Abstract
At the cellular level it is clear that cancer is a genetic disease arising as a clone that expands and grows in an unregulated manner. While it has always been presumed that neoplasia is a consequence of somatic cell mutations, only in the last few years has the magnitude and diversity of these mutations been elucidated by modern DNA sequencing technology. Immunotherapy is the premier biological approach to targeted therapy. Target therapies require targets. In this case the targets are tumor specific or associated antigens, the proteins expressed from these somatic cell mutations. While the immunotherapeutic approach to eliminating cancer was launched with the assumption that cancer cells were homogeneous, the recent genomic understanding of tumor cells indicates that there is both inter- and intra-tumoral heterogeneity. This presentation will discuss the consequences of this new knowledge of tumor cell biology to the immunotherapeutic approach to treating cancer.
Vaccinogen‘s lead product, OncoVAX®, is an active specific immunotherapeutic (ASI), stimulating a patient‘s immune response to autologous tumor cells. Thus, OncoVAX is a targeted, patient specific therapy. To prepare OncoVAX, a patient‘s own tumor is excised, enzymatically dissociated to separate tumor cells from normal tissues, sterilized, and gamma-irradiated (200,000 rads) to render the tumor cells non-dividing and non-tumorigenic, but still metabolically active. According to the OncoVAX protocol the first two injections consist of irradiated tumor cells admixed with fresh-frozen Mycobacteria of the TICE strain of Bacillus Calmette Guérin (BCG). The third and fourth immunization consists of irradiated tumor cells alone. All immunizations are intradermal, the most functional site for vaccinations. TICE strain of BCG is, along with being a vaccine for prevention of tuberculosis, also an approved drug product for the intravesicle treatment of carcinoma in situ bladder cancer and prophylactic prevention of recurrence of superficial papillary bladder cancer.
The patient‘s own cells are the source of the vaccines tumor antigens because of the now, well-recognized phenomenon of tumor heterogeneity. Within the scientific community a serious oversight occurred involving the understanding and utilization of tumor cell biology in cancer treatment strategies. It was due to the under estimation of the consequences of tumor heterogeneity. Cancer is a genetic disease based on a disorder of DNA. The conventional view was that cancer progresses in a linear fashion, with all tumor cells genetically homogeneous at any given point and can be treated with homogeneous therapies. The new technology has directly established the magnitude of tumor cell mutations. The majority of the data on the genomic diversity among and within individual tumors and its consequences to targeted therapies was gathered over the last few years using the second generation sequencing technology.
Both inter-tumoral (among tumors of the same type) and intra-tumoral (within individual tumors) heterogeneity should be expected to have a major impact on homogeneous, combination treatment strategies and the burgeoning industry of personalized medicine based on tumor biopsies and marker proteins. Thus, the view of directing therapy only on the basis of genetic tumor markers is too simplistic and ineffective based on the potential of convergent evolution of the tumor. Targeted therapy requires a functional target. From an immunologic perspective, every one of the mutations presumably gives rise to unique proteins, each of which is non-self. This provides a basis for optimism with regards to autologous tumor vaccines and immunotherapy. There is now direct evidence for inter-tumoral heterogeneity but the genomic findings confirm that the genetic lesions found in the original tumor cells are consistently expressed. This is true in spite of the continuing convergent evolution based on new and multiple mutations in the expanding or metastasizing tumor under treatment. Tumor cell intra-tumoral heterogeneity is not a new concept.Citation1,Citation2 Fidler et al.,Citation3,Citation4 performed clonal selection and transplantation studies in mice over 30 y ago at the National Cancer Institute-Frederick Cancer Research Center (NCI-FCRC), which demonstrated intra-tumoral phenotypic heterogeneity. These studies in a syngeneic mouse model showed that transplantable malignant tumors exhibited diverse biological heterogeneity with respect to tumor invasion and metastasis. This heterogeneity was attributed to two major processes; the selective nature of the metastatic process, and the rapid evolution and phenotypic diversification of clonal tumor cell populations during progressive tumor growth. The latter probably resulting from inherent genetic and epigenetic instability of many clonal populations of cells.
Having been present at the NCI laboratories while this work was being performed and published, and working on immunotherapy in a syngeneic Strain 2 Guinea Pig animal model using a transplantable Line 10 hepatocarcinoma, a poorly immunogenic, progressor tumor, we also gained some insight into some of the tenants of successful immunotherapy.Citation5 Using this model, we learned a variety of fundamental principles by interpreting immunologic and experimental pathological results that were anticipated to be translatable to human cancer. In fact, the model was purposely maneuvered in the experiments toward the conditions confronted with solid tumors in humans. The proof of interpretations, with respect to the development of both effective tumor vaccines and regimens of administration and its translation to the clinic required a decision on the source of the functional antigens and the adjuvant to be used. It was reasoned that if there is intratumoral heterogeneity with respect to phenotypic diversification of clonal tumor cell populations, there is a reasonable possibility of intertumoral antigenic heterogeneity. Therefore, the key decision to use autologous tumor cell vaccines was based on the assumption that the genetic lesions that are found in the original tumor cells are consistently expressed and this would obviate the antigenic diversity in any immunotherapeutic approach. Thirty years later, this strategy has finally gained recognition and direct validation, based on the recent development of second-generation DNA sequencing technologies as applied to the cancer genomes.Citation6,Citation7
To boost the immune response, Vaccinogen includes BCG in the first two injections. BCG is a live but only weakly pathogenic bacterium. Nonetheless, it does generate a local inflammation, inducing a strong immune response by multiple pathways of immune stimulation. In the Strain 2 guinea pig model there was an optimum dose of BCG admixed with tumor cells that was effective. More was not better and often worse. It does not rely on a single mechanism du jour (of which there have been many in immunology through the years), but broadly activates the immune system. In fact 24 h after injection of BCG in guinea pigs and man, there is a massive mobilization of immune progenitor cells in the bloodstream, which results in the “homing” of these cells to the intradermal vaccination site. These immune progenitor cells become activated at the vaccination site to available antigens.
Clinical Strategy
Several phase I/II dose optimization and regimen finding clinical trials were performed with OncoVAX, in stage II and III colon cancer patients. In the 1980s colon cancer treatment was limited to 5fu with trials in combination with leucovorin. Nevertheless as we proceeded to larger, follow-on clinical studies, the next consideration was product manufacturing. Consistent manufacturing of the autologous vaccine in a centralized laboratory presented several rate limiting issues including transportation of patient materials between the clinical and manufacturing sites, a reproducible and cost-effective manufacturing process for individualized therapy on a large scale and automated quality control process to reproducibly release product. The first two issues were evaluated in the first two randomized phase III clinical trials while the last and final issue, a sterile drug product, was put to rest in preparing for the pivotal Phase III study.
From a marketing and distribution perspective an immunotherapeutic process where the vaccine can be manufactured, formulated and administered in each hospital appeared to be desirable. This de-centralized approach was of interest to the Eastern Cooperative Oncology Group (ECOG). They took the challenge to conduct a phase III clinical trial of OncoVAX using this de-centralized approach. However, it was recognized that the quality control and quality assurance that could be achieved through this de-centralized might be difficult with this approach, the degree of difficulty was grossly underestimated.
Only three vaccinations could be consistently produced and often these did not meet the desired specification. The results of the ECOG study were negative in terms of intent-to-treat analysis; however, a subset analysis comparing patients who received vaccines that met specification compared with deficient vaccines showed that the former had significantly improved prognosis.Citation8 When this study of the de-centralized approach was initiated by ECOG, the importance of the inclusion of the fourth, booster, immunization had not yet been realized; the booster immunization was later implemented as part of the study of the centralized manufacturing conducted by the Free University in The Netherlands. This study also differed from the ECOG study in that it utilized a centralized approach to the manufacture, quality control and quality assurance of vaccines in a facility located at the University hospital in Amsterdam. This study also provided for a four-vaccine regimen that had a six-month booster inoculation after the initial three weekly treatments. This required a centralized manufacturing laboratory in logistically reasonable geographical area and some modifications of the practice of medicine by the pathologists to provide the maximum amount of tumor to the manufacturing laboratory, but still allow adequate sample for clinical diagnosis and staging of the patient’s tumor. This four-vaccine study was conducted by Drs. H. Pinedo and Jan Vermorken at the Free University in the Netherlands.Citation9,Citation10 The protocol was essentially the same as the ECOG study, except for the additional booster vaccination in the Amsterdam trial. This multi-center, randomized trial was performed at 12 cooperating hospitals in The Netherlands, and used a central vaccine manufacturing facility at the University Hospital, Vrije Universiteit (Free University), Amsterdam. Colon resections were performed at each of the 12 sites, and all tumor samples were sent to the University Hospital’s vaccine production laboratory for dissociation, cryopreservation, irradiation, and administration.
This study, unlike all of the previous studies, included a six-month booster inoculation after the initial three weekly treatments because a sub-study of one of the phase II trials suggested that immune response begins to wane at 6 mo after the induction vaccinations. In the last two vaccinations, which were BCG negative, positive delayed type hypersenstity (DTH) reactions could be recorded at the site of the intradermal inoculations. A major change perpetrated by the effort to give 4 treatments including both the induction and booster, was that more tumor was required to consistently produce the final drug product with the desired number of live tumor cells. This required larger tumors with the minimum requirement being 3 to 3.5 g. In the stage II patients this forced the patient population into the Stage II (IIA = T3N0M0, IIB = T4aN0M0, IIC = T4bN0M0) disease. Both treated and control patients had vaccines produced before randomization. Subjects with stage II and III colon cancer randomized to the control group (n = 126) received no further treatment after surgical resection and were followed according to scheduled assessments.
For subjects randomized to OncoVAX (n = 128), 28 to 35 d after surgery these patients received three inoculations, the first with tumor cells admixed with BCG and one week later a second identical treatment. The third vaccination one week later was autologous, sterile, irradiated and live tumor cell alone A fourth dose, a booster treatment, was administered six months after surgical resection. The median follow-up in this study was originally 5.3 y and then 5.8 y. The 12 sites enrolled between 5 and 34 subjects each. Randomization into the study was stratified based on TNM stage, tumor location and institution. Patients were well matched with regard to their baseline characteristics.
In the OncoVAX group, 102/128 subjects received all four vaccinations. To determine the extent of DTH reactivity, vaccination sites were measured for indurations 48 h after the third and fourth immunizations. Subjects with an average of the two diameters 5 mm were considered to have effective cellular immunity; 97% of patients after the fourth inoculation achieved effective cellular immunity. While favorable trends were observed, there were no statistically significant differences in recurrence-free survival, overall survival or recurrence-free interval between all subjects, stages I-IV in the control group and those who received OncoVAX. The randomization was stratified so that a prospective analysis by stage of disease was performed. Subjects with stage II disease had both clinically meaningful and statistically significant outcomes in both recurrence free interval and recurrence free survival. When five year event-free rates were measured, clinically and statistically significant outcomes in overall survival were observed.
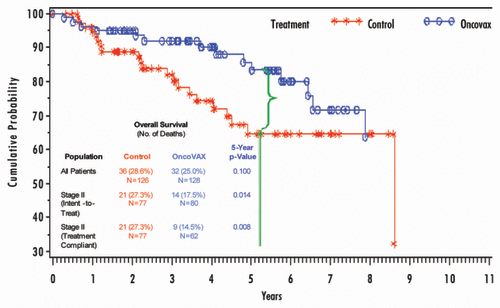
Forty-six TNM stage II patients (29 controls, 17 OncoVAX treatments) were reported as having disease progression or having died during the study. Kaplan-Meier estimates of colon cancer rates show a statistically significant improvement of recurrence free survival in the stage II treated patients (). The percentages after five years of follow-up were 21.3% and 37.7% for the treatment and control groups, respectively. The favorable 16.4% difference represents a 41.4% relative risk reduction of disease progression (5-y survival p = 0.008; log-rank analysis p = 0.018). Thirty-five TNM stage II patients (21 controls, 14 OncoVAX treatments) died during the study. Overall survival showed a statistically significant improvement in the stage II OncoVAX treated patients (17.5%) over those patients in the control group (27.3%) (). The favorable 9.8% difference represents a 33.3% relative risk reduction (5-y survival p = 0.014; log-rank analysis p = 0.074).
Figure 1. Study—recurrence free survival in stage II patients.
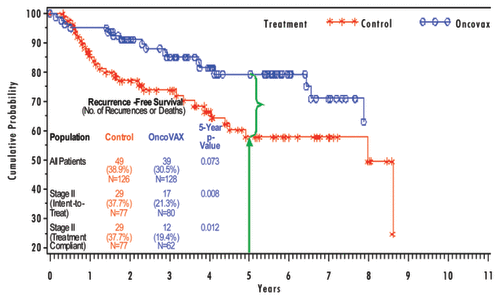
Figure 2. 8701 Study—overall survival in Stage II patients.
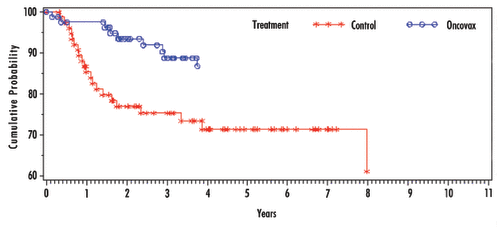
In the intent-to-treat (ITT) population of all patients randomized as stage II, there were 43 recurrences, (). The five-year recurrence free interval p-value (0.010) and the log-rank analysis p-value (0.004) was highly significant, it was discovered in referee pathology diagnosis that this included a proportion of B1 patients (9 control and 4 treated patients). These were excluded in the separate Stage II analysis, the control and OncoVAX treatment groups, respectively. When compared with the control group, the favorable 16% difference represents a 57.1% relative risk reduction in the recurrence of colon cancer in the OncoVAX group (five year survival p = 0.026; log-rank analysis p = 0.008).
Figure 3. 8701 Study—recurrence free interval in Stage II patients.
The control group had a higher percentage of patients with a non-fatal serious adverse event than the OncoVAX group. Thirty-three patients in the OncoVAX group (25.8%) and 46 patients in the control group (36.5%) experienced at least one non-fatal serious adverse event. One serious adverse event was considered related to treatment with OncoVAX. A patient was hospitalized for treatment of a flu-like syndrome and the event resolved nine days later. In addition, treatment with OncoVAX was discontinued in a 71-y old woman who developed 21 x 32 mm ulceration after the second inoculation (from which BCG had been omitted because of adverse events after the first inoculation). The area of ulceration became necrotic and required surgical excision.
In a post-hoc analysis, outcomes of subjects who received all four inoculations were evaluated. In such a case, the combined OncoVAX treated cohort achieved a clinically meaningful and statistically significant outcome in terms of recurrence-free interval and recurrence-free survival. Using this analysis, subjects with stage II disease also achieved a statistically significant difference in overall survival (p = 0.046); 85.5% in the OncoVAX treated group survived vs. 72.7%.
The dialog with the US. Food and Drug Administration (FDA) regarding the approval of OncoVAX, took place between January 2000 and May 31, 2006 and centered on the pharmacological aspects of the drug product. The issues resolved were sterility and complete product characterization studies using flow cytometry. The latter resulted in developing an automated matrix associated potency and identity assay. Finally, the FDA requested a second, confirmatory, randomized controlled phase III trial of OncoVAX in stage II colon cancer. Based on a protocol approved by the FDA, this study will be performed under a Special Protocol Assessment (SPA). An SPA granted by the FDA provides a mechanism for the sponsors and the FDA to reach agreement on, size, execution and analysis of a clinical trial that is intended to form the primary basis for regulatory approval.
The primary endpoint of this pivotal phase III trial is recurrence-free survival (RFS) with an interim and final primary analysis to be made one and three years after following the full enrollment respectively. The study is powered at 90% to detect a 50% increase in recurrence-free survival (RFS) vs. resection only control for final analysis with adjustment for interim analysis. If a robust statistical significance is achieved at the interim analysis, the Biologic License Application can be filed. Past clinical trials using the optimum four immunization regimen will be accepted as supportive studies during the FDA review of the BLA. It must be noted that the period between 2002 and 2005, while we were performing the work described in this text; a number of biotechnology companies attempted and failed in pivotal phase III clinical trials of various platforms of ASI. Some of these studies were performed under FDA granted SPAs. This does not include several other attempts that failed in phase II clinical trials or were put on clinical hold for failing to accomplish pharmacological or manufacturing requirements. The failures of these studies not only negatively affected the economic status of the companies involved, but also have in general negatively affected the status of the field of cancer immunotherapy. The approval of Dendreon’s prostate cancer vaccine “Provenge” proved that the market was enthusiastic about cancer vaccines, only to become disillusioned by the pharmacoeconomics of the product, the deminimis clinical benefit and the failure of management to properly set and meet expectations.
Despite these failed efforts, we remain cautiously optimistic for several reasons. First,OncoVAX is the only immunotherapy platform using autologous tumor cells to treat minimal residual disease. Second, in all of our dose and regimen finding clinical trials, we had randomized surgery only controls and thus more comprehensive clinical data for OncoVAX compared with other immunotherapy products. Finally, we are confident that in all of the attempts to satisfy the compliance aspects of the manufacturing and quality control requirements, we were conscientious to maintain the immunologic essence of the effective vaccine. Major process changes were required to produce sterile autologous tumor cell vaccines. However, we carefully performed a clinical bioequivalence using DTH to the third and fourth (booster) vaccine as a surrogate for immunogenicity, which demonstrated there were comparable DTH responses to those generated by previous non-sterile vaccines. We hope that this critical and careful approach to the clinical development of OncoVAX should allow in the pivotal phase III trial, positive clinical benefit for stage II colon cancer, which remains a true “unmet medical need.”
Related Research Data
References
- Byers VS, Johnston JO. Antigenic differences among osteogenic sarcoma tumor cells taken from different locations in human tumors. Cancer Res 1977; 37:3173 - 83; PMID: 69491
- Pimm MV, Baldwin RW. Antigenic differences between primary methylcholanthrene-induced rat sarcomas and post-surgical recurrences. Int J Cancer 1977; 20:37 - 43; http://dx.doi.org/10.1002/ijc.2910200108; PMID: 71272
- Fidler IJ, Kripke ML. Metastasis results from preexisting variant cells within a malignant tumor. Science 1977; 197:893 - 5; http://dx.doi.org/10.1126/science.887927; PMID: 887927
- Poste G, Doll J, Fidler IJ. Interactions among clonal subpopulations affect stability of the metastatic phenotype in polyclonal populations of B16 melanoma cells. Proc Natl Acad Sci U S A 1981; 78:6226 - 30; http://dx.doi.org/10.1073/pnas.78.10.6226; PMID: 6947225
- Hanna MG Jr., Peters LC, Hoover HC Jr. Immunotherapy by Active Specific Immunization: Basic Principles and Preclinical Studies. Chapter 25, Biologic Therapy of Cancer Principles and Preclinical Studies. Biologic Therapy of Cancer Principles and Practice. Ed. DeVita VT, Hellman S, Rosenberg, SR, p 651, 1991, J. P. Lippincott, Co.
- Gerlinger MR, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012; 366:883 - 92; http://dx.doi.org/10.1056/NEJMoa1113205; PMID: 22397650
- Stratton MR. Exploring the genomes of cancer cells: progress and promise. Science 2011; 331:1553 - 8; http://dx.doi.org/10.1126/science.1204040; PMID: 21436442
- Harris J, Ryan L, Adams G, Benson A, Haller D. Survival and Relapse in Adjuvant Autologous Tumor Vaccine Therapy for Dukes B and C Colon Cancer – ECOG Study 5283. ASCO Abstract 1994.
- Vermorken JB, Claessen AME, van Tinteren H, Gall HE, Ezinga R, Meijer S, et al. Active specific immunotherapy for stage II and stage III human colon cancer: a randomised trial. Lancet 1999; 353:345 - 50; http://dx.doi.org/10.1016/S0140-6736(98)07186-4; PMID: 9950438
- Hanna MG Jr, Hoover HC Jr, Pinedo HM, Finer M. Active Specific Immunotherapy with Autologous Tumor Cell Vaccines for Stage II Colon Cancer: Logistics, Efficacy, Safety and Immunological Pharmacodynamics. Human Vaccines 2006; 2:185 - 191