mAbs, monoclonal antibodies. Importantly, since some of these agents are already included into current therapeutic paradigm and used as a part of standard of care in immunotherapy clinical studies, the effect of these agents on immune response and clinical parameters should be elucidated at sub-population level in order to gain further clinical experience with combination regimens and appropriateness of timing and switching between agents. However there are various clinical issues emerging with this approach: immune response will have to be monitored on regular basis what introduces complexity and additional cost burden for healthcare. In addition, the optimization in posology of approved agents will require complex prospectively planned factorial studies in order to confirm these hypotheses.
In addition to interference of different chemotherapeutic agents in anti-tumor response, different drugs can also influence the immunogenicity of dying tumor. Specific agents can elicit immunogenic cancer cell death, which then stimulates an anticancer immune response that controls residual cancer stem cells. If the chemotherapeutic agent is intrinsically unable to stimulate immunogenic cell death, cancer cells fail to emit immunogenic signals as they die, the immune system fails to perceive such signals, or effector cells are immunosuppressed, therapeutic failure ensues. Large scale clinical studies are required to explore this scenario at pharmacological, immune and histological level and determine agents that induce immunogenic cell death and those that do not.Citation31
There are several emerging antibody technologies such as antibody-drug conjugates (ADCs), bispecific and trifunctional antibodies, domain antibodies, nanobodies, and fragment crystallizable (Fc)-engineered antibodies promise to enhance tumor cytotoxicity via injection into tumor cell some specific toxins (ADC), improving affinity of binding (domain antibodies), engaging T-cell response (bispecific and trifunctional antibodies), and facilitate tissue permeability (domain and nanobodies), and promote pharmacokinetic half-life of the product (via Fc fragment modifications.)Citation26,Citation32 Adcetris (brentuximab vedotin; Seattle Genetics/Takeda) for the treatment of relapsed/refractory Hodgkin's lymphoma and relapsed/refractory systemic anaplastic large cell lymphoma, is the only currently EU and US approved ADC. This, however, is not the only ADC anticipated to be brought to the market during the next five years, as ImmunoGen and Genentech/Roche’s trastuzumab emtansine (pertuzumab) is forecast to launch in 2013. Roche has recently made FDA and EMA submissions for pertuzumab in HER2-receptor positive breast cancer in combination with Herceptin.Citation33,Citation34 The data that support submissions are derived from the pivotal Phase III CLEOPATRA, a randomized, double-blind, placebo-controlled study, which evaluated the efficacy and safety profile of the pertuzumab-based regimen compared with Herceptin and chemotherapy plus placebo in 808 people with previously untreated HER2-positive mBC. The study demonstrated a 6.1 mo improvement in median progression-free survival (PFS) for people who received a pertuzumab-based regimen (pertuzumab combined with Herceptin and docetaxel chemotherapy) compared with those who received Herceptin and chemotherapy alone (median PFS 18.5 vs. 12.4 mo).Citation33,Citation34 Emergence of new antibody technologies which can further enhance highly targeted anti-tumor effect create potential avenues in integrating ADC and cancer vaccines into long-term sustainable combination regimens and provide with incremental gains in PFS/OS, improved quality of life and enhanced safety and tolerability profile.
In melanoma, there has been a recent success in the US and EU approval of vemurafenib (Zelboraf) in patients bearing the V600E activating mutation of BRAF. Vemurafenib has been shown to yield a marked response in more than 50% of patients.Citation35,Citation36 It was developed along with commercially available diagnostic test capable of indentifying V600E activating mutations. Unfortunately, resistance to vemurafenib develops rapidly (< 1 y), creating the necessity for additional therapy. In order to circumvent this problem, a combination with agent like ipilimumab could potentially assist with generating long-lasting protective immunity. Since vemurafinib-induced death of tumors can be expected to release endogenous tumor antigens, it is possible that the small molecule and immunotherapeutic approaches may synergize, with BRAF inhibition acting to help prime de novo T-cell responses that can then be facilitated by the anti-CTLA4. As a consequence, Roche and BMS have reported about mutually agreed plans to initiate development of the combination regimen.Citation37 Interestingly that both, vemurafenib and ipilimumab are currently in the beginning of their life cycle and therefore development of combinations can present with new unique commercial and clinical opportunities.
Regulatory Framework for Development of Cancer Vaccines
To date, an estimated more than 10,000 people have participated in late-stage clinical trials of active cancer immunotherapies. The vast majority of these studies have failed to demonstrate any meaningful efficacy with a large proportion of unsuccessful phase III studies conducted with vaccine candidates that looked quite good in early trials.Citation38
Significant investment and development efforts made by industry and academia resulted in a considerable number of IND submissions and scientific advice interactions between developers and regulatory agencies in the past 20 years. The objective of this review is to provide an overview of most up-to-date regulatory considerations relevant to cancer vaccine products in EU, US, Japan, and some emerging markets.
European Regulatory System
In 1995–2011 y EMA has provided 2553 scientific advice (SA) and protocol assistance procedures across all classes of medicines with 26% of those falling into area of anti-neoplastic and immunomodulatory therapies.Citation39 Between 2006–2011 EMA and FDA also assisted with 17 joint SA and protocol assistance procedures.
In EMA and National authorities SA procedures on cancer vaccine products are driven by oncology experts based at national competent authorities and to a lesser extent by some external experts and advisory boards. Therapeutic cancer vaccines are not subject of consideration by EMA Vaccine Working Party that is focused on preventative vaccines for infectious and communicable diseases. Due to complex and heterogeneous nature of some cancer immunotherapeutics, cell-, virus- and genetherapy-based products might be scrutinised by Committee of Advanced Therapies (CAT) and Genetherapy Working Party functioning within CAT. The quality issues arising on cancer vaccines submitted for full marketing authorization application (MAA) in the EU or products under SA assistance may also be taken for discussions at EMA Biologics Working Party. Coordination of all administrative issues pertaining to SA, including allocation of coordinators and peer-reviewers, requesting the input from different working parties and experts, issuing SA letters etc. is handled by EMA Scientific Advice Working Party (SAWP). Committee of Human Proprietary Medicinal products (CHMP), SAWP, Oncology Working Party and CHMP Scientific Advisory Group on Oncology may independently or jointly recommend on specific needs for further EU guidelines in relation to development of oncology products and specifically cancer immunotherapies. The need for guidelines is dependent on the demand for SA around specific therapeutic areas and EMA experience with evaluation of novel agents. For example, if there are major disagreements between applicants and EMA as well as discordance of views on the scope of the data requirements arising between Rapporteurs and assessors from national agencies, then consideration is given to the development of a guideline that provides the industry and assessors with harmonized framework for clinical data evaluation. Prior such guideline is developed and issued it is preferred to obtain a “real-life” experience with evaluating one or two products coming through MAA submission.
In recent years EMA has experienced several MAA submissions related to cancer immunotherapies: Cerepro for high-grade glioma (2007 and 2010); Advexin for Li Fraumeni syndrome and refractory squamous carcionoma of head and neck (2008), Oncophage for metastatic renal cell carcinoma (mRCC) (2009) and Yervoy for advanced malignant melanoma (2011). There is also currently ongoing EMA submission of Provenge in castrate-resistant prostate cancer which is under evaluation.
MAA for sitimagene ceradenovec (Cerepro) from Ark Therapeutics was reviewed by EMA twice in 2007 and 2010. The product was intended for treatment of high-grade glioma in patients who are eligible for surgery. Major objections in all domains were identified and despite of the company’s appeal, the outcome was negative and resulted in withdrawal of MAA application in 2010. Major clinical issues pertained to the fact that an open pivotal study did not show OS advantage vs. control group. In addition, during the trial, the company changed the primary endpoint in a sequential design from “overall survival” to “time to death or re-intervention,” which is prone to bias by treating physicians. The administration of Cerepro was associated with an increased incidence of adverse and serious adverse events (e.g., hemiparesis, seizures).Citation40,Citation41
In 2008 Gendux Molecular Ltd has withdrawn MAA for Advexin that contains contusugene ladenovec, a genetically modified virus which carries the wild-type p53 gene. Advexin was submitted for orphan designated indication in the treatment of Li-Fraumeni cancer. A separate MAA was submitted by Introgen/Gendux for Advexin for the treatment of recurrent, refractory squamous cell carcinoma of the head and neck. Numerous manufacturing, non-clinical and clinical issues were identified. Clinical benefit in relation to convincing demonstration of survival and progression free status in a limited number of patients treated with Advexin was lacking. Pharmacodynamically, there was no convincing correlation of p53 expression in tumors and clinical response to Advexin. Hence both MAA for Advexin were eventually withdrawn by the company.Citation42
Oncophage (Antigenics) was evaluated by EMA in 2009 in indication of mRCC. A number of major deficiencies in quality, pre-clinical and clinical domains were identified and led to withdrawal of the submission. The lack of statistically and clinically meaningful OS and PFS advantage with Oncophage constituted a major clinical deficiency.Citation43
Finally, in May 2010, the Bristol-Myers Squibb Pharma EEIG submitted an application for marketing authorization for Yervoy (ipilimumab) in patients with advanced melanoma. The CHMP recommended the granting of a marketing authorisation for ipilimumab based on a positive benefit-risk balance. Following this review the European Commission issued a marketing authorization on 13 July 2011 for ipilimumab for the treatment of advanced (unresectable or metastatic) melanoma in adults who have received prior therapy. CHMP members and assessors involved in the review have published EMA experience with evaluation of ipilimumab.Citation44 EMA assessors have placed an emphasis on improvement of overall survival observed in the pivotal study in adult patients with advanced previously treated melanoma receiving ipilimumab monotherapy. In the pivotal phase 3 trials, ipilimumab led to a 3-mo improvement in overall survival, a disease control rate of 28.5%, and 60% of responding patients maintained this response for 2 y. Furthermore, patients who received re-induction with this agent had further disease control. Various limitations in individual sub-populations were acknowledged. For example, less prominent OS advantage was observed in women > 50 y of age. As the subgroups analysis included only small numbers of patients, no definitive conclusions can be drawn from these data. Only limited data on the use of ipilimumab in patients with brain metastases of melanoma and no data in patients with ocular melanoma were available. As no additional safety concerns were expected, the CHMP considered that these should not be an absolute contra-indication for ipilimumab treatment. Finally CHMP has acknowledged a high proportion of immunological reactions, which could be severe or life-threatening, and might involve the gastrointestinal, liver, skin, nervous, endocrine, or other organ systems. Ipilimumab can also confer a risk of autoimmune disorders. Unfortunately, several trials have reported a possible correlation between grade 3 and 4 adverse events with the clinical efficacy of ipilimumab. CTLA-4 inhibition is expected to reduce peripheral self-tolerance and increase the risk of autoimmunity. This suggests that tumor regression is associated with the development of autoimmunity and does imply that enhanced efficacy with anti-CTLA-4 and selectivity cannot be achieved.Citation45 Notwithstanding, with various safety warnings and precautions introduced into the product label and risk minimisation steps proposed by the applicant as a part of risk management plan, overall the benefit-risk balance was viewed favorably.Citation44
Experience with these submissions and parallel development of FDA guidance on clinical considerations for therapeutic cancer vaccines issued in 2011 triggered an addition of the section on cancer immunotherapies in the draft of the currently revised EMA guideline on evaluation of anti-cancer medicinal products in man (CHMP/EWP/433478/2010).Citation46 Principles related to cancer immunotherapies were very broad but conducive to previously reported phenomenon of delayed immunological response with cancer vaccines. The guideline stipulates that “Induction of an effective immune response and clinical response may need more time to develop (delayed effect) compared to classical cytotoxic compounds. Patients may thus experience disease progression prior to the onset of biological activities or clinical effects. Discontinuation of active cancer immunotherapy in case of slow progression may not be appropriate. In these situations a detailed definition of “slow progressive disease” is expected in the study protocol.” It was indicated that OS primary endpoint will be preferred in confirmatory clinical studies. Possible toxicities like induction of autoimmune reactivity (cellular and humoral) and induction of tolerance should be carefully monitored during the clinical development. While neither there is currently designated EMA guideline on development of cancer vaccines nor there is a concept paper announcing on EMA intent to develop one, there are several pertinent guideline documents that provide industry with various methodological tools on immunotherapy products derived from different sources: cells, viruses and antigens. CHMP/BWP/271475/06 which became effective in 2008, outlined principles on potency testing of cell based immunotherapy medicinal products for the treatment of cancer.Citation47 Potency of cell based immunotherapy products can be measured in a number of different assays including in vivo and in vitro test systems. Preferably, a suitable functional potency assay that approximates as close as possible to clinical response should be in place already when material for the first clinical trial is produced and it should be validated prior to phase III clinical trials unless otherwise justified. Where it is impossible to develop a direct measure of biological response, an assay or set of assays evaluating surrogate responses could be proposed. This guideline tackles an issue of product standardization, consistency, stability and release specifications. For example, in order to standardize release and quality controls around autologous and allogeneic cell based vaccines, developers can use a viable cell count and monitor some specific surface expression markers correlating with biological and clinical response.
CHMP guideline on development of human cell-based medicinal products enacted in 2008 (EMEA/CHMP/410869/2006)Citation48 and CAT guideline on risk-based approach applied to Advanced Therapy Medicinal Products (EMA/CAT/CPWP/686637/2011)Citation49 released in 2011 provide with some general principles in relation to all cell based therapies including those that are used for immunotherapy of cancer. CHMP recommended risk-based approach in the overall evaluation of the product development. Criteria for risk may include the following: the origin of cells, ability to proliferate and/or differentiate; ability to initiate an immune response (as target or effector); level of cell manipulation (in vitro/ex vivo expansion/activation/differentiation/genetic manipulation/cryo-conservation); mode of administration (e.g., ex vivo perfusion, local or systemic surgery); duration of exposure or culture (short to permanent) or life span of cell; combination product (cells and bioactive molecules or structural materials); availability of clinical data on or experience with similar products. For instance, risk of cell migration and distribution is perceived to be higher with undifferentiated dividing allogeneic and autologous cells vs. well differentiated and unable to migrate cell lines. In the former case, non-clinical distribution studies, tissue biopsies and monitoring for tumorigenicity markers are likely to be required. A set of surface markers that well characterize cell derived product and indicate stability of cell population is critical in determining release specifications and stability.
CAT/CHMP has acknowledged limitations of non-clinical studies with cell-based products and recommended to perform essential proof-of concept studies in relevant animal species and some in vitro studies if animal studies are unfeasible or not-practical. CHMP has also issued guidelines on quality, pre-clinical and clinical development of genetherapy products; ICH considerations on oncolytic viruses; quality, non-clinical and clinical issues relating specifically to recombinant adeno-associated viral vectors; quality, pre-clinical and clinical aspects of medicinal products containing genetically modified cells. These guidelines are relevant to gene- and virally based therapies for cancer.Citation50 There are some specific requirements to evaluate viral shedding in systemic circulation and secreted fluids. In conjunction with administration of virally based products, developers will need to evaluate, quantify, and minimise environmental and transmission risks posed to environment, caregivers, health practitioners and family members. It is however clear that the degree of acceptance for potential and identified risks as well as uncertainties around the product will be much higher with cell-based and genetherapy products intended for treatment of life-threatening oncology indications as opposed to life style or chronic degenerative diseases.
However, despite that EU regulatory framework around advanced cell therapies has outpaced legislation developed and enacted by FDA in past 10 years, there are still several gaps in guidelines issued by EMA in relation to cancer immunotherapies. Cancer vaccines represent a heterogeneous group of products and there is no designated guideline on standardized antigen-based vaccines or adjuvants used for cancer vaccine therapies. Methodologies for evaluation of new adjuvants and vaccine candidates communicated in EMA guidelines on new adjuvants (EMEA/CHMP/VEG/134716/2004Citation51 and EMEA/CHMP/VWP/244894/2006)Citation52 and vaccines (EMEA/CHMP/VWP/164653/05)Citation53 are of relevance only to products intended for communicable diseases and given for preventative purposes to healthy subjects rather than for therapeutic purposes in patients with malignancies. Therefore further fine-tuning of EMA regulatory framework around cancer immunotherapies is required. More open and flexible pathways for evaluation of cancer immunotherapies developed for orphan indications or settings where measurement of disease is particularly challenging (e.g., ovarian cancer or hematological malignancies). It should be also recognized that with limitations of pre-clinical data, a “tick-boxing approach” should not be universally applied and leverage should be given to situations when the main evidence of efficacy is derived from clinical models.
Contentious and complex issues are expected to be openly raised by developers during SA procedures. SA should be contemplated early in clinical development process and most certainly prior phase III study will commence. Global development program could be reviewed by EMA and FDA in so-called parallel SA procedures to simultaneously gauge the view from both agencies. Oncology is named as one of the area eligible for parallel SA route. If the request is submitted in a synchronised manner to the FDA and the EMA, similar procedural timelines allow for discussion before the final decision is reached by each agency. This exercise is not intended to provide a combined or joint advice from the two regulatory authorities, but is an opportunity for increased dialog. Each agency will provide their independent advice to the developer. It is also possible to request SA from CHMP with participation of Health Technology assessment bodies.
FDA Requirements on Cancer Vaccines
FDA reported that in 2010 in excess of 1400 active investigational files were handled by FDA Offices and Departments. Oncology drug developers are increasingly required to consider use of different biomarkers in development of targeted therapies. FDA Critical Path initiative which was launched in 2004 has proposed this paradigm but, according to a 2008 review, only 3% of clinical trials have incorporated a novel biomarker of efficacy into their clinical trial design and there are significant delays with development of diagnostic companion tests.Citation27
FDA has kept a considerable interest to cancer immunotherapies through participation in numerous workshops and conferences, e.g., FDA/NCI Co-Sponsored Workshop on Cancer Vaccines and Immunotherapy in 2007 and 2nd World Cancer Vaccine Congress in Boston in 2011 and other events.Citation54,Citation55 From FDA perspective, cancer vaccines fall into category of products evaluated by Office of Cellular, Tissue and Gene Therapy products at CBER (Center for Biologics Evaluation and Research). Immunotherapy products that include adjuvants, nanoparticles and other non-cell derived components may require involvement of CDER (Center of Drug Evaluation and Research), CDRH (Center for Devices and Radiological Health), Office of Combination products (OCP) and Office of Drug Safety (ODS). Combination of cancer vaccines with other agents into single formulation poses particular challenges for developers as CMC data may reside in Master Files or cross-referenced files may not be accessible to applicants. In addition, FDA cannot discuss or divulge CMC issues without holder’s authorization.Citation55 Therefore it is crucial that applicants seek a comprehensive and transparent exchange of the data on in-licensed components from licensor’s CMC files.
For the approval of a Biologics License Application (BLA), it is critical that sufficient evidence of effectiveness is available so that both the sponsor and the FDA can adequately complete the benefit/risk (B/R) assessment of the new molecular entity (NME) (21CFR 314.126). In addition, the product should be of acceptable safety (21CFR 314.126) and the product label would define an appropriate patient population and provide with an adequate information enabling safe and effective use of the product 21CFR 201).Citation56 Section 505(d) of the FD&C Act, as well as Section 351 of the Public Health Service Act, indicate that new drugs and biologics should establish substantial evidence of clinical effectiveness through means of “adequate and well-controlled studies.” The base assumption is that since the term studies is plural, two or more controlled randomized clinical trials are required to establish efficacy. Specifically in oncology, there are many scenarios (and many past examples) where FDA rendered a single pivotal study sufficient for approval. The case for adequacy of a single study as well as a qualification for accelerated evaluation and approval should be made on the basis of advantages seen with the product in extending PFS and OS (as per phase I–III studies), gains observed in evaluation of patient-reported outcomes and quality of life; and favorable effect on established surrogate and composite endpoints.Citation57 With potential limitations and caveats in clinical data, sponsors might be prepared to seek a conditional approval route with ways to generate further clinical data supporting clinical benefits via post-approval commitments.Citation58,Citation59
The experience of FDA with evaluation of cancer immunotherapies included a number of submissions with negative outcomes: negative advisory board in 2001 for Melacine (Coriza) in advanced melanoma; rejection of Introgen’s Advexin p53 therapy for recurrent, refractory head and neck cancer in 2008 and others. Notably, some cancer vaccine developers did not attempt to submit BLAs in USA while they have tried MAA submissions in EU, e.g., Oncophage (Antigenics) for mRCC was never attempted for BLA submission.
The roller-coaster of immunotherapies was finally rewarded by two long-awaited successes. Sipuleucel-T (Provenge) from Dendrion was approved in 2010 for asymptomatic or minimally symptomatic metastatic castrate resistant (hormone refractory) advanced prostate cancer. Three pivotal phase 3 trials have shown an improvement in OS, the largest of which was the IMPACT study which unlike the previous studies was powered to detect an improvement in OS. This study has demonstrated the 4.1-mo improvement in median OS. Notably, Dendrion has currently submitted Provenge in Europe and the evaluation outcome in EU will not be necessarily in line with the one in USA. There are various issues associated with the magnitude of OS effect in comparator arm in Provenge’s study which could have contributed to a better separation of OS effect. In addition, the treatment paradigm utilized in IMPACT does not reflect current therapeutic landscape in EU, e.g., with lack of exposure to docetaxel-containing regimens.
In line with EMA decision, in 2011 FDA approved ipilimumab (Yervoy) for advanced melanoma. FDA has requested risk-evaluation and mitigation strategy (REMS) for ipilimumab in order to elucidate, control, minimize and prevent various safety risks (including those of autoimmunity) reported with Yervoy. Biovest International is currently seeking BLA submission for BiovaxID for treatment of follicular lymphoma.Citation60 Several phase III studies were approved by FDA. including Epeus’s program for Rexin-G genetherapy in pancreatic cancer, osteosarcoma and soft tissue sarcoma.Citation61
Having reviewed a plethora of cancer vaccine products in different stages of development (around 124 IND submission),Citation55 FDA has initially issued draft and in October 2011 released a final guidance on clinical considerations for therapeutic cancer vaccines. The guidance does not cover adoptive immunotherapies or vaccines intended for prevention of cancers.Citation62
Core messages from FDA guidanceCitation62 include the following:
Given relatively favorable safety profile with cancer immunotherapeutics and saturable dose-response curve, classical dose escalation studies are not appropriate for cancer vaccines and accelerated or continuous escalation regimens might be explored.
Exploratory phase I–II studies are extremely useful in evaluating cellular immunological responses, the pattern of ORR, dose-dependent relationships with surrogate outcome (e.g., skin test of delayed hypersensitivity reaction) and the rate of disease recurrence, isolating patient subgroups which benefit the most (e.g., HLA, NK and immunological biomarker-stratified methodologies).
The type of schedule and route of administrations are particularly relevant for evaluation of the efficacy.
Throughout an entire development, immune response should be evaluated, validated and correlated with any observed clinical outcomes. Therefore appropriate assays should be developed, validated and bridged, if necessary.
Different disease settings might be pursued and development decisions should take into account the length of time required to establish delayed immune response and capture its effect on PFS/OS, with consequential considerations for resource burden in planning of an appropriate phase III study.
The choice of patient population should be as homogenous as possible. This is of particular significance in trials with autologous vaccines due to inherent feature of the product heterogeneity.
Clinical models used should be sufficiently sensitive to demonstrate clinical benefit.
Phase II studies should be conducted in controlled fashion to yield maximum of the information on “Go” and “No-Go” decisions.
Limitations with use of ORR and PFS due to immunotherapy-mediated effect on tumor swelling, cell infiltration and remodelling. Alternative definitions of disease progression can be utilized if subjects still meet study-related eligibility criteria, do not show deterioration in performance scores or quality of life and there is no dose-limiting toxicity.
Use of biomarkers and development of combination therapies were discussed.
Based on these recommendations, cancer vaccine developers are expected to generate a substantial pre-phase III package to convince FDA on suitability of the subsequent BLA for accelerated approval or sufficiency of a single pivotal phase III study. Numerous interactions staggered throughout development process with particular important EOP2 and post-phase III meetings as well as ad hoc meetings can be requested by developers from FDA in order to establish a collaborative and cohesive dialog with the Agency. Due to some differences in methodology of assessment and reported regulatory outcomes in the EU and US, some developers can opt for a parallel SA interaction which can be requested from both agencies in a synchronised manner.
Situation in Japan
Modern cancer immunotherapy started in Japan in the 1970s following its introduction in western countries. First, the use of non-specific immunopotentiators was introduced. They included stabilized bacteria, fungi and bacterial components. Thus, some of the various glucans studied obtained government approval for medical use ().Citation63
Table 1. Non-specific immunopotentiators with anti-cancer activity approved by the Department of Health, Labor and Welfare of JapanCitation63
In the 1980s, due to profound progress in the field of genetic engineering, recombinant cytokines became readily available. This situation promoted the use of cytokines in the therapy of cancer; they included interferons, interleukins, tumor necrosis factor and colony stimulating factors, some of which have gained government approval for use in cancer therapy. Then, in the latter half of the 1980s, immuno-cell therapy was introduced in Japan.Citation63 Currently, two major fields of this therapy are autologous activated lymphocyte therapy (adoptive immunotherapy) and dendritic cell vaccination therapy, both of which include different variations. At present, at least 15 large public medical institutions are carrying out autologous activated lymphocyte therapy. Approximately a half of them have the approval from the PMDA as “highly advanced medical technology.” Also, more than 25 large medical institutions are carrying out dendritic cell vaccination therapy. In 1999, the Seta Clinic was established in Tokyo as the first private and leading clinic specializing in autologous immuno-cell therapy, followed by several other clinics.
Autologous cell-based products are exempt from central regulations and governed by local hospital authorities. For example, autologous cancer vaccine derived from patient tumor during the surgery and exposed to a minimal manipulation prior to re-administration into the patient can be regulated as surgical and medical intervention procedure. It can be performed under informed consent given by the patient and agreed between treating physician and the patient in relation to potential risks and benefits. Autologous and manipulated cells such as dentritic cell suspensions are also covered by local institutional ethics committee approval and institutional manufacturing license in dendritic cell therapies. Therefore for some cell-based immunotherapeutics, hospital contract and local ethical committee’s approval might be sufficient from regulatory and legal perspectives. Cell therapy must be either fully covered by insurance or by private cover without options available for mixed payment sources. If even a single drug from the combination is not reimbursed, the patient will have to cover the cost of entire treatment regimen.Citation64
Low regulatory barriers around autologous cell therapies contrast with those in South Korea, where all advanced cell therapies qualify as medicinal products and subject to regulations by Korean FDA. Japan represents an attractive destination for medical tourism associated with cell based therapies. The number of foreign patients visiting Japan to obtain cell based therapies was projected to be 1.6 million by 2010.Citation65 However many experts are concerned with GMP standards on producing of autologous advanced cell therapies in some academic institutions and hospitals. For example, concurrent manipulations with cells derived from different patients within the same bio-incubator, co-culturing of human cells with animal cells are common and pose potential health risks.Citation66 Establishment of institutional GMP framework for academic and hospital-related centers in relation to advanced therapies is considered mandatory in improvement of quality and safety safeguards around emerging complex autologous cell therapies.
Drug regulation is Japan is governed by Pharmaceuticals and Medical Device Agency (PMDA) established in 2004 by integrating three review-related bodies. PMDA has been working under a five-year plan first announced in 2007 to address criticism of review delays and inconsistent quality and lack of transparency. According to new PMDA figures, the target mean review time for new drug applications was reduced from 16 mo in total (including stoppages to resolve questions) in 2010, to 11.7 mo respectively, based on data up to the end of October 2011. Combination products vaccines containing adjuvants, genetherapy products for treatment of cancer and allogeneic cell-based immunotherapies are classified as medicinal products and fall under PMDA regulatory authority. Two closely interacting Biologics Offices within PMDA are involved in evaluation of cancer immunotherapies: genetherapies (Biologics 1), cancer vaccines and cell advanced therapies (Biologics 2). Developers can seek scientific advice and continuous dialog with PMDA via interactions held from pre-phase 1 until the dossier submission.
There is no designated cancer vaccine guideline issued by PMDA. However there are some broad and pertinent guidance documents which are of relevance to cancer immunotherapies: Notification No.266 (28 Mar. 2001): Handling and Use of Cell Therapies; Quality and Safety of Human Cells/Tissue-based Products; Notification No.0208004 (8 Feb. 2008): Quality and Safety of Autologous Human Cells/Tissue-based products; Notification No.0912007 (12 Sep. 2008)—Quality and Safety of Allogeneic Human Cells/Tissue-based products; No.0327025 (27 Mar. 2008): Manufacturing and Quality Control of Autologous Human Cells/Tissue-based Products; No.1062 (15 Nov. 1995, revised in 2002–2004): Quality and Safety of Gene-therapy Products.Citation67 Guidelines on development of novel vaccines and adjuvants are currently under development.
Japanese market remains extremely attractive for novel oncology therapies due to unmet medical need remaining in some common forms of cancer (gastric, pancreatic, hepatocellular, lung, head and neck tumors) and premium price reimbursement for biologics. Therefore it is expected that Japan will remain one of the pivotal markets included in clinical development and commercialization plans for upcoming cancer vaccines.
Rest of world
South Korean model for biotechnology innovation is now becoming a success story to follow. In 2006, Korean Government formulated “Bio-Vision 2016” (year 2006–2016) to acquire competitive source technologies and expand industrial infrastructure. It is intended that South Korean biotechnology sector will propel the country into a position of global leadership, through strengthening of collaboration between Ministry of Health, Korean FDA, health insurance / reimbursement bodies and innovation oversight committees. As a future growth engine industry, Korean market of biotechnology industry is expected to reach $60 billion in 2015.
As a consequence of “Biovision-2016” program, South Korean government has been working very closely with Korean FDA to enhance domestic R&D innovation, manufacturing and commercialization of new biotechnology products, including biosimilars, cell advanced therapies and vaccines.Citation68 A series of regulatory pathways were set up by the Korean Food and Drug Administration (KFDA) around manufacturing and clinical requirements for approval of biotechnology products with minimal data and using abbreviated and conditional routes.
KFDA Notification No. 2010–50 provides with principles for approval of cell derived products, genetherapies and bridging approach in utilizing foreign clinical data for approval in Korea. Some of KFDA guidelines were inspired by framework of conditional approval established by EMA. As a consequence, South Korea has seen several cancer immunotherapy products approved conditionally () and there are numerous products in development phase.
Table 2. Cancer vaccine products approved in South Korea.Citation69,Citation70
There is currently no designated cancer vaccine guideline in South Korea, Singapore, China, India, Brazil and Russia. In these countries cancer vaccines fall under provision of legislation for biologics or cell and tissue product approval. In addition, some pertinent principles from country-specific guidelines on evaluation of oncology products can be drawn to assist developers in approval process.
Gene-therapy product Rexin-G (replication-incompetent, pathotropic, tumor matrix (collagen)-targeted retrovector encoding an N-terminal deletion mutant of the cyclin G1 gene) from Epeius has been approved in Philippines as stand-alone therapy for the treatment of all chemotherapy-resistant solid tumors.Citation61 Two cancer oncovirus technologies, such as Gendicine (Shenzhen SiBiono Genetech) and Oncorine (Shanghai Sunway Biotech) were approved and successfully commercialized in China indicating that it is possible to achieve commercial success with premium-priced products in emerging markets ().
Table 3. Cancer oncoviral immunotherapies approved in ChinaCitation71-Citation73
Both products were approved by Chinese State FDA on the basis of open label clinical studies with various flaws in internal and external validity of results. The evaluation of clinical efficacy was based on ORR rather than on more robust clinical endpoints such as OR or PFS. Therefore it is highly unlikely that these two products will be available in EU and US markets as this has been confirmed by rejection of Advexin genetherapy submission by FDA and EMA in 2008.
Manufacturing and Product Development
Common pitfalls in manufacturing of biotechnology products were detailed elsewhere.Citation74 In a typical development program for a biotechnological or cell advanced therapy product, the manufacturing process is developed in parallel to the molecular or cellular characterization and this link has to be kept throughout all development phases. It is pivotal that appropriate potency assays or assays enabling to determine cell profile are developed as early as possible in the development in order to evaluate product and process consistency, and link emerging clinical data with quality attributes for the product. Early manufacturing development also heavily depends on establishing process control parameters, definition and quality of raw and auxiliary materials used, including viral clearance and viral validation methodology.
For cell derived immunotherapeutics key factors for successful development GMP manufacturing of cells of autologous or allogeneic nature include the following:
The source of cells, including master cell banks, where appropriate, is established and well characterized.
Cell population of sufficient homogeneity, purity, reproducible behavior, and pathogen-free.
Minimal batch-to-batch variation.
Production processes can be thoroughly characterized to ensure optimal performance.
Molecular characterization studies reveal similar expression of either surface markers or genetic signatures to demonstrate controllability of the product and show how these markers change through cell transition/differentiation.
Regulatory approval is likely to be successful when the CMC data are sufficiently comprehensive to appreciate that developers fully understand their product, can determine a margin for specification criteria and can relate quality attributes established during late stage development with the efficacy and safety features.
An ultimate goal of successful cancer vaccine is to simplify manufacturing and ensuing that product is brought closer to patients through supply and distribution network. Therefore from commercial perspective, development of off-shelf product is much more attractive as the number of manufacturing sites can be considerably minimized. Issues such as stability and cold-chain requirements should be explored throughout product development with appropriate manufacturing, distribution, and storage vendors identified and validated as necessary. Cell derived products will invariably require higher investment in order to create a lean supply chain. Requirement for multiple GMP-compliant manufacturing sites will most likely confine the product to a specific well optimized market rather than allowing a global outreach. Finally, the manufacturing costs should be kept to a minimum and leveraging reimbursement and pricing across both developed and emerging markets. Overreliance on manufacturing outsourcing of complex products such as cancer vaccines may not provide with cost-saving solutions and could pose risks on increased dependency from CMOs, product variability, and lacking in-house CMC expertise and knowledge in the product. Therefore in-house CMC expertise should be thoroughly maintained in order to mitigate potential regulatory and supply issues.
Clinical Development Considerations
Animal models used for investigation of cancer immunotherapies have extremely limited role in predicting the efficacy of vaccine candidates in human. In particular, for some antigen-specific cancer vaccines the target antigen may not have a close homolog in animals. In situations where reliable predictive animal models to assess immunological toxicity and activity are not available and the development of knockout and transgenic models is cumbersome, in vitro cell based models utilizing normal human and tumor tissues. The minimal size of pre-clinical package and necessity of individual animal models should be negotiated early with regulatory authorities. If vaccine contains adjuvants or other chemically or biotechnologically derived immunomodulators, a battery of appropriate toxicology, gene-toxicity, local tolerance and other studies will be expected. Provided that methodology for testing clinical, immunological and other biomarkers is established and sufficiently optimized, an early testing of vaccine candidate in phase I or proof-of-concept human study is strongly recommended.
Concepts laid into FDA guidance have partially originated from the Cancer Vaccine Clinical Trial Working Group (CVCTWG), which suggested completely new approach to cancer vaccine trial design in which the three-phase structure of product development is replaced with a two-stage model.Citation75 In this model, the first stage of development would be a proof-of-principle trial with the objective of determining safety, dose and schedule, and the demonstration of biological activity, the later incorporating appropriate immune and molecular markers.
This early phase of clinical investigation may include as minimum as 20 patients in a homogenous, sensitive and well-defined population and should be performed in an adjuvant setting, or one without rapidly progressive disease, to allow vaccines adequate time to induce biological activity and elicit immunological response. Evidence of biological activity defined as effect of the vaccine on clinical, molecular or immune response markers should be demonstrated to provide a sound rationale for commencing subsequent stages of development. According to Hoos et al.Citation75,Citation76 successful proof-of-principle trials support a more flexible, expeditious and focused clinical developmental process with early and informed decision making. Core recommendations on use of most sensitive, sufficiently lengthy and homogenous clinical settings are reflected in FDA guidance and have been indirectly confirmed by recent regulatory successes of ipilimumab and sipuleucel-T. As opposed to CVCTWG recommendations, it is now apparent that an early POC study should be conducted in a controlled fashion allowing for more accurate judgment on the magnitude and trajectory of biological response. Numerous cancer vaccines failed in phase III studies were upstaged on the basis on erroneous evaluations of uncontrolled phase II studies.
The identification and validation of appropriate immunological biomarker is crucial in development of cancer vaccines. Typical roadmap for implementing immune-response evaluation as a biomarker has been outlined by Ogino et al.Citation77 and consists of steps illustrated in .
Figure 4. Roadmap for the development of immune-response related biomarker in cancer vaccine program.
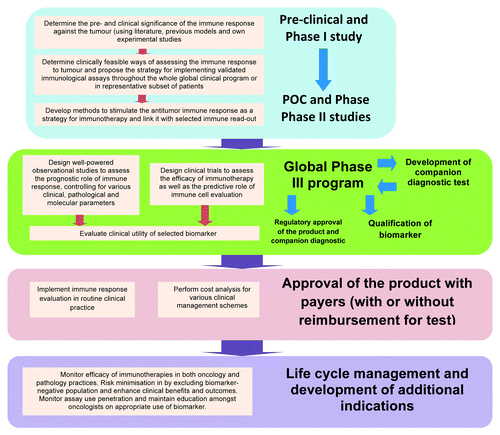
Apart from clinical validation of the immune-response related biomarker via randomized controlled and non-randomized observational studies, an early (pre-phase III) partnership with diagnostic company is required to initiate development and implementation of companion diagnostic test throughout phase III. Developers should consider cost-effectiveness, clinical utility and clinical feasibility of biomarker testing. If the test is very cumbersome (requires unique set of technical skills), prohibitive in terms of cost to oncology health care and difficult to commercialize, it is likely that reimbursement bodies will displace these costs on developers or conclude on lack of cost-effectiveness. Poor technical feasibility of biomarker testing will most certainly reduce sales penetration and will require additional investment in training of health care practitioners and laboratory staff. Finally, it was reported that for some previously approved biomarker tests, such as KRAS testing, which is mandatory for selection of patients with metastatic colon cancer in terms of eligibility for treatment with anti-EGFR monoclonal antibodies cetuximab and panitumomab, technical feasibility and inconsistent implementation of KRAS test procedures across EU might have posed risks for inappropriate and off-label use of products.Citation78 These findings prompted EU experts to develop EU Quality Assurance program and issue pathology guidelines on optimal methodologies for KRAS testing in the EU.
During phase III planning, an adequate and pre-specified definition of progression and response are vital. Since cancer vaccines can cause tumor swelling, infiltration and tumor fibrosis, some patient’s tumors may progress, increase their volume and give rise to new lesions (some possibly due to initial inflammation), which may be followed by a delayed response. As a consequence, PFS may not be a suitable clinical endpoint in cancer vaccine studies.Citation75,Citation79 A typical example includes studies with PSA-TRICOM vaccine (prostate-specific antigen plus a TRIad of COstimulatory Molecules; PROSTVAC) consisting of a priming vaccination with recombinant vaccinia- (rV-)PSA-TRICOM and booster vaccinations with recombinant fowlpox-(rF-) PSA-TRICOM. The efficacy of PSA-TRICOM has been evaluated in 2 phase II clinical trials in patients with metastatic hormone-refractory prostate cancer. In the first multicenter clinical trial, 122 patients with Gleason scores of ≤ 7 were randomized 2:1 to receive PSA-TRICOM plus GM-CSF (n = 82) vs. an empty-vector placebo (n = 40). A vaccinia-based vector was used as prime, followed by 6 boosts with a fowlpox-based vector. Vaccinated patients had a greater 3-y OS compared with the placebo arm (30% vs. 17%, respectively) and an improvement in median OS of 8.5 mo (24.5 mo vs. 16 mo, respectively; p = 0.016). However there were no differences in PFS between arms.Citation80
It is therefore recommended that the definition of progression should account for clinically relevant parameters such as patient’s performance status and/or global quality of life and not for volumetric or tumor-measurable criteria. However disease measurement should not be abandoned as long-term volumetric disease stabilization also should be considered as a measure of clinical activity.Citation75,Citation81 Continued treatment beyond progression under certain conditions can be summarized in blinded safety listings and monitored by independent data safety monitoring board (DSMB). The sponsor may need to plan regular DSMB safety reviews on e.g., 3–6-monthly basis within the timeframe of expected delayed immune response (12–24 mo from the onset of vaccination). Studies with cancer immunotherapeutics can be irreversibly compromised by various confounding factors arising due to use of rescue medications, concomitant therapies and crossovers as a consequence of reported disease progression. Crossovers and concomitant therapies were previously reported to abolish any clinically and statistically significant differences in overall survival for several anticancer agents: bortezomib for multiple myeloma, lapatinib for advanced or metastatic breast cancer and bevacizumab, sorafenib, sunitinib and temsirolimus for renal cell carcinoma.Citation82,Citation83 Nevertheless, the advantage in PFS gain was still preserved but was not always sufficient from regulatory perspective. Crossovers can also contribute to reduced separation in OS. In the pivotal registration IMPACT trial with sipuleucel-T, control subjects who demonstrated objective disease progression were offered three infusions of an autologous cellular therapy produced from cells frozen at the time of control product generation. An exploratory analysis was performed to estimate how autologous therapy impacted the OS benefit of sipuleucel-T. It was found that post-progression treatment with sipuleucel-T may have extended the survival of control subjects in the IMPACT study. Adjusting for use of autologous rescue therapy resulted in an increase in median OS benefit with sipuleucel-T from 4.1 mo to 7.8 mo.Citation84 These results suggest that crossover reduced the prominence of Sipuleucel T-driven favorable effect on OS.
In view of these findings, it was proposed that regulatory and HTA bodies should hold discussions with companies to increase joint understanding as to those instances in which PFS is a good predictor of OS and those in which it is not.Citation83 Advice from regulatory agencies and NICE on these matters could be reviewed with companies through the advisory/consultancy process that has been established. However, as reliance upon these data are common among agencies across the developed world, a coordinated and international approach may be the best way to tackle these issues.
The risk of various confounding factors and impact of crossovers is especially high in oncology studies of long duration and in tumors of relatively favorable prognosis (e.g., renal, ovarian, thyroid, bladder, etc.). Considering that cancer vaccines are unlikely to yield significant improvements in PFS, it is especially critical that the risk of crossovers and imbalanced use of concomitant therapies is minimised as much as possible through adequate protocol planning. From ethical perspective it is important that patients from placebo group will have access to the treatment which seems to show an improvement in clinical outcomes. However due to delayed effect of immunotherapeutics on PFS and long duration of studies required to demonstrate OS gains, the magnitude of the clinical benefit might not be so apparent. Therefore a careful consideration should be given to the crossover eligibility. The impact of any potential imbalances on OS/PFS evaluation should be elucidated using pre-specified statistical analyses and modeling. Investigators should be adequately trained how to differentiate temporary worsening in disease vs. steady progression related deterioration. Sponsors should pre-define consistent characteristics constituting clinically relevant progression based on features agreed by patient and/or caregiver and treating physician. Whenever possible and feasible, a verification of underlying delayed immune response can be supported using tumor biopsy specimens and biomarker assays performed using peripheral blood samples. Correlation between immune assays and PFS/OS serve as one of the pivotal gate rules at the transition between phase II and III.
For example, Stimuvax (BLP25) which is currently studied in unresectable stage III NSCLC patients by Merck Serono-Oncothyreon, has shown a favorable albeit not statistically significant effect on OS only in patients with stage IIIB locoregional NSCLC, for whom the median survival time for the L-BLP25 arm has not yet been reached at the time of reporting results compared with 13.3 mo for the control arm.Citation85 Favorable effect on OS was specifically confined to patients with Stage III NSCLC who represent a target group for ongoing phase III development by Merck Serono and Oncothyreon. However the evaluation of immune responses in different subgroups was not reported and remain unclear whether such an effect might be explained by features of immune response in less advanced locoregional disease or it might be influenced by stage-specific differences in clinical management, e.g., use of chemotherapy and radiotherapy interventions.
As described in FDA guidance, immunotherapy may induce novel patterns of antitumor response not captured by Response Evaluation Criteria in Solid Tumors (RECIST) or World Health Organization (WHO) criteria. Clinical protocols for investigation of cancer immunotherapies may utilize pre-specified adjusted response criteria for endpoints such as response rate or delayed PFS, which more comprehensively capture all response patterns.Citation75 Altered statistical models describing PFS hazard ratios as a function of time and recognizing differences before and after separation of curves may be prospectively employed for designing phase III trials but should be mutually agreed between the sponsor and regulatory authorities.
Increasingly, developers plan a global regulatory strategy in order to achieve product positioning in multiple regions. However the feedback from different agencies can considerably vary complicating the implementation of “blended” or harmonized development and regulatory strategy. It was reported that due to variation in oncology standards of care across EU and different approach to evaluation of products, national competent authorities from individual European countries have previously provided with very contrasting recommendations with regards to clinical development program, namely study design and comparator use (communication at DIA-2011). Since cancer vaccines qualify for a mandatory centralised route of submission via EMA, it is recommended to approach for EMA Scientific Advice as early as possible in the clinical development in order to obtain more harmonized and consistent advice. Subsequent to phase III results reporting, it is sensible to approach several competent authorities (especially key players at CHMP such as Germany, UK, Netherlands, France, Sweden, and Spain) and seek their interest in acting as Rapporteur or Co-rapporteur for the proposed EMA submission. Any significant differences in advice given by FDA and EMA should be mitigated via open dialog and negotiation with authorities.
An analysis in 2010 indicated that for oncology drugs, the success rate from 1993 to 2004 was 20%, but the risk that these drugs may not gain marketing approval or were unsuccessful in phase III trials was higher for oncology drugs relative to other therapeutic areas (55% success rate in phase III trials, compared with an overall success rate of 64%).Citation86 It is thought that there might be interplay of different causes for high attrition rate in late-stage oncology clinical trials during last decade. Apart from dose-finding choices and regulatory challenges, various suboptimal strategic and operational decisions might explain it, e.g., lack of translation between early ORR effects and immunological read-outs and OS, cost-driven planning of the study power, suboptimal quality of results due to study execution in new emerging territories etc. For instance, an underpowered study for cancer vaccine may not only lead to failure in achieving mature data sets with sufficient separation in OS but also could compromise favorable trends in immunologically stratified patient subpopulations. In addition, some unexpected factors may risk an entire trial. For example, an unusually high rate of placebo response was reported in some Eastern European countries due to overly close relationships between patients and caregivers with study monitors and principal investigators resulting in inadvertent unmasking of given treatment.Citation87 Traditionally, some nations such as Russian or Chinese regard medical practitioner as highly competent and prestigious profession. For that reason, the investigator-subject relationships are usually built on trust and mutual understanding. These factors in combination with low migration rates can considerably enhance patient recruitment and retention but needs to be carefully managed with regards to treatment concealment. It is important to remember that administration of some cancer vaccines might be accompanied with local hypersensitivity or infusion reactions which may unmask the treatment throughout lengthy clinical study. Therefore apart from double blind, various operational techniques can assist in maintaining the integrity of the study: rotation of observers, concealment of injection sites from principal investigators, separation of observers dealing with systemic assessment of patient status and collection of questionnaires from the staff involved in product administration and safety monitoring.
Specifically for global multi-country and multi-regional oncology trials, the mean anticipated PFS/OS in control group might be significantly shorter in a subset of patients from developing countries as opposed to patients from US and EU due to often suboptimal and underdeveloped standards of current oncology care in some regions such as Asia. This issue might be particularly important in a single pivotal registration study with significant share of patient population from Asian region. Although statistically significant gain in OS/PFS in such a study might look inadequate in comparison to expected gains with other approved therapies or expected survival time in countries with advanced oncology care. For example, in Sorefenib trials in patients with hepatocellular carcinoma, gain in median OS was from 7.9 mo to 10.7 mo and from 4.2 mo to 6.5 mo in US/EU and Asian studies, respectively.Citation88 If these trials would have been combined into a single study, the appraisal of OS gain and consequential cost-effectiveness benefit would have been particularly challenging. In similar situations more than one pivotal study might be required for global registration purposes.
In summary, a host of commercial, manufacturing, regulatory, clinical, laboratory and operational issues need to be addressed during a typical clinical development path for a novel cancer vaccine product demanding careful and continuous risk evaluation and considerable expertise from developers.
Note
The views expressed in this article are the personal views of the author and may not be understood, interpreted, or quoted as being made on behalf of, or reflecting the position of any other companies, agencies or parties cited in this article.
References
- Copier J, Dalgleish A. Whole-cell vaccines: A failure or a success waiting to happen?. Curr Opin Mol Ther 2010; 12:14 - 20; PMID: 20140812
- Copier J, Dalgleish AG, Britten CM, Finke LH, Gaudernack G, Gnjatic S, et al. Improving the efficacy of cancer immunotherapy. Eur J Cancer 2009; 45:1424 - 31; http://dx.doi.org/10.1016/j.ejca.2008.12.017; PMID: 19167214
- Copier J, Ward S, Dalgleish A. Cell based cancer vaccines: regulatory and commercial development. Vaccine 2007; 25:Suppl 2 B35 - 46; http://dx.doi.org/10.1016/j.vaccine.2007.06.041; PMID: 17916462
- Dalgleish AG. Therapeutic cancer vaccines: why so few randomised phase III studies reflect the initial optimism of phase II studies. Vaccine 2011; 29:8501 - 5; http://dx.doi.org/10.1016/j.vaccine.2011.09.012; PMID: 21933695
- Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011; 480:480 - 9; http://dx.doi.org/10.1038/nature10673; PMID: 22193102
- Vergati M, Intrivici C, Huen NY, Schlom J, Tsang KY. Strategies for Cancer Vaccine Development. J Biomed Biotechnol 2010: 13 p.
- Datamonitor. Dendreon Corporation PharmaVitae Emerging Biopharma Company Analysis, 2012. www.datamonitor.com
- FDA. FDA Prescribing Information for Provenge, 2010. Available at: http://www.fda.gov/downloads/BiologicsBloodVaccines/CellularGeneTherapyProducts/Approved-Products/UCM210031.pdf. Accessed: April 12, 2012.
- Sims RB. Development of sipuleucel-T: Autologous cellular immunotherapy for the treatment of metastatic castrate resistant prostate cancer. Vaccine 2011; Available online 25 November 2011 at http://www.sciencedirect.com/science/article/pii/S0264410X11018548. Accessed on April 11, 2012.
- Sonpavde G, Di Lorenzo G, Higano CS, Kantoff PW, Madan R, Shore ND. The role of sipuleucel-T in therapy for castration-resistant prostate cancer: a critical analysis of the literature. Eur Urol 2012; 61:639 - 47; http://dx.doi.org/10.1016/j.eururo.2011.10.027; PMID: 22036643
- Datamonitor. Pipeline Insight: Therapeutic Cancer Vaccines, 2009. www.datamonitor.com
- Neller MA, López JA, Schmidt CW. Antigens for cancer immunotherapy. Semin Immunol 2008; 20:286 - 95; http://dx.doi.org/10.1016/j.smim.2008.09.006; PMID: 18951039
- Snook AE, Eisenlohr LC, Rothstein JL, Waldman SA. Cancer mucosa antigens as a novel immunotherapeutic class of tumor-associated antigen. Clin Pharmacol Ther 2007; 82:734 - 9; http://dx.doi.org/10.1038/sj.clpt.6100369; PMID: 17898707
- Alpizar YA, Chain B, Collins MK, Greenwood J, Katz D, Stauss HJ, et al. Ten years of progress in vaccination against cancer: the need to counteract cancer evasion by dual targeting in future therapies. Cancer Immunol Immunother 2011; 60:1127 - 35; http://dx.doi.org/10.1007/s00262-011-0985-7; PMID: 21479639
- Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Res 2012; 22:457 - 72; http://dx.doi.org/10.1038/cr.2012.13; PMID: 22357481
- Nishizawa S, Hirohashi Y, Torigoe T, et al. A novel tumor-associated antigen, DNAJB8 is highly expressed in cancer stem-like cells / cancer initiating cells of renal cell carcinoma, and is potent target for immunotherapy. J Urol 2012; •••:187
- McMillin DW, Delmore J, Weisberg E, Negri JM, Geer DC, Klippel S, et al. Tumor cell-specific bioluminescence platform to identify stroma-induced changes to anticancer drug activity. Nat Med 2010; 16:483 - 9; http://dx.doi.org/10.1038/nm.2112; PMID: 20228816
- Haabeth OA, Lorvik KB, Hammarström C, Donaldson IM, Haraldsen G, Bogen B, et al. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat Commun 2011; 2:240 - 52; http://dx.doi.org/10.1038/ncomms1239; PMID: 21407206
- Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 2009; 30:1073 - 81; http://dx.doi.org/10.1093/carcin/bgp127; PMID: 19468060
- Santos N, Wenger JB, Havre P, Liu Y, Dagan R, Imanirad I, et al. Combination therapy for renal cell cancer: what are possible options?. Oncology 2011; 81:220 - 9; http://dx.doi.org/10.1159/000333470; PMID: 22085914
- Datamonitor. Therapy area series: Product Profiles. Prostate Cancer 2011. www.datamonitor.com
- National Comprehensive Cancer Network. Guideline of treatment of prostate cancer, 2011; Available from: http://www.nccn.org/professionals/physician_ gls/pdf/prostate.pdf Accessed on April 13, 2012.
- Solomon B, Bunn PA Jr.. Adjuvant chemotherapy for non-small cell lung cancer. Cancer Invest 2007; 25:217 - 25; http://dx.doi.org/10.1080/07357900701206281; PMID: 17612931
- Pfisterer JS, Berek A, Casado B, Cwiertka K, Pinter T, Pluzanska A, et al. Randomized double-blind placebo-controlled international trial of abagovomab maintenance therapy in patients with advanced ovarian cancer after complete response to first-line chemotherapy: The Monoclonal Antibody Immunotherapy for Malignancies of the Ovary by Subcutaneous Abagovomab (MIMOSA) trial. J Clin Oncol 2011; 29
- Schmidt C. Ovarian cancer treatments on the horizon. J Natl Cancer Inst 2011; 103:1284 - 5; http://dx.doi.org/10.1093/jnci/djr343; PMID: 21852261
- Yervoy FDA. (ipilimumab): Risk Evaluation and Mitigation Strategy (REMS) - Severe Immune-Mediated Adverse Reactions, 2011; Available from: http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm249770.htm Accessed on April 13, 2012.
- Datamonitor. Pipeline Insight: Small Molecule Targeted Cancer Therapies, 2010. www.datamonitor.com
- FDA. Guidance for Industry: Co-development of Two or More Unmarketed Investigational Drugs for Use in Combination, 2010; Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM236669.pdf
- Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol 2012; 90:85 - 94; http://dx.doi.org/10.1038/icb.2011.100; PMID: 22124371
- Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer 2012; 12:237 - 51; http://dx.doi.org/10.1038/nrc3237; PMID: 22437869
- Zitvogel L, Kepp O, Kroemer G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol 2011; 8:151 - 60; http://dx.doi.org/10.1038/nrclinonc.2010.223; PMID: 21364688
- Alley SC, Okeley NM, Senter PD. Antibody-drug conjugates: targeted drug delivery for cancer. Curr Opin Chem Biol 2010; 14:529 - 37; http://dx.doi.org/10.1016/j.cbpa.2010.06.170; PMID: 20643572
- Datamonitor. PharmaVitae Emerging Technologies Series: New Antibody-Based Technologies, 2011. www.datamonitor.com
- Roche. Roche announces positive results of pivotal Phase III study with pertuzumab in HER2-positive metastatic breast cancer, 2011; Available from: http://www.roche.com/media/media_releases/med-cor-2011-12-08.htm Accessed on April 14, 2012.
- Zelboraf EMA. 2012; Available from: http://www.ema.europa.eu/ ema/index.jsp?curl=pages/medicines/human/medicines/002409/human_med_001544.jsp&mid=WC0b01ac058001d124&murl=menus/medicines/medicines.jsp Accessed on April 14, 2012.
- FDA. FDA approves Zelboraf and companion diagnostic test for late-stage skin cancer, 2011; Available from: http://www.fda.gov/NewsEvents/ Newsroom/PressAnnouncements/ucm268241.htm Accessed on April 13, 2012.
- Clinicaltrials.gov search, 2012; Available from: http://www.clinicaltrials.gov Accessed on 11, April 2012.
- Goldman B, DeFrancesco L. The cancer vaccine roller coaster. Nat Biotechnol 2009; 27:129 - 39; http://dx.doi.org/10.1038/nbt0209-129; PMID: 19204689
- EMA. Scientific advice and protocol assistance, 2012; Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Other/2012/03/WC500124212.pdf Accessed on April 13, 2012.
- Ark Therapeutics. Cerepro (sitimagene ceradenovec) – treatment for brain cancer (malignant glioma). Ark Therapeutics, 2009; Available from: http://www.arktherapeutics.com/main/products.php?content=products_cerepro Accessed on April 10, 2012.
- Cerepro EMA. 2007; Available from: http://www.ema.europa.eu/ema/index.jsp?curl=/pages/medicines/landing/wapp_search.jsp&mid=WC0b01ac058001d128&source=homeMedSearch&category=human&keyword=cerepro&isNewQuery=true. Accessed on April 14, 2012.
- Advexin EMA. 2008; Available from: http://www.ema.europa.eu/ema/ index.jsp?curl=pages/medicines/human/medicines/000919/wapp/Initial_authorisation/human_wapp_000042.jsp&mid=WC0b01ac058001d128&source=homeMedSearch&category=human Accessed on April 14, 2012.
- EMA. EMA/CHMP/775076/2009: Withdrawal assessment report for Oncophage. 2009; Available from: http://www.ema.europa.eu/docs/en_GB/ document_library/Application_withdrawal_assessment_report/2010/03/WC500075459.pdf Accessed on April 13, 2012.
- Hanaizi Z, van Zwieten-Boot B, Calvo G, Lopez AS, van Dartel M, Camarero J, et al. The European Medicines Agency review of ipilimumab (Yervoy) for the treatment of advanced (unresectable or metastatic) melanoma in adults who have received prior therapy: summary of the scientific assessment of the Committee for Medicinal Products for Human Use. Eur J Cancer 2012; 48:237 - 42; http://dx.doi.org/10.1016/j.ejca.2011.09.018; PMID: 22030452
- Bakacs T, Mehrishi JN, Szabó M, Moss RW. Interesting possibilities to improve the safety and efficacy of ipilimumab (Yervoy). Pharmacological Research. 2012; Available from: http://www.sciencedirect.com/science/article/pii/S1043661812000643 Accessed on April 13, 2012.
- EMA. CHMP/EWP/433478/2010: Need to revise the guideline on the evaluation of anticancer medicinal products in man. 2010; Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000406.jsp&mid=WC0b01ac0580034cf3 Accessed on April 14, 2012.
- EMA. EMEA/CHMP/BWP/271475/2006: Guideline on potency testing of cell based immunotherapy medicinal products for the treatment of cancer. 2008; Available from www.ema.europa.eu Accessed on April 12, 2012.
- EMA. CHMP/410869/06: Human cell-based medicinal products. 2008; Available from: http://www.ema.europa.eu/ema/index.jsp?curl =pages/regulation/general/general_content_000405.jsp&mid=WC0b01ac058002958a Accessed on April 14, 2012.
- EMA. EMA/CAT/CPWP/686637/2011: Committee for Advanced Therapies (CAT) Draft guideline on the risk-based approach according to Annex I, part IV of Directive 2001/83/EC applied to Advanced Therapy Medicinal Products. 2011; Available from www.ema.europa.eu Accessed on April 12, 2012.
- EMA. EMEA/CHMP/GTWP/587488/2007 Rev. 1: Committee for the Medicinal Products for Human Use (CHMP) Reflection paper on quality, non-clinical and clinical issues related to the development of recombinant adeno-associated viral vectors. 2007; Available from www.ema.europa.eu Accessed on April 12, 2012.
- EMA. EMEA/CHMP/VEG/134716/2004: Adjuvants in vaccines for human use. 2005; Available from: http://www.ema.europa.eu/ema/index.jsp?curl =pages/regulation/general/general_content_000407.jsp&mid=WC0b01ac 058002958b&jsenabled=true Accessed on April 14, 2012.
- EMA. EMEA/CHMP/VWP/244894/2006: Explanatory note on immunomodulators for the guideline on adjuvants in vaccines for human use. 2006; Available from: http://www.ema.europa.eu/ema/index.jsp?curl =pages/regulation/general/general_content_000407.jsp&mid=WC0b01ac 058002958b&jsenabled=true Accessed on April 14, 2012.
- EMA. EMEA/CHMP/VWP/164653/05: Clinical evaluation of new vaccines. 2007; Available from: http://www.ema.europa.eu/ema/index.jsp?curl =pages/regulation/general/general_content_000407.jsp&mid=WC0b01ac 058002958b&jsenabled=true Accessed on April 14, 2012.
- Oh S. Cell-Based Medicinal Products for Global Market: FDA Perspectives CAT-ESGCT Workshop ESGCT Annual Meeting, Brighton, UK. 2011; Available from: http://www.ema.europa.eu/docs/en_GB/document_library/ Presentation/2012/02/WC500122168.pdf Accessed on April 3, 2012.
- Witten C. FDA and Cancer Vaccine Development. 2008; Available from: http://www.fda.gov/downloads/biologicsbloodvaccines/newsevents/ workshopsmeetingsconferences/ucm102978.pdf Accessed on April 11, 2012.
- FDA. Code of Federal Regulations. Title 21 Food and Drugs. Part 314.126. 2008; Available from: http://edocket.access.gpo.gov/cfr_2008/ aprqtr/pdf/21cfr314.126.pdf Accessed on April 3, 2012.
- FDA. Guidance for industry: clinical trial endpoints for the approval of cancer drugs and biologics. 2007; Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM071590.pdf Accessed on April 3, 2012.
- Chabner BA. Early accelerated approval for highly targeted cancer drugs. N Engl J Med 2011; 364:1087 - 9; http://dx.doi.org/10.1056/NEJMp1100548; PMID: 21428763
- Coutant D, Riggs D, Hoffman EV. Substantial Evidence: When Is a Single Trial Sufficient for Approval and Promotion?. Drug Inf J 2011; 45:253 - 63; http://dx.doi.org/10.1177/009286151104500306
- Biovest. Biovest. 2012; Available from: www.biovest.com Accessed on April 13, 2012.
- Gordon EM, Hall FL. Noteworthy clinical case studies in cancer gene therapy: tumor-targeted Rexin-G advances as an efficacious anti-cancer agent. Int J Oncol 2010; 36:1341 - 53; http://dx.doi.org/10.3892/ijo_00000619; PMID: 20428757
- FDA. Guidance for Industry: Clinical Considerations for Therapeutic Cancer Vaccines. 2011; Available from: http://www.fda.gov/downloads/ biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/vaccines/ucm278673.pdf Accessed on April 11, 2012.
- Egawa K. Immuno-cell therapy of cancer in Japan. Anticancer Res 2004; 24:5C 3321 - 6; PMID: 15515427
- Heike Y. Cellular therapies in Japan. 2010; Available from: http://www.google.co.uk/#hl=en&site=&source=hp&q=heike+and+cell+therapies+in+japan&oq=heike+and+cell+therapies+in+japan&aq=f&aqi=&aql=&gs_l=hp.3...1279l12964l0l13322l55l55l11l0l0l0l125l2622l41j2l43l0.frgbld.&bav=on.2,or.r_gc.r_pw.,cf.osb&fp=24a658667a23338a&biw=1344&bih=658. Accessed on April 11, 2012.
- Deloitte. Medical tourism: consumers in search of value. 2008; Available from: http://www.deloitte.com/assets/Dcom-unitedStates/Local%20Assets/Docume nts/ us_chs_MedicalTourismStudy(3).pdf Accessed on April 15, 2012.
- Maekawa T, Kimura S, Kasai Y. Development of Novel Advanced Cell and Gene Therapy and GMP-Controlled Cell Processing. JMAJ 2005; 48:81 - 4
- Narita M. Current status of Current Japanese Regulation and Development of Biologics. 2011; Available from: http://www.pmda.go.jp/english/past/2009bio_sympo/file/Keynote_Speech_Narita_(PMDA).pdf Accessed on April 13, 2012.
- Landsdell M. Innovative drug discovery in emerging markets. Business Insights, 2011.
- Hong S. Korean perspective on biologics regulation. 2008; Available from: http://www.pmda.go.jp/english/past/pdf/06.Dr.Hong.pdf Accessed on April 13, 2012.
- KFDA. Recent trends and Korea's cases in the development of gene therapy products KFDA. 2008; Available from: http://www.bioin.or.kr/upload.do?cmd=download&seq=7676&bid=industry. Accessed on April 5, 2012.
- Cancer therapy China. Gene Therapy (Gendicine). 2009; Available from: http://www.cancertherapychina.com/index.php?option=com_content&view=article&id=84&Itemid=23 Accessed on April 10, 2012.
- Datamonitor. Stakeholder Opinions: Gene Therapy. Gene therapy to deliver after 20 years of clinical development, 2009. www.datamonitor.com
- Datamonitor. Oncology Market Access in China Market bolstered by rising incidence and improved regulatory environment, 2011. www.datamonitor.com
- Schneider CK, Schäffner-Dallmann G. Typical pitfalls in applications for marketing authorization of biotechnological products in Europe. Nat Rev Drug Discov 2008; 7:893 - 9; http://dx.doi.org/10.1038/nrd2728; PMID: 18974748
- Hoos A, Eggermont AM, Janetzki S, Hodi FS, Ibrahim R, Anderson A, et al. Improved endpoints for cancer immunotherapy trials. J Natl Cancer Inst 2010; 102:1388 - 97; http://dx.doi.org/10.1093/jnci/djq310; PMID: 20826737
- Hoos A, Parmiani G, Hege K, Sznol M, Loibner H, Eggermont A, et al, Cancer Vaccine Clinical Trial Working Group. A clinical development paradigm for cancer vaccines and related biologics. J Immunother 2007; 30:1 - 15; http://dx.doi.org/10.1097/01.cji.0000211341.88835.ae; PMID: 17198079
- Ogino S, Galon J, Fuchs CS, Dranoff G. Cancer immunology—analysis of host and tumor factors for personalized medicine Nature Rev. Clin Oncol 2011; 8:711 - 9
- van Krieken JH, Jung A, Kirchner T, Carneiro F, Seruca R, Bosman FT, et al. KRAS mutation testing for predicting response to anti-EGFR therapy for colorectal carcinoma: proposal for an European quality assurance program. Virchows Arch 2008; 453:417 - 31; http://dx.doi.org/10.1007/s00428-008-0665-y; PMID: 18802721
- Di Lorenzo G, Buonerba C. Kidney cancer: Overall Survival is an unsuitable primary clinical endpoint. Nature Urology 2010; 7:367 - 8; http://dx.doi.org/10.1038/nrurol.2010.84
- Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al, IMPACT Study Investigators. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010; 363:411 - 22; http://dx.doi.org/10.1056/NEJMoa1001294; PMID: 20818862
- Wolchok JD, Hoos A, O’Day S, Weber JS, Hamid O, Lebbé C, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res 2009; 15:7412 - 20; http://dx.doi.org/10.1158/1078-0432.CCR-09-1624; PMID: 19934295
- NICE. Renal cell carcinoma - bevacizumab, sorafenib, sunitinib and temsirolimus: appraisal consultation document. 2008; Available at: http://www.nice.org.uk/guidance/index.jsp?action=article&o=41473 Accessed on April 13, 2012.
- Garau M, Shah K, Towse A, Wang Q. Assessment and appraisal of oncology medicines: does NICE’s approach include all relevant elements? What can be learnt from international HTA experiences? Report for the Pharmaceutical Oncology Initiative Group (POI). 2009; Available from: http://www.ohe.org/publications/article/assessment-and-appraisal-of-oncology-medicines-33.cfm Accessed on April 13, 2012.
- Gomella L, Nabhan C, DeVries T, Whitmore J, Frohlich M. Estimating the overall survival benefit of sipuleucel-T in the IMPACT trial accounting for crossover treatment in control subjects with autologous immunotherapy generated from cryopreserved cell. J Urol 2012; 187:E278 - 9; http://dx.doi.org/10.1016/j.juro.2012.02.765
- Butts C, Murray N, Maksymiuk A, Goss G, Marshall E, Soulières D, et al. Randomized phase IIB trial of BLP25 liposome vaccine in stage IIIB and IV non-small-cell lung cancer. J Clin Oncol 2005; 23:6674 - 81; http://dx.doi.org/10.1200/JCO.2005.13.011; PMID: 16170175
- DiMasi JA, Feldman L, Seckler A, Wilson A. Trends in risks associated with new drug development: success rates for investigational drugs. Clin Pharmacol Ther 2010; 87:272 - 7; http://dx.doi.org/10.1038/clpt.2009.295; PMID: 20130567
- Reinisch W, Sandborn WJ, Hommes DW, D’Haens G, Hanauer S, Schreiber S, et al. Adalimumab for induction of clinical remission in moderately to severely active ulcerative colitis: results of a randomised controlled trial. Gut 2011; 60:780 - 7; http://dx.doi.org/10.1136/gut.2010.221127; PMID: 21209123
- Nexavar EMA. 2012; Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000690/human_med_000929.jsp&murl=menus/medicines/medicines.jsp&mid=WC0b01ac058001d124&jsenabled=true Accessed on April 15, 2012.