Abstract
Today the task of cancer vaccine developers is not only to excel in cancer immunology and art of conducting immunotherapy trials but also in the analysis and forecasting the cost-effectiveness of the final product. This article reviews methodology used by EU health-technology bodies in the appraisal of new therapies based on economic and clinical values and different budgetary uncertainties. Increasingly, new oncology treatments were able to access EU market only under provision of risk-sharing agreements with payers and examples of such agreements are given here. Cancer vaccine developers should consider early collection of patient reported outcomes in order to project additional clinical and economic value with immunotherapy. Furthermore, early interaction with different stakeholders including patient organizations, physicians and payer bodies can facilitate market access.
Cost of Oncology Healthcare
According to the World Health Organization, the global burden of cancer is expected to grow from 10 million new cases in 2000 to 15 million in 2015. Based on current projections, cancer deaths will continue to rise, with nine million people estimated to die from the disease in 2015, and more than 11 million in 2030. Globally, cancer is the leading cause of death of those under 85, and because it disproportionately affects the elderly, oncology expenditure will become an even greater concern in future as a result of the aging population. In addition, some forms of cancer are now manifesting as “chronic disease” due to early diagnosis, improved sequential treatments and clinical outcomes. Therefore the cost burden is deemed to increase proportionally with increase of therapies applied sequentially due to premium pricing for medicines used in second- and third lines of treatments.
The National Institutes of Health (NIH) estimates that the overall costs of cancer in 2007 were $226.8 billion: $103.8 billion for direct medical costs (total of all health expenditures) and $123.0 billion for indirect mortality costs (cost of lost productivity due to premature death).Citation1 Although the sales of both conventional and targeted oncology drug therapies are expanding, the majority of sales growth is attributed to an increasing uptake of targeted cancer therapies. The 2008 market for targeted oncology therapies was approximately $25 billion with US sales in excess of $10 billion—an almost 2-fold increase since 2005. An unprecedented growth in sales of oncology drugs stimulated by historically low regulatory and reimbursement barriers for entry onto the market resulted in significant increase of oncology share in the total pharma market. In 2000, oncology drugs accounted for approximately 3.5% of global pharmaceutical market sales. By 2006, this figure had increased to over 5.5% and it in 2011 it reached 7%. This trend is directly linked to the significant and rapid penetration of certain targeted therapies such as Rituxan, Herceptin, Gleevec, Avastin, Erbitux, Sutent and Nexavar etc. It is also a result of the high prices enjoyed by these drugs and their analogs.Citation2 Generally, price is not directly linked to the incidence of a particular cancer, meaning that a rare cancer does not automatically warrant an expensive drug. However, a clearer relationship between price and incidence can be seen for biologicals compared with small molecules. Recently approved targeted drugs with new mechanisms of actions and/or biologicals have benefited from high prices, with annual treatment costs of $20,000–40,000 becoming increasingly common.Citation3 Provenge has been approved in the USCitation4 with a price tag of $93,000 per a course of treatment. With the growing number of patients receiving such therapies, expanding competitive landscape and the high profitability of these drugs (gross margins for biological are often 80–90% of sales), healthcare systems will not be able to absorb these vast costs in a sustainable fashion. With cost containment policies being pursued in most industrialised countries, cancer drugs are expected to come under increasing pricing pressure in the near future.
Several factors will contribute to the growing pressure on cancer drug prices in particular:
• Rapid growth of cancer treatment spending as part of a total and oncology-related healthcare expenditure;
• The expected increase in combinations of costly drugs. If such combinations are approved, the higher prices will clearly become unsustainable for healthcare systems;
• Potentially disproportionate distribution of healthcare resources due to targeted therapies: biomarker positive patient population might represent a minor share of the market in some cases yet requiring a large proportion of resources;
• Growth of maintenance treatments for cancer (similar to long-term cancer immunotherapeutic regimens). Some forms of cancer will masquerade as chronic disease;
• Scarcity of economic resources and healthcare budgets;
• The increasing number of products resulting from competition among a greater number of players;
• Emergence of cost-effective discounted generic copies and biosimilar versions of biologics.
Payers: Changing Dynamics
It is not uncommon for small or mid-size biotechnology companies with products in early stages of development to defer the pricing and reimbursement issues until product launch or even delegate the responsibility to a marketing partner. However the value of a sound pricing strategy can translate directly into commercial strategy, company's royalty fee or acquisition price and ultimately into profitability. Failure to account for reimbursement barriers can drive companies back to the bargaining table or even worse, out of business entirely. Cancer vaccine developers should be especially vigilant in evaluating pricing environment for oncology agents throughout the globe and make appropriate adjustment to their pricing plans in order to avoid developing efficacious and safe yet non-reimbursable and non-profitable products.
The most comprehensive way to evaluate cost-effectiveness, clinical and economic values associated with new products is through health technology assessments (HTA), as conducted by UK organizations such as NICE and IQWiG. Most European countries (and indeed global healthcare systems) are influenced by the decisions made by NICE and IQWiG. This is because the basic principles of an HTA apply to all stakeholders, regardless of their specific methodologies. For example, payers in Spain will carefully study NICE's verdict on a given therapy (within a given patient population) via an STA (single technology appraisal) or MTA (multiple technology appraisal).Citation5 Therefore there is an element of HTA “eco-system” which is beneficial in terms of avoiding duplication of efforts/resources for industry, HTA bodies and payers. However there are still numerous issues associated with cross-country collaborations in making HTA decisions. Various HTA bodies are concerned only with the national level of implementation of their guidance while the approval for many oncology products is granted at pan-European level. Cross-HTA body collaboration is really needed to enhance the robustness of value driven principles in HTA assessment and holistic appraisal of values for all different EU stakeholders.
Although not all countries follow a formal HTA-based system, most of countries use the component concepts in varying degrees. Payers are responsible for ensuring prudent and principled use of scarce economic resources, whereas healthcare providers and patients legitimately want access to technologies from which they could benefit. Although all pricing and reimbursement systems employ the same concepts and tools, their manner of appraisal may differ considerably. The healthcare systems of UK, France, Germany, Italy and the US have been taken as examples to describe the variation in approaches and simultaneously convey the commonalities. The commonalities are evident given the common concerns faced by payers in all major markets. These include several types of risk: budget-based risk related to over-extending fixed budgets; population-based risk related to funding therapies for patients that will not benefit the majority; longitudinal risk: paying for treatment failures/multiple sub-optimal therapies over time.Citation5
There are fundamental differences in payer environment in the US and EU, which were summarized in . Some of these differences are linked to payers’ infrastructures and some others are related to institutional and healthcare budget features. The threshold for quality-adjusted value of life (QALY) employed by US payers is around $100,000 vs. the NICE-recommended £30,000. Therefore US oncology market is saturated with some costly oncology treatments which were seemingly not-cost effective in EU environment. With increased scarcity of economic and financial resources due to EU economic crisis, EU HTA evaluations on costly oncology treatments are expected to have a greater scrutiny and higher threshold for refusal than in years before.
Table 1. Differences in payer environments in US and EU.Citation5
In England, the National Institute for Health and Clinical Excellence (NICE) is responsible for conducting appraisals and developing guidelines for new technologies. In Scotland, Wales and Northern Ireland the key HTA bodies are: the Scottish Medicines Consortium (SMC), the All Wales Medicines Strategy Group (AWMSG) and the National Centre for Pharmacoeconomics (NCPE) respectively. All HTA bodies except NICE require the review of all new drugs which enter the market. Patients’ rights in relation to access to NICE-approved drugs are to be included in a new NHS constitution. NICE has faced considerable criticism for denying patients’ access to drugs in the NHS, especially some oncology drugs for orphan indications (e.g., sorafenib for hepatocellular and renal carcinoma; sunitinib for renal carcinoma etc.). To overcome this hurdle, companies have developed risk-sharing type schemes in order to gain a positive recommendation from NICE, or to secure funding for a drug at the local level, particularly in the case of high cost drugs.Citation5 More recent reforms of National Health Service (NHS) in the UK will result in decentralization of payment decisions aiming to shift some authority to downstream decision makers (such as general practitioners). NICE will increasingly take a mediating and expert advisory role in HTA assessment rather than ruling an entire process for value assessment. In the future, NICE may play a pivotal role in establishing risk-sharing schemes and negotiating between the NHS and the pharmaceutical industry over drug prices, capturing all values of healthcare innovation; refocusing attention on clinical best practice and quality standards. The UK government has confirmed that patients in England will benefit from a £200 min per year cancer drug fund to improve access to cancer drugs between April 2011 until March 2014. Via the fund, cancer patients will be able to apply to a panel of doctors in their area for funding for drugs that have not yet been approved by NICE or have been turned down because they are not deemed cost-effective. GP consortia will decide on how the government's new £200 min (€228 min) per year cancer drugs fund is spent. Cancer Fund might serve as an instrument for reimbursement for some promising cancer immunotherapeutics which have shown favorable efficacy data in phase II–III clinical studies.
In France, the reimbursement decision is driven by considerations of the seriousness of the disease or condition rather than by cost-effectiveness evaluation. The product is evaluated by an independent scientific committee, prior to price negotiations. This Transparency Committee, named after the European Transparency Directive, assesses the therapeutic value, or clinical benefits of a drug, and compares it with existing therapies. Clinical effectiveness and budget impact are used to determine prices, with cost-effectiveness having no current focus. Drugs are evaluated against two sets of complex criteria: their therapeutic value and added therapeutic value. The Transparency Committee assesses the product’s medical benefit (SMR) and improvement in medical benefit (ASMR) ratings compared with therapeutic equivalents for each of the drug’s indications. SMR rating is used to provide the basis for the reimbursement decision. The ASMR rating is the most important factor in determining the product’s price. The main problem in assessing this added therapeutic value is the time it takes, as well as the lack of proper clinical trials against alternative products on the market. Products seeking reimbursement must undergo assessment by the Transparency Commission before a price is set. In order to speed up the pricing and reimbursement process, manufacturers of drugs going through the centralized marketing authorization procedure have been permitted to submit a pre-application dossier to the CT and The Economic Committee for Health Products (CEPS) since 2004. The pre-application dossier may be submitted prior to the official granting of marketing authorization. Manufacturers must, however, still submit the standard application dossier as soon as marketing authorization has been granted. In next decade, French healthcare system will see increasing controls and regulation, chiefly due to the health insurance budget deficit, which remains of crucial importance. Regulation of biotechnology drug spending will be closely monitored and regulated by regional bodies such as OMEDIT, increasing regional influence in regulating hospital drug spending.Citation5
Historically, German IQWiG evaluated effectiveness evidence in relation to the nature and severity of the disease. The German healthcare has experienced significant deficits for public health insurers, which were expected to reach €15 billion in 2011. In addition to premiums for insurance rising to 15.5% of employee’s gross pay (from 14.9%), health insurers are also to set co-payments for employees to pay for general practitioner visits and spending cuts are to be imposed on doctors, and clinics. However, the most dramatic changes are in relation to the pricing of branded pharmaceuticals, where free market pricing means that spending is among the highest in Europe (€38.2 billion in 2009), with a 5.3% rise in pharmaceutical expenditure in 2009.Citation5
Budget deficit has triggered AMNOG Arzneimittelmarkt-Neuordnungsgesetz legislation in 2011. Under this legislation, all new drugs must be evaluated by Federal Joint Committee (GBA) for clinical benefit to determine whether they will be placed within a fixed price set or if they can negotiate price directly with insurers based on their cost-effectiveness. Cost-effectiveness arguments will become even more central to the value assessment process of a new drug application by GBA in Germany, especially related to the rapid clinical effectiveness assessment for new drugs within three months of receiving marketing authorization.Citation5 In some cases IQWIG might be invited by GBA to carry out HTA evaluation but in majority of situations GBA will directly mandate its decisions. A dossier will be required by both the GBA and IQWiG which demonstrates effectiveness against a comparator. Additional reference comparator-controlled studies are also mandatory, with the GBA being able to exclude those drugs from statutory reimbursement if they fail to deliver these. Price estimates of new drugs are likely to fall regardless of their incremental clinical benefit. AMNOG legislation will undoubtedly bring down costs of all drugs, as a consequence of price negotiation and risk-sharing schemes. Some companies may not risk launching innovative drugs if they cannot prove clinical or cost benefits. This is especially pertinent considering that the definition of benefit has not been set yet. In addition, AMNOG may negatively affect the rest of the European market which traditionally uses high German prices for referencing purposes. Critically, German market was historically viewed as one of the most attractive in the EU in terms of product positioning. With reimbursement pressures and requirements for cost-effectiveness studies in Germany, commercial returns from EU market will be less attractive.
Norway’s SLV uses reimbursement criteria that are established in law and include disease severity. This formally recognized in the Norwegian pharmacoeconomic guidelines which recommend the use of cost-value analysis to ensure that utilities encapsulate concerns for giving priority to the worst off. Sweden’s LFN (now TLV) makes reimbursement decisions assessing manufacturer’s evidence including disease severity. Other countries take broader societal issues into consideration. For example, Ireland’s HIQA assesses the quality of life, quality of end of life, social and ethical issues and social benefits. In Portugal, the principles governing reimbursement decisions are necessity and social justice. Spain’s pricing and reimbursement decisions are informed by disease severity and duration and by “social and therapeutic usefulness.”Citation5 Due to considerable budget deficit in Spain, Portugal and Ireland, it is expected that regional pricing control over costly biotechnology treatments will be further scrutinised limiting access of new products.
Principles of HTA Evaluations and Potential Impact on Cancer Vaccines
In the context of the modern day healthcare system, a biopharmaceutical intervention may add two types of value: clinical value, which relates to the improvement in patient outcomes brought about by the therapeutic intervention, and economic value which refers to the net positive financial impact on the healthcare system.
Crucially for biopharma companies investing in oncology R&D, value-based medicine requires that value components are built into drug-development programs early. New drugs need to demonstrate not just clinical efficacy and safety but also economic and social value proof points throughout drug development. Ideally, clinical, economic and humanistic evidence can be generated to corroborate a product’s value proposition from multiple angles to multiple stakeholders. This compendium of evidence needs to then be customized and translated to each stakeholder category to optimally communicate value to all customers. The value proposition for different stakeholders and for the society as a whole should be well presented in health-technology submissions to various payers’ bodies.Citation6
By focusing exclusively on clinical evidence of efficacy and safety, but not overall value, evidence based medicine may either overestimate, or underestimate, the value of a cancer intervention. For example, in oncology, the primary focus will be on the clinical benefit arising from an additional time of life gained from a cancer treatment. The quality of that extended life is often not quantitatively evaluated, yet in many circumstances the total value of the remaining life may be significantly decreased by the cancer treatment. Patients’ life expectancy may increase from 7 to 10 mo and the “value accrued over remaining life” can be calculated in terms of a QALY. The QALY measure aims to give an idea of how many extra months or years of life (of reasonable quality) a patient might expect to gain as a result of treatment. Drug treatment costs per QALY can then be calculated. The QALY tool applied to a life-threatening condition therefore combines a measure of improvement in survival with any changes in QoL and then determines the treatment cost to gain that QALY. If that treatment significantly compromises the QoL (for example, by inducing severe adverse effects, increasing the risk of infection, or causing impaired appetite and increased fatigue) those extra 3 mo of life will yield a much lower QALY score. In such circumstances, evidence-based medicine can overestimate the value of a cancer therapy. On another hand, there might be instances of underestimating the value of an intervention. Treatments with no additional effect on OS compared with the standard treatment might be deemed of low benefit by EBM, yet could offer better QoL to the patient through having fewer side effects, or by eliminating the need for frequent monitoring, hospital visits for infusions, or reducing the move to a second and third-line therapy at a later stage in disease progression. These factors could result in an overall positive value for the new treatment despite no clinical survival benefit.Citation6 It is anticipated that given the safety profile is beneficial, but the impact on PFS is less clear, cancer immunotherapies could fall into the latter group and can be easily underestimated in terms of the total clinical and economic value. Therefore cancer vaccine developers should consider different types of models capturing all types of clinical and economic values, including gains arising from favorable safety, reduction in use of cytotoxic regimens and surgeries, improved quality of life and various social values associated with enhanced well-being.
The core principles of HTA evaluation include the following:Citation5
• Clinical benefit — The short and long-term comparative effectiveness, side effects, and interactions with other therapies when used in the community;
• Adoption and diffusion — The number of eligible patients in a population, rate of uptake and whether the technology replaces or is added to current practice; and economic impact–the overall cost (both direct and indirect) to the health system, taking into account any cost savings that may be realized.
• Value for money — the utility placed on health gains achieved and the opportunity costs of those health gains (i.e., the other services forgone).
Measures that indicate clinical and economic value of a novel cancer vaccine will potential include features illustrated on .Citation5
Figure 1. Values for reimbursement from payer perspective. (A) measure of clinical value determined with cancer immunotherapy. (B) measures that indicate economic value of a drug.
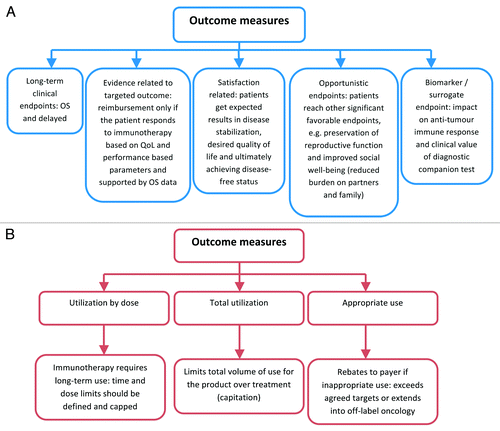
The less information available from the clinical program in relation to clinically and economically attributed values in a targeted oncology indication, the greater the uncertainty and, in turn, risk of an error in HTA. Consequently, HTA body is likely to issue a refusal in circumstances when potential risk to the budget and the number of uncertainties around values outweigh claimed benefits ().
Figure 2. Uncertainties facing payers in assessing product value at launch.Citation5
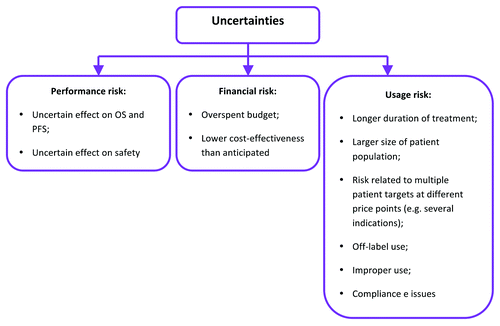
NICE has previously reviewed uncertainties and limitations of tools employed in evaluation of cost-effectiveness appraisal of new medicines. It was acknowledged that OS will remain a major clinically relevant endpoint for oncology drugs. The role of PFS is less clear due to lack of predictive relationship between PFS and OS. NICE recommended that developers should approach HTA bodies and MHRA for joint consultations in instances in which PFS is a good predictor of OS and those in which it is not. Cancer immunotherapeutics are not anticipated to have a profound favorable effect on PFS except for a potential to extend PFS during the phase when a delayed immune response will reach the peak. Due to complexity of the clinical effect and controversy surrounding the role of PFS in evaluation of cancer vaccines, it is crucial that companies would approach HTA bodies for consultation prior planning phase III studies. Further limitations in relation to QALY use in oncology relate to the following issues:
• The sensitivity of the QoL tools in measuring changes in health status and inability to pick up changes in some individual domains;
• Validity of the underlying constant proportional trade-off assumption used when eliciting health states values with the time trade-off method;
• Possible discrepancies between public and patients’ values of health states.Citation7,Citation8
One of the ways forward will be the development and validation of patient reported outcome (PRO) evaluation tools which could capture more subtle yet clinically important changes in manifestation of symptoms. The effect of the treatment on PRO could be translated in clinical and economic terms. PRO refers to any outcome that is directly derived from patient report without interpretation by the health provider, and can include assessment of symptoms, functional status, general health perception and health-related quality of life. Symptoms are considered most proximal to the disease process, and can be caused by either disease or treatment; the symptom burden may be estimated from the severity of individual or combined symptom(s) most associated with a disease or treatment, as reported by the patient’s perception of the impact of these same symptoms on their daily life. In December 2009, FDA issued its finalized Guidance on PRO measures, which provides stringent but general (that is, not disease specific) guidelines related to use of PRO instruments as effectiveness endpoints in clinical trials for the support of new drug applications for labeling claims.Citation9 PRO instruments will be especially useful in development of new immunotherapeutics as cancer vaccines are expected to show more profound effect on cancer-related symptoms, e.g., fatigue, loss of appetite and gastrointestinal symptoms, sleep disturbance etc. One of the successful regulatory examples on how PRO instrument was validated and assisted in product approval, was development of JAK 1 and 2 inhibitor ruxolitinib (Jakafi) in treating patients with myelofibrosis. A new PRO instrument for evaluation of myelofibrosis was developed and implemented in phase III studies yielding favorable effect of the product on clinically important symptoms such as abdominal discomfort, nocturnal sweats, bone pain, itching and early satiety.Citation10 This resulted in FDA approval of the product in November 2011. It is expected that PRO evaluation will especially relevant in some forms of cancers: effect on abdominal discomfort associated with malignant ascites in ovarian tumors; respiratory symptoms and nocturnal discomfort in patients with lung cancer; pruritis and abdominal discomfort in patients with hepatocellular carcinoma etc.Citation11 Development of PRO instruments for different forms of cancer not only can assist in obtaining additional labeling claims in the USA but also in capturing additional clinical, social and economic values and enhancing favorable cost-effectiveness profile of new cancer immunotherapies.
Safety and enhanced compliance with cancer vaccines is another area for capturing clinical value. Many patients suffering from advanced cancer are either refusing to go through aggressive chemotherapy regimens or not fully compliant with treatments. Younger patients with better prognosis are often less compliant than older patients. Non-adherence with some oncology treatments was reported in up to 50% of patients. On another hand, elderly patients with cancer tend to develop more safety, intolerance and drug-drug interaction related issues and therefore remain at higher risk of incompliance and consequential deterioration in quality of life.Citation12 Cancer immunotherapies provide with opportunities of improving prognosis and quality of life among elderly patients and groups with high rates of non-adherence. Self-administered (especially via subcutaneous route) cancer vaccines may alleviate oncology staffing shortages, especially those of nurses and pharmacist. Thus, implementation of cancer immunotherapies may provide with resource savings.
HTA Review of Ipilimumab
Real-life examples with reimbursement of cancer vaccines remain very limited. Ipilimumab is an antineoplastic human monoclonal antibody which acts as a T-cell potentiator that specifically blocks the inhibitory signal of CTLA-4 (CD152) resulting in T-cell activation, proliferation, and lymphocyte infiltration into tumors and so prompting cancer cell death. Ipilimumab has been often attributed to cancer immunotherapeutics due to presence of adjuvant gp100 in its composition and relationship between inhibition of CTLA-4 and immunological tolerance toward the tumor.
In July 2011, EMA recommended the granting of a marketing authorisation for ipilimumab (Yervoy) for the treatment of advanced (unresectable or metastatic) melanoma in adults who have received prior therapy. CHMP members and assessors involved in the review have published EMA experience with evaluation of ipilimumab.Citation13 In the pivotal phase 3 trials, ipilimumab led to a 3-mo improvement in overall survival, a disease control rate of 28.5%, and 60% of responding patients maintained this response for 2 y. Furthermore, patients who received re-induction with this agent had further disease control. Various limitations in individual sub-populations were acknowledged but did not preclude the conclusion on favorable risk-benefit profile.Citation13 Subsequently, Ipilimumab was approved by HTA bodies in Germany, Sweden and Austria.
Around 400–500 UK patients with advanced melanoma require second line treatment such as ipilimumab each year. The UK NICE has recognized ipilimumab as a “step change” in the treatment of advanced melanoma, according to draft guidance published on October 14 2011 and subsequent review. Despite this, ipilimumab was not considered to be a cost-effective use of NHS resources due to uncertainty over its long-term benefits and, potential overestimation of OS gains presented in manufacturer’ economic model. Ipilimumab proposed at a cost of £20,000 per dose, or £80,000 per patient, was deemed to be too costly in light of uncertainties and risks for NHS. NICE estimated the most likely incremental cost-effectiveness ratio to be between £54,000 and £70,000 per quality adjusted life year. In fact NICE evaluation group applied further corrections and amendments to the manufacturer’s model and arrived to conclusion that the calculated incremental cost-effectiveness ration exceeding £96,000 per QALY gained, suggesting that the true ratio is likely to be greater than that obtained with the revised by BMS base case analysis.Citation14
NICE has also expressed concerns over uncertainties around unapproved comparator (gp100) employed in a pivotal study, lack of prominent clinical differences between the effect of 3 mg and 10 mg doses, and detrimental impact of adverse effects that might affect the patient's quality of life (diarrhea, rash, fatigue, nausea, vomiting, decreased appetite, abdominal pain and risk of autoimmune disease). NICE acknowledged that the trial results indicated that Yervoy could potentially be very effective, but in a small number of patients. The appraisal committee accepted that around 30% of patients treated with Yervoy would have improved survival, and just 10% would experience long-term benefits. Therefore NICE has concluded that a lack of appropriate biomarker (e.g., HLA subtype) to predict patients who are likely to benefit is a major deficiency in current Ipilimumab submission.
In response to NICE, BMS might conduct further retrospective subset analysis to identify a suitable biomarker, but this could prove time consuming and may not provide with statistically robust results. Re-submission of the new economic model based on biomarker-stratified population might increase the probability of success in similar fashion to prior U-turn of NICE on HTA re-submission of cetuximab (Erbitux) in patients with metastatic colorectal cancer carrying wild type KRAS.Citation14 Most importantly, there is an expectation that manufacturer will propose a risk-sharing scheme which would mitigate various financial and economic risks and uncertainties for UK health care. Meanwhile, ipilimumab is available through a number of regional cancer drugs funds, which were set up by UK coalition government to address poor uptake of newer oncology medicines in England.
Risk-sharing Schemes
With increasing competition in crowded oncology space and ballooning healthcare budget in most developed countries, it is invariable that level or risk and uncertainty associated with HTA appraisals will be growing resulting in higher number of non-reimbursed or not universally reimbursed medicines. In order to mitigate this scenario, many developers entered in negotiations with HTA bodies as well as local payers and proactively offered various risk-sharing schemes which would alleviate budgetary risks and uncertainties. Some of the examples of oncology agents reimbursed under risk-sharing agreements are summarized in the .
Table 2. Outcome-based pricing agreements for oncology products.Citation14
The common features of risk-sharing schemes are as follows:
• There is either persistent discount or discount applied at the onset or maintenance phase of the treatment;
• There is a cap for amount of reimbursed product;
• There are strict criteria for treatment eligibility under approved indication along with criteria for response, non-response and treatment discontinuation.
• There is often free provision of the drug in some circumstances, possibly triggered not only by pressure from payers but also by willingness of manufacturers to collect long-term efficacy and safety outcomes (e.g., registries or post-marketing safety studies) requested by regulatory authorities.
Of course, risk sharing schemes may not be necessary in all oncology cases. It is thought that highly innovative product with demonstrable overwhelming effect on OS gains and significant clinical and economic impact in uncontested oncology indication could qualify for premium price reimbursement. Recently, German GBA has approved reimbursement scheme for anti-fibrotic agent pirfenidone (Esbriet) for idiopathic pulmonary fibrosis with price around Euro 38000–40000K per year of treatment based on unique clinical benefit accompanied by OS gain, which “is currently not quantifiable but will be estimated by measuring further long-term outcomes.”Citation15
In conclusion, payers around the globe — and particularly in the EU are adopting policies which require companies to demonstrate improved health outcomes before approving high-priced drugs for coverage. This trend has resulted in a number of risk-sharing agreements in recent years, and we can expect the incidence and variety of these arrangements to increase. However, though the adoption of such schemes is spreading in cancer treatments, it remains to be seen whether they are sustainable for rare oncology diseases. Indeed, the cost-effectiveness gains achieved by both parties sharing the risk might well be outweighed by the very high per capita administrative costs associated with the follow-up of the patients involved. With payers more conscious of costs of rare diseases and big pharma's growing interest in niche markets, the era of large product surpluses is likely to end. To succeed, approval from the regulatory authorities is no longer enough; pharmaceutical companies will increasingly need to convince payers of the cost-effectiveness of their products. This implies shifting resources from traditional commercial teams to pinpointing subgroups of patients and providers, investing in data collection and cost-effectiveness expertise and being more aware of payers' constraints.
Value for Different Stakeholders
Market access to expensive therapies is often influenced by specialty medical societies and patient advocacy groups in both formal and informal ways. In addition to their other functions, such groups represent the financial interests of their constituents to entities such as the American Medical Association (AMA) and Centers for Medicare and Medicaid Services (CMS), both of which have responsibility for various components of the coding and payment systems. Working closely with an appropriate specialty medical society can help build support between the clinicians that will use the drug in the new indication product. For example, efforts by oncology related patient advocacy groups have played a role in the launch of the £200 min Cancer Fund in the UK. Any dual pricing initiative must keep the incentives of such lobbying groups in mind.
The value equation for an oncology product may also be changed by assessing the impact of therapy in particular patient subgroups, including non-responding or underserved patient sub-populations. Such evidence has already been persuasive to regulators and can appeal to payers channelling therapies and their costs toward the patients most likely to benefit from therapy. Despite that positive results from post-hoc analysis of population subgroups from negative or non-significant studies are unlikely to be the basis for approvals, an identification of favorable trends and building subsequent economic models addressing under-served patient segments and backing-up this approach with risk-sharing might be accompanied with greater probability for success. Importantly, the definition of value differs between various stakeholder groups. Self-administered cancer vaccines may allow patients to be treated as outpatients, where they assume greater responsibility for their own treatment. Many patients in developed and developing markets will bear a substantial portion of the costs of treatment directly. As a group, patients with cancer are keen to understand their options and play a role in making value decisions over treatment choice. As patients assume more responsibility for administration and payment for cancer drugs, it will be imperative to assess the drivers and obstacles to medication adherence.Citation6 Patients will have to be educated about long-term advantages of cancer vaccines in order to appreciate the clinical rationale and the value of treatment. HTA submissions should be aimed at communicating product value not just for clinicians, regulators and policy makers but also for patients and patient-support groups who are often engaged in HTA appraisals. From clinician perspective, cancer regression and OS remain to be highly desirable clinical outcomes. Since cancer vaccines may not provide with consistent effect on PFS and ORR, clinicians will require education around clinical benefits derived from OS and PRO-driven outcomes. The move toward value-based medicine may in time affect regulatory decision making, with regulation and HTA-type processes coming closer together as part of drug approvals. Patient-reported outcomes will be increasingly incorporated into product labels in US and other countries. In the future, payers will be paying premium prices only for products deemed to be truly innovative and providing with impressive OS gains. From governmental perspective, cancer is a major killer and a global health care problem which cannot be ignored. Healthcare planning will invariably consider innovative treatments for cancer which could assist in reducing burden of the disease.
Conclusions
Even if the sponsor follows advice from key opinion leaders (KOLs), monitors competitive landscape, thoroughly plans manufacturing and clinical decisions, consults with regulatory agencies, and starts with a clear destination in mind (e.g., the targeted desired draft launch label or the target product profile), there remains the possibility that the product won’t reach approval by regulatory or payer authorities. Clinical development path can be significantly influenced by budgetary pressures and decisions, unexpected clinical findings in own and/or external programs, portfolio priorities, re-organisational changes, mergers and acquisitions, changes in competitive landscape, pricing and reimbursement climate etc. Today the task of cancer vaccine developers is not only to excel in cancer immunology and art of conducting immunotherapy trials but also to perfect in commercial wizardry of analyzing and re-evaluating internal rate of returns and projected commercial attractiveness of the product. Requirements imposed by payers and reimbursement bodies on data demonstrating the cost-effectiveness, the breadth of relevant clinical outcomes, regionally appropriate data sets and comparative efficacy of novel products against locally approved comparators add an additional layer of hurdles on the path to the market. Pricing pressures arising in increasingly competitive oncology market can easily erode profit margins and therefore need to be taken into account in determining internal rates of return. Conservative methodologies should be employed in order to account for potential price reductions imposed by reimbursement bodies. Re-evaluations throughout development cycle are recommended in order to determine commercial attractiveness of the program. Since the assessment of cost and relative clinical effectiveness of new oncology agents is performed by bodies which are independent to regulatory authorities, joint consultations/advice should be sought as early in development process as possible. Most importantly, value-driven methodologies should prompt cancer vaccine developers to collaborate closely with all oncology stakeholders in order to capture different values and pathways for the product reimbursement.
Disclaimer
The views expressed in this article are the personal views of the author and may not be understood, interpreted, or quoted as being made on behalf of, or reflecting the position of any other companies, agencies or parties cited in this article.
References
- American Cancer Society. (2011) Cancer Facts and Figures 2011. Atlanta, American Cancer Society.
- The cancer market outlook to 2016. Business Insights 2011 http://www.researchandmarkets.com/reports/1839878/the_cancer_market_outlook_to_2016
- Desdouits F, Delaporte L, Parnis S. Roadmap for success in oncology. Script 2008 1: 1-11. http://www.researchandmarkets.com/reports/1198415/roadmap_for_success_in_oncology
- FDA. (2010) FDA Prescribing Information for Provenge. Available at: http://www.fda.gov/downloads/BiologicsBloodVaccines/CellularGeneTherapyProducts/Approved-Products/UCM210031.pdf. Accessed: April 12, 2012.
- Misra G. (2011) Reimbursement in Europe and Indication-Specific Pricing. Business Insights.
- Huber B, Doyle J. (2010) Oncology Drug Development and Value-based Medicine. Available from: http://www.quintiles.com/information-library/white-papers/oncology-drug-development-and-value-based-medicine/. Accessed on April 13, 2012.
- Barton GR, Sach TH, Doherty M, Avery AJ, Jenkinson C, Muir KR. An assessment of the discriminative ability of the EQ-5Dindex, SF-6D, and EQ VAS, using sociodemographic factors and clinical conditions. Eur J Health Econ 2008; 9:237 - 49; http://dx.doi.org/10.1007/s10198-007-0068-z; PMID: 17605057
- Garau M, Shah K, Towse A, Wang Q. (2009) Assessment and appraisal of oncology medicines: does NICE’s approach include all relevant elements? What can be learnt from international HTA experiences? Report for the Pharmaceutical Oncology Initiative Group (POI). Available from: http://www.ohe.org/publications/article/assessment-and-appraisal-of-oncology-medicines-33.cfm Accessed on April 13, 2012.
- FDA. (2009) Guidance for Industry: Patient-reported outcome measures: Use in medical product development to support labeling claims. http://www.ispor.org/workpaper/FDA%20PRO%20Guidance.pdf Accessed on April 13, 2012.
- Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012; 366:799 - 807; http://dx.doi.org/10.1056/NEJMoa1110557; PMID: 22375971
- Pardanani A, Vannucchi AM, Passamonti F, Cervantes F, Barbui T, Tefferi A. JAK inhibitor therapy for myelofibrosis: critical assessment of value and limitations. Leukemia 2011; 25:218 - 25; http://dx.doi.org/10.1038/leu.2010.269; PMID: 21079613
- Banna GL, Collovà E, Gebbia V, Lipari H, Giuffrida P, Cavallaro S, et al. Anticancer oral therapy: emerging related issues. Cancer Treat Rev 2010; 36:595 - 605; http://dx.doi.org/10.1016/j.ctrv.2010.04.005; PMID: 20570443
- Hanaizi Z, van Zwieten-Boot B, Calvo G, Lopez AS, van Dartel M, Camarero J, et al. The European Medicines Agency review of ipilimumab (Yervoy) for the treatment of advanced (unresectable or metastatic) melanoma in adults who have received prior therapy: summary of the scientific assessment of the Committee for Medicinal Products for Human Use. Eur J Cancer 2012; 48:237 - 42; http://dx.doi.org/10.1016/j.ejca.2011.09.018; PMID: 22030452
- Bruce F. (2011) “Step change” Yervoy from BMS not cost effective for NICE: may need biomarker test. Available from: http://www.scripintelligence.com/home/Step-change-Yervoy-from-BMS-not-cost-effective-for-NICE-may-need-biomarker-test-322459 Accessed on April 13, 2012.
- Intermune (2012) Esbriet (pirfenidone) Benefit Granted by German Federal Joint Committee (GBA). Available from: http://www.marketwatch.com/story/esbriet-pirfenidone-benefit-granted-by-german-federal-joint-committee-g-ba-2012-03-15 Accessed on April 17, 2012.