Abstract
Human cytomegalovirus (CMV) establishes a lifelong persistent infection characterized by periods of latency and sporadic viral replication and is a major infectious cause of birth defects following congenital infection. Currently, no licensed vaccine is available that would prevent CMV infection. In an effort to develop a prophylactic CMV vaccine, the effects of different formulations, immunization routes and delivery devices on the immunogenicity of plasmid DNA (pDNA)-based vaccines were evaluated in rabbits and mice. Compared with PBS- and poloxamer-based formulations, significantly higher antibody responses were obtained with pDNA formulated with Vaxfectin®, a cationic lipid-based adjuvant. With low vaccine doses, the intradermal (ID) route resulted in higher antibody responses than obtained when the same dose was administered intramuscularly (IM). Since the IM route allowed injection of larger volumes and higher doses than could be administered at a single ID site, better antibody responses were obtained using the IM route. The needle-free injection system Biojector® 2000 and electroporation devices enhanced antibody responses only marginally compared with responses obtained with Vaxfectin®-formulated pDNA injected IM with a needle. A single-vial Vaxfectin® formulation was developed in a dosage form ready for use after thawing at room temperature. Finally, in a GLP-compliant repeat-dose toxicology study conducted in rabbits, single-vial Vaxfectin®-formulated vaccines, containing pDNA and Vaxfectin® up to 4.5 mg and 2 mg/injection, respectively, showed a favorable safety profile and were judged as well-tolerated. The results support further development of a Vaxfectin®-formulated pDNA vaccine to target congenital CMV infection.
Introduction
Human cytomegalovirus (CMV) is a betaherpesvirus that establishes a lifelong persistent infection characterized by periods of latency and sporadic viral replication. In the US, CMV seroprevalence increases gradually with age, from 36% in 6- to 11-y-olds to 49% in 20- to 29-y-olds and to 91% in those aged ≥ 80 y.Citation1 For the vast majority, CMV infection is asymptomatic and does not pose a serious health problem. However, CMV is an important pathogen for individuals who become immunocompromised, including solid organ and hematopoietic cell transplant recipients, and is a major infectious cause of birth defects following congenital infection.Citation2,Citation3 CMV is the most common intrauterine infection in the US, with debilitating effects such as mental retardation, hearing or vision loss, and cerebral palsy, and has a 10–12% mortality rate in infants who are symptomatic at birth.Citation4,Citation5 Worldwide, ~0.6% of infants are born with congenital CMV infectionCitation6; in the US, ~40,000 infants are born annually with congenital infection and between 5,000 and 9,000 children suffer permanent sequelae.Citation5,Citation7,Citation8
The overall disease burden to the healthcare system from congenital CMV infections has been estimated at over $300,000 per infected child.Citation9 Because of the cost savings in medical care that could be realized by an effective universal vaccine, the Institute of Medicine has ranked CMV in the highest priority category of infectious disease vaccines that need to be developed in the U.SCitation5; despite this, no vaccine candidate has advanced beyond phase 2 testing and a high unmet need still exists.
Antibody responses directed to CMV envelope glycoproteins appear to be critical for the prevention of fetal CMV infection by blocking transplacental transmission; several studies in humans and animal models have demonstrated the importance of antibodies in decreasing or preventing CMV infection and disease.Citation2,Citation4 Vaccine development efforts have largely focused on glycoprotein B (gB), a major target for CMV neutralizing antibodies which block cell attachment to and penetration of fibroblasts.Citation10 A recombinant gB subunit vaccine containing the squalene-based adjuvant MF-59 has demonstrated 50% efficacy at preventing maternal infection,Citation11 providing proof-of-concept for inclusion of this antigen in a vaccine. Other vaccine candidates that have been or are being tested in clinical trials include live-attenuated or chimeric CMV strains, recombinant viral vectors expressing CMV antigens including poxvirus and alphavirus-based vectors, and plasmid DNA (pDNA)-based vaccines.Citation3,Citation4,Citation7,Citation9
Plasmid DNA vaccines represent an attractive platform because they are noninfectious, can induce both cellular and humoral immune responses against target antigens without inducing antivector responses that may limit boosting, and have been shown to be well tolerated and safe in numerous clinical trials. There are currently three pDNA vaccines licensed for veterinary useCitation12-Citation14 and several pDNA products for human use have reached Phase 2 or Phase 3 testing.Citation15-Citation18 A therapeutic CMV pDNA vaccine is advancing to Phase 3 testing for the prevention of CMV reactivation in hematopoietic stem cell transplant recipients; this vaccine consists of a poloxamer-formulated bivalent product containing plasmids expressing CMV gB and phosphoprotein 65 (pp65).Citation17,Citation19-Citation21 The vaccine provided significant reductions in several measures of CMV viremia in addition to marked increases in pp65 T-cell frequencies after transplantation in a recently completed Phase 2 proof-of-concept study.Citation17 While this vaccine represents an encouraging prospect for controlling CMV replication in transplant recipients, a different formulation may be important for preventing congenital CMV infection, particularly one which may maximize generation of antibody responses.
In this report, we evaluate the effect of different formulations, immunization routes, delivery devices, and vaccination schedules on the immunogenicity of pDNA-based CMV vaccines in rabbits and mice. Studies primarily focus on achieving high antibody responses with a gB-encoding plasmid vaccine, but also test a bivalent product incorporating pp65 to enhance the breadth of the immune responses to the vaccine. Two formulations were evaluated because they have previously demonstrated: (1) enhanced immune responses compared with PBS formulations in animal models; (2) favorable safety, tolerability, and both humoral and cellular immunogenicity profiles with plasmids in Phase 1 and/or Phase 2 clinical trials; and (3) feasibility for development as single vial formulations. One of the formulations evaluated includes CRL1005/BAK; a poloxamer CRL1005 combined with benzalkonium chloride (BAK).Citation17,Citation19-Citation21 The second formulation includes a cationic lipid-based adjuvant; Vaxfectin®.Citation22-Citation25
Results
Evaluation of formulations, routes and devices
For practical reasons, ID administration and delivery devices were tested in rabbits rather than in mice. The two formulations evaluated were pDNA formulated either with CRL1005/BAK or Vaxfectin®. Compared with PBS-formulated vaccines, CRL1005/BAK has previously been shown to enhance immune responses with a bivalent CMV vaccine,Citation20 and Vaxfectin® has been shown to enhance the immunogenicity of several antigens encoded by pDNA.Citation24 In order to achieve sufficient statistical power, at least six rabbits were included per group.
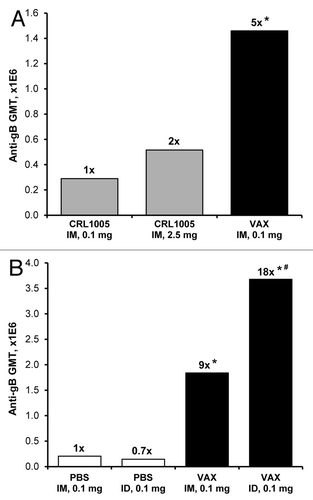
Both IM and ID immunization routes were evaluated with each formulation. In some groups, vaccines were injected using the Biojector® 2000 needle-free injection system, a device currently used to deliver licensed human vaccines. Immunization of rabbits with Vaxfectin®-formulated pDNA encoding CMV gB (VR-6365) resulted in significantly stronger antibody responses than obtained when the same or a 25-fold higher pDNA dose formulated with CRL1005/BAK was injected IM (), demonstrating a substantial dose-sparing effect. Furthermore, gB-specific antibody levels in rabbits vaccinated with Vaxfectin® formulations were 9- to 25-fold higher than obtained with PBS-formulated VR-6365 injected either IM or ID, respectively ().
Figure 1. Evaluation of vaccine formulations in rabbits. (A) On Day 0 and 21, rabbits (n = 6 per group) received unilateral IM (500 µl/leg) injections of either 0.1 mg or 2.5 mg of VR-6365 formulated with CRL1005/BAK (CRL1005) administered with Biojector® 2000. A third group received unilateral IM injections of 0.1 mg of VR-6365 formulated with Vaxfectin® (VAX) administered with needle and syringe. Day 42 serum samples were assayed for gB-specific antibody responses using ELISA. The bars represent geometric mean titers (GMT). Fold increases in antibody responses compared with the group immunized with 0.1 mg of CRL1005/BAK formulations are indicated above each group. * Significantly different from groups vaccinated with CRL1005/BAK formulations (p ≤ 0.01). (B) On Day 0 and 21, rabbits (n = 6 per group) received either unilateral IM (500 µl/leg) or ID (100 µl/site) injections of 0.1 mg VR-6365 formulated either with PBS or Vaxfectin® (VAX) administered with Biojector® 2000. Day 42 serum samples were assayed for gB-specific antibody responses using ELISA. The bars represent geometric mean titers (GMT). Fold increases in antibody responses compared with the group injected with PBS formulations IM are indicated above each group. * Significantly different from groups vaccinated with PBS formulations (p < 0.01). # Significantly different from group vaccinated with Vaxfectin® formulation injected IM (p < 0.05).
When mice (n = 15 per group) were injected IM with 2.5 µg of VR-6365 (1.25 µg pDNA/50 µl/leg) on Day 0, 20 and 126, and antibody responses were determined three weeks after the second and third immunizations, the results showed that formulating VR-6365 with Vaxfectin® produced a statistically significant (p < 0.001) 5- to 6-fold increase in gB-specific geometric mean titers (GMT) compared with VR-6365 formulated with PBS (Day 41: 33,779; range 6,400 to 102,400 vs. 7,352, range 800 to 25,600. Day 145: 93,360; range 25,600 to 409,600 vs. 16,127, range 6,400 to 102,400; GMT and range of titers in Vaxfectin® and PBS groups, respectively).
Since Vaxfectin®-formulated VR-6365 resulted in stronger antibody responses than obtained with PBS or CRL1005/BAK formulations, further evaluation of IM and ID immunization routes focused on Vaxfectin® formulations. Dose-response studies in rabbits showed a dose-dependent increase in gB-specific antibody responses, both when VR-6365 was administered either IM or ID with Biojector® 2000 () or when injected IM using needle and syringe (). With low vaccine doses, stronger antibody responses were obtained with ID immunization route than with IM route. When a 10 µg dose of Vaxfectin®-formulated VR-6365 was administered ID, significantly higher gB-specific responses were obtained than when the same dose was administered IM using Biojector® 2000, and were comparable to responses obtained with a 10-fold higher dose injected IM with needle and syringe (). With the 100 µg dose of Vaxfectin®-formulated VR-6365 administered with Biojector® 2000, however, IM and ID immunization routes resulted in comparable gB-specific responses (). One potential drawback associated with ID immunization is the relatively small volume that can be administered at one site, thus limiting the amount of vaccine delivered with a single ID injection. In rabbits and humans, ID injections are restricted to approximately 100 µL per site, whereas 1 mL volume can be administered IM as a single injection. When rabbits were immunized using a single ID (100 µg vaccine per 100 µL) or IM (500 µg vaccine per 500 µL) injection given with Biojector® 2000, significantly higher gB-specific antibody levels were obtained using the IM route, which allowed delivery of a 5-times larger vaccine dose ().
Figure 2. Dose responses and time courses of antibody responses in rabbits immunized with Vaxfectin®-formulated vaccine injected either IM or ID. On Day 0 and 21 (arrows), rabbits (n = 6 per group) received either a single IM (500 µL per muscle) or ID (100 µL per site) injection of various doses of Vaxfectin®-formulated VR-6365 administered with Biojector® 2000 (B2000). One group of rabbits was vaccinated with a 100 µg dose of Vaxfectin®-formulated VR-6365 injected IM with needle and syringe. Day 42 serum samples were assayed for gB-specific antibody responses with ELISA using recombinant human CMV gB protein, and geometric mean titers (GMT) for each group are shown in panel (A). Temporal changes in gB-specific antibody responses in select groups were analyzed using commercial ELISA plates pre-coated with CMV antigens, and the results are shown in panel (B). # Significantly different from group which received a 10 µg dose of vaccine IM with Biojector® 2000 (p < 0.01). * Significantly different from groups which received a 100 µg dose of vaccine either IM or ID with Biojector® 2000 (p < 0.02). + Significantly different from group which received a 100 µg dose of vaccine ID with Biojector® 2000 (p < 0.05).
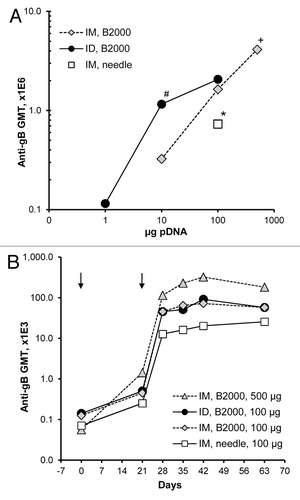
Table 1. gB-specific antibody responses in rabbits immunized with Vaxfectin®-formulated monovalent and bivalent vaccinesa
The Biojector® 2000 device was well tolerated when used either for IM or ID vaccine delivery in unanaesthetized rabbits. It did not increase antibody responses compared with needle injections when a 100 µg dose of VR-6365 formulated with PBS was delivered IM (data not shown). When VR-6365 was formulated with Vaxfectin® and injected IM using Biojector® 2000, however, gB-specific antibody levels were 2-fold higher (p = 0.019, Wilcoxon rank sum test) compared with those obtained following needle and syringe injections (), demonstrating that the use of Biojector® 2000 produced a modest but significant enhancement in gB-specific antibody responses with Vaxfectin® formulations. Time course studies in rabbits with Vaxfectin®-formulated VR-6365 demonstrated a substantial increase in antibody responses after boost injections, and that gB-specific antibody levels were sustained at these levels at least until Day 63, i.e., for 42 d after the second injections ().
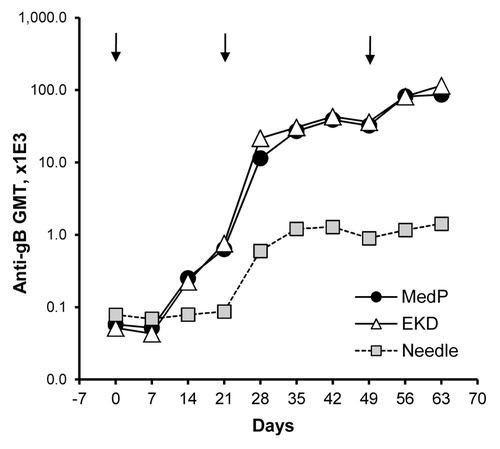
A second pDNA delivery method, electroporation (EP), was evaluated to explore whether a more complicated vaccine administration approach may be warranted by providing enhanced immunogenicity over formulated pDNA injected with or without Biojector® 2000. Intramuscular injections of a 500 µg dose of PBS-formulated VR-6365 followed by EP resulted in accelerated kinetics of antibody responses, and increased gB-specific antibody levels approximately by 30-fold at Day 42, compared with IM injection without EP (). The third EP-assisted immunization resulted in a further 2- to 3-fold increase in antibody responses and, at Day 63, gB-specific antibody levels in electroporated groups were 60- to 80-fold higher than in rabbits vaccinated using conventional needle and syringe injections without EP. Constant-voltage and constant-current EP devices resulted in a comparable enhancement in antibody responses (). However, similar gB-specific antibody levels were obtained in rabbits with three vaccinations with a 500 µg dose of PBS-formulated VR-6365 injected IM with needle and syringe followed by EP (), compared with levels obtained with just two vaccinations with a 100 µg or 500 µg dose of Vaxfectin®-formulated VR-6365 injected IM using Biojector® 2000 without EP ().
Figure 3. Time courses of antibody responses in rabbits receiving electroporation-assisted vaccination with PBS-formulated vaccine. On Day 0, 21 and 49 (arrows), rabbits (n = 6–12 per group) received a single IM injection of 500 µg of VR-6365 in 0.5 mL PBS administered with needle and syringe. After injections, muscles were electroporated using either a constant-voltage (MedPulser® DNA Delivery System, MedP) or a constant-current (Electrokinetic Device, EKD) device. Vaccine in the control group was administered without electroporation (Needle). Temporal changes in gB-specific antibody responses were analyzed using commercial ELISA plates pre-coated with CMV antigens, and geometric mean titers (GMT) for each group are shown.
Bivalent CMV gB+pp65 vaccine
A prophylactic vaccine to protect against congenital CMV infection will likely require not only gB, but also one or more additional antigens to maximize efficacy. Plasmid VR-6368 encoding the CMV major tegument phosphoprotein pp65 has previously been tested in animal models and in transplant recipients, and found to be a highly immunogenic T cell antigen.Citation17,Citation20 Immune responses to gB and pp65 antigens were therefore characterized when administered as a bivalent product formulated with Vaxfectin®.
Injecting rabbits with a 1 mg dose of Vaxfectin®-formulated bivalent vaccine, consisting of 1:1 mass ratio of plasmids VR-6365 and VR-6368 encoding CMV gB and pp65, respectively, resulted in significantly higher gB-specific antibody responses than obtained with a 0.1 mg dose of the bivalent vaccine ().
Mice were used to assess antibody as well as T-cell responses that were induced with the bivalent vaccine. Injecting mice with Vaxfectin®-formulated vaccine resulted in a statistically significant 4-fold increase in gB-specific antibody responses at Day 42 compared with mice injected with PBS-formulated vaccine (). After Day 42, antibody responses in mice injected with Vaxfectin® formulations declined, but remained higher than in mice injected with PBS formulations. At Day 208, i.e., approximately six months after the second injections, gB-specific responses in mice administered the Vaxfectin®-formulated vaccine were still 2-fold higher than in mice administered the PBS-formulated vaccine ().
Figure 4. Durability of immune responses in mice. On Day 0 and 21, mice received IM injections with needle of a bivalent vaccine consisting of 2.5 µg of VR-6365 encoding gB and 2.5 µg of VR-6368 encoding pp65 formulated either with PBS or Vaxfectin® (VAX). (A) Day 42 (n = 12) and 208 (n = 6) serum samples were assayed for gB-specific antibody responses with ELISA using recombinant human CMV gB protein. The bars represent geometric mean titers (GMT). Fold increases in antibody responses compared with the group immunized with PBS formulations are indicated above each Vaxfectin® group. * Significantly different from group vaccinated with PBS formulations (p < 0.001). (B) On Day 42 (n = 6) and 208 (n = 6), gB- (hatched bars) and pp65-specific (crosshatched bars) T-cell responses in mice immunized with Vaxfectin®-formulated vaccine were measured using the IFN-γ ELISPOT assay. Data are presented as the number of antigen-specific IFN-γ producing T-cells, designated as spot forming units (SFU) per million splenocytes (SFU/106 cells).
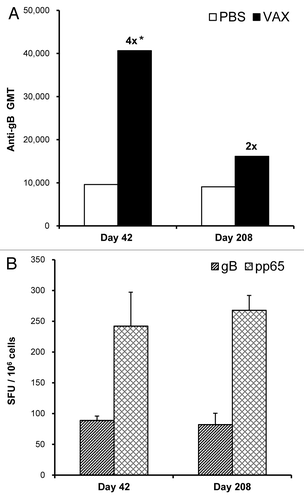
The number of antigen-specific IFN-γ producing T-cells was comparable in mice injected either with Vaxfectin® or PBS formulations as measured by an IFN-γ ELISPOT assay (data not shown). When mice were injected on Day 0 and 21, and T-cell responses were measured on Day 42 and 208, i.e., three weeks and approximately six months after the second injections, respectively, the results showed no apparent decline in the number of gB- or pp65-specific IFN-γ secreting splenocytes (), suggesting that immunization with the bivalent CMV pDNA vaccine resulted in durable cellular immune responses.
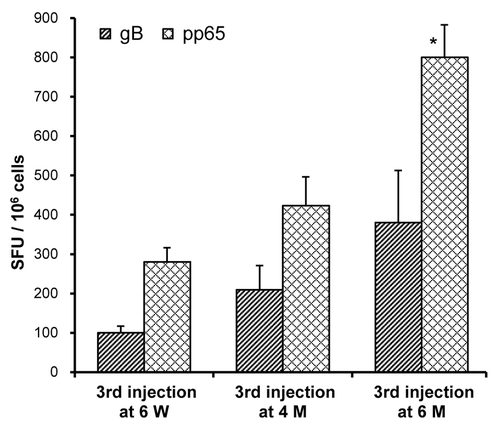
In order to assess the effect of different injection regimens on immune responses, mice were injected with Vaxfectin®-formulated bivalent vaccine on Day 0 and 21. A third injection was administered either at six weeks, at four months or at six months. ELISPOT assays performed three weeks after the third injection showed that increasing the time interval between the second and the third injection enhanced cellular immune responses, as determined by comparing the number of antigen-specific IFN-γ producing T-cells (). A similar trend was observed with the PBS-formulated bivalent vaccine (data not shown). Increasing the time interval between the second and the third injection did not appear to potentiate antibody responses in these mice since gB-specific antibody titers were comparable in serum samples collected either three weeks after the second or the third dose. However, the third injection of Vaxfectin®-formulated bivalent vaccine still induced significantly higher antibody responses than obtained with three injections with PBS-formulated vaccine (p < 0.05, data not shown).
Figure 5. Effect of immunization regimen on T-cell responses in mice. On Day 0 and 21, mice (n = 5–6 per group) received IM injections with needle of a bivalent vaccine consisting of 2.5 µg of VR-6365 encoding gB and 2.5 µg of VR-6368 encoding pp65 formulated with Vaxfectin®. The third immunization was performed either on Day 42, i.e., at six weeks (6 W), on Day 126, i.e., at four months (4 M) or on Day 188, i.e., at six months (6 M). Three weeks after the third immunization, gB- (hatched bars) and pp65-specific (crosshatched bars) T-cell responses were measured using the IFN-γ ELISPOT assay. Data are presented as the number of antigen-specific IFN-γ producing T-cells, designated as spot forming units (SFU) per million splenocytes (SFU/106 cells). * Significantly different from groups which received the third immunization either at six weeks or at four months (p < 0.01).
Single-vial Vaxfectin® formulation
In the studies described thus far, Vaxfectin® formulations were prepared as multiple-vial (MV) formulations. Multiple-vial vaccines are adequate for early phase clinical product testing despite the requirements of several mixing steps by a pharmacist prior to administration. Because this format is clearly not the preferred product configuration, a single-vial (SV) formulation was developed, that could be filled in advance and stored frozen until the day of use (see Materials and Methods). Comparability analysis of the MV and SV formulations included tests for appearance, pH, pDNA concentration, concentration of the GAP-DMORIE and DPyPE lipids, determination of the percent supercoiled pDNA by HPLC, analysis of in vitro transcriptional expression using real-time polymerase chain reaction assay, and particle size analysis. These tests demonstrated that the two formulations were analytically comparable (data not shown). The MV and SV vaccine formulations were evaluated for immunogenicity by administering each formulation IM on Days 0 and 21 to rabbits (unilateral injections into vastus lateralis; 1 mg in 1 mL per muscle), and antibody responses in Day 42 serum samples were analyzed. The serum gB-specific antibody titers in the group administered the MV formulation ranged from 102,400 to 1,638,400 with a geometric mean titer of 631,690, compared with the group administered the SV formulation, which ranged from 204,800 to 1,638,400 with a geometric mean titer of 688,862. The results for the SV formulation were not significantly different from those obtained with the MV formulation (p = 0.782; Wilcoxon rank-sum test), demonstrating that similar antibody responses were obtained in rabbits with the two formulations.
Repeat-dose toxicology studies
Both Vaxfectin®-formulated bivalent vaccines (see Materials and Methods) showed a low potential for toxicity and were judged as well-tolerated. The major test article-related findings, as would be expected for adjuvanted vaccines, were localized inflammatory reactions at injection sites. This was observed macroscopically as erythema and/or edema, and microscopically as mononuclear and/or polymorphonuclear cell infiltrates. Both findings were limited in scope and resolved with time (within a few days in the case of the macroscopic observations). Increases in a limited set of systemic markers (e.g., circulating leukocytes and serum C-reactive protein) reflected the local inflammatory responses and, like those reactions, were both limited in scope and transient.
The extent of test article-related inflammation was for the most part equivalent for both formulations tested. We had previously tested a MV formulation of 1 mg pDNA/1 mg Vaxfectin® administered using a Biojector® 2000 device in a GLP repeat-dose toxicology study and found a comparable safety profile (data not shown). The lowest dose formulation (1 mg/1 mg) did show the greatest injection site reactogenicity among the three formulations, but this was likely due to the slight physical trauma associated with needle free delivery observed in other GLP toxicology studies.Citation26 The favorable safety data generated from these doses would support human testing at up to any of the pDNA/lipid doses tested here.
Discussion
Currently no vaccine or treatment is available to prevent congenital CMV infection. It has become increasingly clear that a successful vaccine against congenital CMV must be effective not only in seronegative women experiencing primary infection, but also in seropositive women experiencing nonprimary infections because the latter accounts for most disease burden.Citation27 A recent study conducted in CMV seropositive women immunized at 0, 1, and 6 mo with recombinant gB protein adjuvanted with MF-59 demonstrated that boosting of naturally-acquired antibody and CD4+ T-cell responses can be achieved.Citation28 This same vaccine was found to provide 50% efficacy against primary maternal CMV infection in seronegative women,Citation11 indicating both the importance of including gB in a CMV vaccine and the need for incorporating one or more additional antigens. While the efficacy endpoint in the study was maternal infection,Citation11 an appropriate endpoint for an ultimate pivotal trial remains to be definedCitation7 and would ultimately need to consider both seronegative and seropositive subjects.
In this report, the effect of different immunization routes, delivery devices and formulations on the immunogenicity of pDNA-based CMV vaccines was evaluated in rabbits and mice. The study primarily used gB as a model antigen, as preclinical and clinical data have demonstrated the importance of gB antibody responses in neutralizing entry of CMV into fibroblasts and, therefore, gB quite likely will be one of the components included in a congenital CMV vaccine. A prophylactic CMV vaccine may have to generate antibody responses that also neutralize viral entry into cell types other than fibroblasts, such as epithelial cells.Citation29,Citation30 Other antigens, in addition to gB, may thus have to be evaluated, either to broaden the humoral immune responses and/or to induce T-cell responses; vaccination of guinea pigs with UL83 antigen alone, a guinea pig homolog of human CMV pp65 phosphoprotein, has provided protection in a guinea pig CMV challenge model.Citation31 Antigen selection for the final multivalent CMV vaccine candidate, however, was beyond the scope of this study.
Most inactivated and subunit vaccines licensed in the United States are administered IM, while most live attenuated vaccines are given by the subcutaneous route; there are relatively few vaccines administered via ID, intranasal or oral routes.Citation32 In recent years, skin has gained interest as delivery site for both protein and DNA vaccines because it is easily accessible and contains a large number of professional antigen presenting cells (APCs). A number of clinical trials have demonstrated that ID delivery of a reduced dose of licensed influenza and rabies vaccines have resulted in equivalent or better immune responses compared with the standard dose delivered by the IM route.Citation32-Citation34 Some trials, however, have shown mixed results with no consistent benefit demonstrated with the ID delivery route. Making a conclusion whether one immunization route is better than the other is further complicated by the fact that only few studies have directly compared identical antigen amounts delivered by the ID and the IM routes.Citation35,Citation36 In this study, IM and ID immunization routes were evaluated in rabbits; a subcutaneous route was not explored since this is the least effective for pDNA-based vaccines.Citation37,Citation38 Significantly higher antibody titers were obtained in rabbits when a low dose of Vaxfectin®-formulated pDNA vaccine was injected ID, compared with the same dose administered IM. In fact, a 10-fold dose-sparing effect was observed with a low vaccine dose administered ID. With a high vaccine dose, however, better antibody responses were obtained with the IM route, which allowed injection of a larger volume than could be administered at a single ID site. While the data presented here with needle or needle-free injection suggest that ID is a viable route for pDNA vaccination, the IM route offers wider dosing range options and a simpler injection procedure.
The use of ID immunization route also introduces challenges for vaccine development. In addition to being more technically demanding to administer by needle and syringe, ID vaccines are constrained by smaller volumes delivered to a single site (usually 100 µL) and must therefore contain more concentrated formulations. ID immunization can also carry an increased incidence of local adverse events,Citation36 which may be of further concern if novel adjuvants are used. In order to make dermal vaccine administration more practical, several devices and delivery technologies are being evaluated, including needle-free jet injectors, microneedle and micro-injection systems, particle-mediated epidermal delivery devices, as well as transcutaneous patches.Citation32,Citation33,Citation39-Citation42
Needle-free jet injectors force liquid through a tiny orifice to create a fine, high-pressure cutting stream that penetrates the skin to deliver vaccine to ID, IM and/or subcutaneous locations. Compared with needle and syringe injections, jet injectors have been shown to enhance the immunogenicity of pDNA vaccines in preclinical studies,Citation22,Citation32,Citation43 possibly by exposing more APCs to the antigens due to better tissue dispersion of the vaccine, combined with increased transfection and antigen production.Citation44,Citation45 In the current study, Vaxfectin®-formulated VR-6365 injected IM using Biojector® 2000 increased gB-specific antibody levels in rabbits 2-fold compared with needle and syringe injections. Several Phase 1 trials have tested nonadjuvanted pDNA vaccines administered only by the Biojector® 2000 device without direct comparison to needle deliveryCitation46; one Phase 1 trial reported improved immune responses with jet injection.Citation47 While the preclinical studies reported here were ongoing, Phase 1 trials with Vaxfectin®-formulated influenza H5 hemagglutinin pDNA vaccines administered IM either with needle and syringe or with Biojector® 2000 were initiated. The magnitude of HI titers and percentage of responders in these studies did not appear to be augmented by the use of the Biojector® 2000 compared with standard needle and syringe delivery; in addition, the reactogenicity rates appeared higher with the device.Citation25 A careful balance must therefore be achieved between enhanced immunological performance and acceptable reactogenicity profiles to warrant the use of a device in addition to an adjuvant.
Electroporation is another device-dependent strategy being explored to enhance the immunogenicity of pDNA-based vaccines by improving delivery to muscle myofibers. Applying brief electrical pulses to the tissue after pDNA injections is believed to induce temporary pores in the cell membrane, thus allowing the plasmid to enter cells more readily. This in turn may result in increased antigen production. In addition, tissue damage induced by EP and subsequent recruitment of APCs combined with enhanced inflammatory responses may contribute to the increased immunogenicity of pDNA vaccines delivered by EP as compared with conventional needle and syringe injections.Citation48-Citation51 In the current study, EP-assisted vaccination accelerated the kinetics of antibody responses and substantially increased gB-specific antibody levels in rabbits compared with PBS-formulated VR-6365 injected IM without EP, in agreement with previous studies performed using other antigens.Citation48-Citation51 Constant-voltage and constant-current devices resulted in a comparable enhancement in antibody responses. Interestingly, similar gB-specific antibody levels were obtained in rabbits which received IM injections of PBS-formulated VR-6365 followed by EP, compared with rabbits which received Vaxfectin®-formulated VR-6365 using Biojector® 2000, indicating that the use of EP may not result in substantially stronger responses than what can be achieved with adjuvant-formulated pDNA vaccine. Despite the data demonstrating the potential for EP to enhance the immunogenicity of PBS-formulated pDNA vaccines, there remain concerns regarding the cost, complexity and tolerability associated with its use, especially for indications that require immunizations of healthy subjects.
Vaxfectin® is a synthetic adjuvant with a straightforward, scalable synthesis of the two constituting lipids. Analytical assays have been developed to ensure lot-to-lot consistency during lipid manufacturing, as well as during complexation with pDNA to create drug product. Nonclinical studies conducted in a variety of animal models using multiple disease indications and plasmid constructs have demonstrated that, relative to nonadjuvanted pDNA, Vaxfectin® can significantly increase antibody and T-cell responses to pDNA-expressed antigens, provide dose-sparing, and enhance protection by increasing survival or reducing viral load in challenge models.Citation24 Unlike electroporation and jet injectors, Vaxfectin® does not increase transgene expression in muscle when formulated with pDNA.Citation22,Citation52 In this regard, Vaxfectin® formulations also differ from CRL1005/BAK formulations, which have been shown to increase transgene expression in the injected muscle, compared with PBS-formulated pDNA, and thus may enhance immunogenicity of pDNA vaccines by enhancing the delivery of the pDNA into cells.Citation19 Compared with PBS-formulated vaccines, Vaxfectin® formulations have been shown to induce a transient elevation in local and systemic levels of certain cytokines and chemokines in mice, and to increase cellular infiltrates in the injected muscle.Citation23,Citation52,Citation53
In this study, cellular immune responses in mice injected with Vaxfectin®-formulated bivalent vaccine were enhanced when the time interval between the second and the third dose increased. This finding is in an agreement with previous reports demonstrating that the immunogenicity of pDNA vaccines improved in miceCitation54,Citation55 and nonhuman primatesCitation38,Citation56 by lengthening the rest period between injections.
In GLP-compliant toxicology and biodistribution studies, Vaxfectin®-formulated trivalent H5N1 influenza pDNA vaccine displayed a well-tolerated safety profile in rabbits.Citation26 The primary observations made were mild inflammatory responses at injection sites, indicative of a local reactogenicity, accompanied by a limited set of associated clinical pathology changes. These responses resolved in a few days to weeks, and were consistent with the responses expected after IM vaccination. Biodistribution and integration studies followed the fate of Vaxfectin®-adjuvanted pDNA vaccines using quantitative PCR. Plasmids were primarily localized at injection sites, where levels declined steadily over a 2-mo period. The risk of genomic integration was found to be negligible.Citation26
To date, Vaxfectin® has been tested clinically in nearly 100 normal healthy subjects with pDNA vaccines expressing influenza antigens with doses escalating from 0.1 to 1 mg of total pDNA per injection twice over three weeks. In these Phase 1 trials, Vaxfectin® exhibited a favorable safety and tolerability profile; no vaccine-related clinical or laboratory serious adverse events were reported and all events resolved within a few days. Furthermore, a Vaxfectin®-formulated influenza H5 hemagglutinin pDNA vaccine elicited both functional antibody responses in 47–67% of the subjects after two doses, and positive IFN-γ-producing T-cell responses in 75–100% of the subjects.Citation25 These serological results were in the range of those reported for protein-based H5 vaccines.Citation57 It is conceivable that plasmids encoding more immunogenic antigens, given at higher doses (> 1 mg) and more times (≥ 3), may afford even higher immune response rates and must be evaluated on an individual basis.
Because of the tropism of human CMV and therefore the lack of any animal challenge model to test human vaccine candidates, one limitation of the current studies was that immunogenicity, not protection, was the endpoint examined. However, immunogenicity is a practical endpoint for evaluation because of the large number of different variables to test including formulation, route, devices, etc. Guinea pigs can be infected by guinea pig CMV (gpCMV) but this would permit testing only of gpCMV antigens, not human CMV antigens. Plasmid DNA vaccines encoding the gpCMV homologs of human CMV gB and pp65 antigens have been tested for protective efficacy as preconception vaccines in guinea pigs infected during the third trimester with gpCMV.Citation58 Improved pregnancy outcomes, including reductions in maternal viral loads, reduced pup mortality, and/or reduced congenital infection in surviving pups, have been demonstrated using gB pDNA delivered by gene gun,Citation59 adjuvanted recombinant gB protein,Citation60 or a pp65-expressing alphavirus-based vaccine.Citation31 Importantly, the latter study indicates that a vaccine eliciting only T-cell responses in the absence of neutralizing antibodies favorably impacts congenital CMV transmission. Collectively, these preclinical proof-of-concept studies using either gB or pp65 alone suggest improved vaccine performance may be achievable with a bivalent combination formulated with an adjuvant which can augment both antibody and Th1 responses, such as Vaxfectin®.Citation24
In this report, significantly higher antibody responses were obtained in rabbits and mice with Vaxfectin®-formulated pDNA encoding CMV gB, compared with PBS- or poloxamer-based formulations. Although the needle-free injection system Biojector® 2000 and electroporation devices enhanced antibody responses, the enhancement was marginal compared with responses obtained with Vaxfectin®-formulated pDNA injected IM with a needle. Similar antibody levels were obtained with two Vaxfectin® formulations; a single-vial frozen formulation prepared in advance in bulk, and a multiple-vial formulation prepared on the day of immunization. A single-vial product configuration is ideal not only because of decreased production costs, but because it maximizes final product consistency while simplifying vaccine administration. In a GLP-compliant repeat-dose toxicology study conducted in rabbits, single-vial Vaxfectin®-formulated vaccines, containing pDNA and Vaxfectin® up to 4.5 mg and 2mg/injection, respectively, showed a favorable safety profile and were judged as well-tolerated, adding to the growing safety database for Vaxfectin®-formulated pDNA-based vaccines. These preclinical results suggest that Vaxfectin®-formulated pDNA vaccine injected IM with conventional needle and syringe may provide a practical and cost-effective approach for administering a vaccine targeting congenital CMV infection.
Material and Methods
Materials and formulations
Vaxfectin® consists of a 1:1 molar mixture of a cationic lipid GAP-DMORIE and a neutral co-lipid DPyPE. GAP-DMORIE [(± )-N-(3-aminopropyl)-N,N-dimethyl-2,3-bis(cis-9-tetradecenyloxy)-1-propanaminium bromide] was synthesized as previously described.Citation22 DPyPE (1,2-diphytanoyl-sn-glycero-3- phosphoethanolamine) was purchased from Avanti Polar Lipids, Inc. Vaxfectin® was prepared as dried lipid film by mixing equimolar chloroform solutions of GAP-DMORIE and DPyPE. The chloroform was evaporated under a stream of nitrogen and the lipid-containing glass vials were placed under vacuum overnight to remove solvent traces. For most rabbit studies and all mouse studies, Vaxfectin® formulations were prepared on the day of administration as multiple-vial (MV) formulations, as previously described.Citation22,Citation61 Each vial, containing 2.18 mg of Vaxfectin®, was resuspended by adding 1 mL of 0.9% sodium chloride and the vial was vortexed for 5 min. The Vaxfectin® liposomes were then added to an equal volume of pDNA prepared in another vial at 2 mg/mL in 0.9% sodium chloride, 20 mM sodium phosphate, pH 7.2, and the vial was mixed by gentle inversion. The final formulation contained 1 mg/mL pDNA and 1.09 mg/mL Vaxfectin® in 0.9% sodium chloride, 10 mM sodium phosphate at a final pDNA (phosphate):cationic lipid molar ratio of 4:1. The MV formulations were used for immunizations either at the 1 mg pDNA/mL concentration, or were further diluted to the required concentration in phosphate-buffered saline (PBS, 0.9% sodium chloride + 10 mM sodium phosphate, pH 7.2).
Immunogenicity of single-vial (SV) Vaxfectin® formulations, which were prepared in advance and stored frozen until the day of administration, was evaluated in rabbits. A Vaxfectin® dried lipid film was resuspended in 20 mM sodium chloride and then formulated with the pDNA in 20 mM sodium chloride, 20 mM sodium phosphate and 17% sucrose (1:1 volume of Vaxfectin®:pDNA). The resulting final formulation contained 1 mg/mL pDNA and 1.09 mg/mL Vaxfectin® in 20 mM sodium chloride, 10 mM sodium phosphate, pH 7.2, and 8.5% sucrose. Thus the components of the SV formulation were similar to those in the MV formulation, with the exception of two modifications: sucrose was added as a cryoprotectant to prevent aggregation following thawing of the frozen formulation, and the amount of sodium chloride was decreased from 150 mM to 20 mM to maintain isotonicity of the formulation. Single-use vials were filled with aliquots of the formulations and frozen between -10°C and -30°C until the day of use. The MV and SV vaccine formulations were evaluated for immunogenicity by administering each formulation intramuscularly with needle and syringe on Days 0 and 21 to rabbits (8 rabbits per group, 1 mg pDNA/1 mL/muscle), and antibody responses in Day 42 serum samples were analyzed.
The nonionic tri-block copolymer (poloxamer) CRL1005 was obtained from Organichem, and the cationic surfactant BAK (50% solution, BTC 50® NF) from Ruger Chemical Co., Inc. Poloxamer vaccine formulations (CRL1005/BAK) were prepared as previously described.Citation19,Citation20 Briefly, the required concentration of pDNA in PBS was stirred on ice and the required amount of poloxamer CRL1005 was added using a positive displacement pipette. The solution was stirred on ice until the poloxamer dissolved and then the required concentration of BAK dissolved in PBS was added. The solution was thermocycled through the cloud point (7–12°C) several times to ensure homogeneity, filter sterilized through a Millipore Steriflip disposable vacuum filtration system (Millipore) at 4°C, and stored frozen. Prior to injections, the vaccine was thawed at ambient temperature and, if required, diluted to the required pDNA concentration with PBS above the cloud point of CRL1005.
Plasmid constructs
Plasmid constructs expressing human CMV antigens were produced based on the published viral sequences of strain AD169, as previously described.Citation20 VR-6365 encodes the extracellular domain (amino acids 1–713) of the human CMV gB antigen lacking the transmembrane and cytoplasmic domains of the full-length glycoprotein to facilitate secretion, but retains the native signal sequence. VR-6368 encodes a mutated form of the CMV pp65 antigen in which amino acids 435RKRK438 have been deleted to abolish putative kinase activity. Both antigens were cloned into VR-10551 mammalian expression vector, which contains the human CMV immediate early 1 promoter-enhancer, CMV intron A, and a synthetic rabbit β-globin consensus poly A signal.Citation20
In vivo procedures in mouse studies
All animal procedures were approved by the Vical Institutional Animal Care and Use Committee (IACUC), and complied with the standards set forth in the ‘Guide for the Care and Use of Laboratory Animals’ (Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council National Academy Press, Washington, D. C., 1996) and the Animal Welfare Act and Animal Care Regulations (http://www.aphis.usda.gov/ac/publications.html).
BALB/cByJ mice (6- to 10-week old females, Jackson Laboratories) were immunized by intramuscular (IM) injection. Bilateral IM injections into the quadriceps muscle (50 µl/leg) were performed with needle and syringe as previously described.Citation22,Citation62 Mice were bled via the ophthalmic venous plexus prior to the first injection, and periodically during the study as indicated. Sera were stored at -20°C until assayed for gB-specific antibodies by enzyme-linked immunosorbent assay (ELISA). Approximately three weeks after the last immunization, groups of mice were euthanized by injection with Sleepaway (Fort Dodge Animal Health), splenocytes were harvested and antigen-specific T-cell responses were measured by interferon-gamma (IFN-γ) enzyme-linked immunospot (ELISPOT) assay.
In vivo procedures in rabbit studies
Injections with needle or Biojector® 2000
New Zealand White rabbits (1.5–3.5 kg females, Harlan, Indianapolis, IN), received VR-6365 formulated either with PBS, CRL1005/BAK or Vaxfectin®. Unilateral IM injections in the right quadriceps (500 µL per muscle) were administered either with needle and syringe or with a Biojector® 2000, a CO2-powered needle-free injection system (Bioject Medical Technologies, Inc., Tualatin, OR). Biojector syringe #2 was used for the IM injections with the Biojector® 2000. Intradermal (ID) vaccinations with the Biojector® 2000 were performed in the skin area overlaying the quadriceps (1 site per rabbit, 100 µL per site) using Biojector syringe #2 fitted with an intradermal spacer. Before injections, the injection site was shaved and cleaned with alcohol. Identical boost injections were administered on Day 21. Rabbits were bled from the ear vein on Day -1 (prebleed), 21 and 42, and serum anti-gB titers were determined with ELISA.
Electroporation (EP)-assisted vaccinations
New Zealand White rabbits (1.8–2.8 kg females) were anesthetized using a Ketamine/Xylazene cocktail administered intravenously. The injection site was shaved and cleaned. Rabbits received a single unilateral IM injection of 500 µg VR-6365 in 0.5 mL PBS administered in the right triceps brachii caput longum (TBCL) muscle using a 1cc tuberculin syringe fitted with a 21G 2” needle on Day 0. Identical vaccinations were performed on Day 21 in the left and on Day 49 in the right TBCL muscle. Approximately 80 sec after the vaccine was injected, muscles were electroporated using either a constant-voltage (MedPulser® DNA Delivery System, Inovio Biomedical Corporation) or a constant-current (ADViSYS electrokinetic device, EKD, ADViSYS, Inc.) device. Vaccine in the control group (no EP) was administered in the TBCL muscle of anaesthetized rabbits using similar 1cc tuberculin syringes fitted with a 21G 2” needle.
With MedPulser®, two constant-voltage square electric pulses of 106 V of 60 msec duration each (nominal field strength 246 V/cm) were administered using 0.5 cm square gold plated four needle arrays (needle length 1.0 cm). With EKD, sterile 5-needle electrode arrays, in which the stainless steel electrodes were 1.0 cm apart in diameter, were used for EP. The guide disk of the array was adjusted so that the penetration depth of the electrodes was approximately 1.0 cm. After the array was inserted into the muscle, vaccine was administered through a central injection port located at the top of the array. The penetration depth of the injection needle was adjusted so that the bevel of the needle did not extend beyond the electrode array. The injection needle was removed, and the muscles were electroporated with three 0.6 A pulses (52 ms/pulse, 1 sec between pulses, constant-current pulse pattern #5).Citation63,Citation64 With both devices, a new electrode array was used for each rabbit muscle.
Repeat-dose toxicology studies
To assess the toxicity potential of SV Vaxfectin®-formulated vaccines, a good laboratory practices (GLP)-compliant repeat-dose toxicology study was conducted in New Zealand White rabbits (2.7–3.5 kg, n = 20 per group, evenly divided by sex). Rabbits received a bivalent vaccine (1:1 mass ratio of VCL-6365 and VCL-6368) formulated with Vaxfectin®, or PBS as a control, delivered as 1 mL unilateral IM injections with needle and syringe on Days 0, 21, and 42 (alternating limbs on subsequent injections). Two SV Vaxfectin® formulations were tested containing either 3 mg pDNA/2 mg Vaxfectin®, or 4.5 mg pDNA/1 mg Vaxfectin® (mg of total pDNA formulated with mg of total lipid, respectively). Animals were followed for up to 85 d and evaluated for clinical signs (including injection site reactogenicity), ophthalmology, body weight, food consumption, clinical pathology (hematology, coagulation and clinical chemistry), gross pathology (at necropsy), and histopathology as previously described.Citation26
gB antibody ELISAs
To detect serum gB-specific immunoglobulin G (IgG) antibodies, 96-well plates were coated overnight at 2–8°C with recombinant full-length human CMV gB protein purified from transfected Chinese hamster ovary cells (Austral Biologicals) at a concentration of 2 µg/mL. Antibody levels, reported as endpoint titers, were determined as previously described.Citation20 Serum gB antibodies were undetectable in all samples collected from rabbits and mice before vaccination (prebleeds) and tested at the starting dilution of 1:100.
Unless otherwise stated in the text, gB-specific antibody responses were determined using ELISA plates coated with recombinant human CMV gB protein as described above. Antibody responses in some serum samples were analyzed using ELISA plates included in the CMV IgG Enzyme Immunoassay Test Kits (BioCheck, Inc.). These commercially available ELISA plates precoated with human CMV antigens were less sensitive than ELISA plates coated with recombinant gB protein, and they were only used to monitor temporal changes in antibody responses in rabbits immunized with VR-6365.
IFN-γ ELISPOT assays
Three weeks after the last immunization, T-cell responses to CMV antigens in vaccinated mice were determined by quantifying the number of splenocytes secreting IFN-γ in response to antigen-specific stimulation, as previously described.Citation20 To quantify pp65-specific responses, splenocytes were seeded in quadruplicates at a density of 1x106 cells per well, and cells were stimulated with two separate pools of overlapping peptides (15mer peptides overlapping by 11 amino acids) that together spanned the entire pp65 protein. Separate plates were prepared for pp65 peptide pool I (consisting of peptides #1–68) and for pp65 peptide pool II (consisting of peptides #69–137). For gB-specific T-cell responses, splenocytes were stimulated either with gB peptide pool I (consisting of peptides #1–88, 15mer peptides overlapping by 11 amino acids) or peptide pool II (consisting of peptides #89–176). Final concentration of each peptide was 5 µg/mL, and culture media was supplemented with 1 U/mL of IL-2. Data were presented as the number of spot forming units (SFU) produced in response to antigen-specific stimulation per million cells plated (SFU/106 cells). Total antigen-specific T cell responses were derived by combining the spot counts obtained with peptide pool I with the counts obtained with pool II.
Statistical analysis
Statistical comparisons were performed using a non-parametric Wilcoxon rank-sum test (SAS version 9.1, SAS Institute). Differences were considered statistically significant when the p value was less than 0.05.
| Abbreviations: | ||
| APC | = | antigen presenting cell |
| B2000 | = | Biojector® 2000 |
| BAK | = | benzalkonium chloride |
| CMV | = | cytomegalovirus |
| EKD | = | electrokinetic device |
| ELISA | = | enzyme-linked immunosorbent assay |
| ELISPOT | = | enzyme-linked immunospot |
| EP | = | electroporation |
| gB | = | glycoprotein B |
| GLP | = | good laboratory practices |
| GMT | = | geometric mean titer |
| gpCMV | = | guinea pig cytomegalovirus |
| IACUC | = | Institutional Animal Care and Use Committee |
| ID | = | intradermal(ly) |
| IgG | = | immunoglobulin G |
| IM | = | intramuscular(ly) |
| IFN-γ | = | interferon-gamma |
| MV | = | multiple-vial |
| PBS | = | phosphate-buffered saline |
| pDNA | = | plasmid DNA |
| pp65 | = | phosphoprotein 65 |
| SFU | = | spot forming units |
| SV | = | single-vial |
| TBCL | = | triceps brachii caput longum |
Acknowledgments
We thank Dawn Bellinger and Thomas Hilgert for assistance with the animal studies, and Alice Chu for performing the statistical analysis. We also thank Bioject Medical Technologies for providing the Bioject® 2000 device and syringes for the study, as well as Inovio Biomedical Corporation and ADViSYS Inc. for supplying the electroporation devices and electrodes.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Staras SA, Dollard SC, Radford KW, Flanders WD, Pass RF, Cannon MJ. Seroprevalence of cytomegalovirus infection in the United States, 1988-1994. Clin Infect Dis 2006; 43:1143 - 51; http://dx.doi.org/10.1086/508173; PMID: 17029132
- Crough T, Khanna R. Immunobiology of human cytomegalovirus: from bench to bedside. Clin Microbiol Rev 2009; 22:76 - 98; http://dx.doi.org/10.1128/CMR.00034-08; PMID: 19136435
- Landolfo S, Gariglio M, Gribaudo G, Lembo D. The human cytomegalovirus. Pharmacol Ther 2003; 98:269 - 97; http://dx.doi.org/10.1016/S0163-7258(03)00034-2; PMID: 12782241
- Plotkin SA. Cytomegalovirus vaccines. In: Plotkin SA, Orenstein WA, Offit PA, eds. Vaccines: Saunders Elsevier, 2008:1147-54.
- Stratton KR, Durch JS, Lawrence RS. Vaccines for the 21st Century: A Tool for Decisionmaking. Washington, D.C.: Institute of Medicine, National Academy Press, 1999.
- Kenneson A, Cannon MJ. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol 2007; 17:253 - 76; http://dx.doi.org/10.1002/rmv.535; PMID: 17579921
- Adler SP. Human CMV vaccine trials: what if CMV caused a rash?. J Clin Virol 2008; 41:231 - 6; http://dx.doi.org/10.1016/j.jcv.2007.11.008; PMID: 18096431
- CDC. (Centers for Diseases Control and Prevention web site, last updated on July 28, 2010). Cytomegalovirus (CMV) and Congenital CMV Infection - Trends and Statistics. Retrieved January 17, 2012 from: http://www.cdc.gov/cmv/trends-stats.html.
- Arvin AM, Fast P, Myers M, Plotkin S, Rabinovich R, National Vaccine Advisory Committee. Vaccine development to prevent cytomegalovirus disease: report from the National Vaccine Advisory Committee. Clin Infect Dis 2004; 39:233 - 9; http://dx.doi.org/10.1086/421999; PMID: 15307033
- Britt WJ, Vugler L, Butfiloski EJ, Stephens EB. Cell surface expression of human cytomegalovirus (HCMV) gp55-116 (gB): use of HCMV-recombinant vaccinia virus-infected cells in analysis of the human neutralizing antibody response. J Virol 1990; 64:1079 - 85; PMID: 2154594
- Pass RF, Zhang C, Evans A, Simpson T, Andrews W, Huang ML, et al. Vaccine prevention of maternal cytomegalovirus infection. N Engl J Med 2009; 360:1191 - 9; http://dx.doi.org/10.1056/NEJMoa0804749; PMID: 19297572
- Cui Z. DNA vaccine. Adv Genet 2005; 54:257 - 89; http://dx.doi.org/10.1016/S0065-2660(05)54011-2; PMID: 16096015
- Kutzler MA, Weiner DB. DNA vaccines: ready for prime time?. Nat Rev Genet 2008; 9:776 - 88; http://dx.doi.org/10.1038/nrg2432; PMID: 18781156
- Liu MA. DNA vaccines: an historical perspective and view to the future. Immunol Rev 2011; 239:62 - 84; http://dx.doi.org/10.1111/j.1600-065X.2010.00980.x; PMID: 21198665
- Bedikian AY, Richards J, Kharkevitch D, Atkins MB, Whitman E, Gonzalez R. A phase 2 study of high-dose Allovectin-7 in patients with advanced metastatic melanoma. Melanoma Res 2010; 20:218 - 26; PMID: 20354459
- Chowdhery R, Gonzalez R. Immunologic therapy targeting metastatic melanoma: allovectin-7. Immunotherapy 2011; 3:17 - 21; http://dx.doi.org/10.2217/imt.10.89; PMID: 21174553
- Kharfan-Dabaja MA, Boeckh M, Wilck MB, Langston AA, Chu AH, Wloch MK, et al. A novel therapeutic cytomegalovirus DNA vaccine in allogeneic haemopoietic stem-cell transplantation: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Infect Dis 2012; 12:290 - 9; http://dx.doi.org/10.1016/S1473-3099(11)70344-9; PMID: 22237175
- Matijevic M, Hedley ML, Urban RG, Chicz RM, Lajoie C, Luby TM. Immunization with a poly (lactide co-glycolide) encapsulated plasmid DNA expressing antigenic regions of HPV 16 and 18 results in an increase in the precursor frequency of T cells that respond to epitopes from HPV 16, 18, 6 and 11. Cell Immunol 2011; 270:62 - 9; http://dx.doi.org/10.1016/j.cellimm.2011.04.005; PMID: 21550027
- Hartikka J, Geall A, Bozoukova V, Kurniadi D, Rusalov D, Enas J, et al. Physical characterization and in vivo evaluation of poloxamer-based DNA vaccine formulations. J Gene Med 2008; 10:770 - 82; http://dx.doi.org/10.1002/jgm.1199; PMID: 18425981
- Selinsky C, Luke C, Wloch M, Geall A, Hermanson G, Kaslow D, et al. A DNA-based vaccine for the prevention of human cytomegalovirus-associated diseases. Hum Vaccin 2005; 1:16 - 23; PMID: 17038834
- Wloch MK, Smith LR, Boutsaboualoy S, Reyes L, Han C, Kehler J, et al. Safety and immunogenicity of a bivalent cytomegalovirus DNA vaccine in healthy adult subjects. J Infect Dis 2008; 197:1634 - 42; http://dx.doi.org/10.1086/588385; PMID: 18444883
- Hartikka J, Bozoukova VV, Ferrari M, Sukhu L, Enas J, Sawdey M, et al. Vaxfectin enhances the humoral immune response to plasmid DNA-encoded antigens. Vaccine 2001; 19:1911 - 23; http://dx.doi.org/10.1016/S0264-410X(00)00445-X; PMID: 11228361
- Reyes L, Hartikka J, Bozoukova V, Sukhu L, Nishioka W, Singh G, et al. Vaxfectin enhances antigen specific antibody titers and maintains Th1 type immune responses to plasmid DNA immunization. Vaccine 2001; 19:3778 - 86; http://dx.doi.org/10.1016/S0264-410X(01)00090-1; PMID: 11395213
- Sullivan SM, Doukas J, Hartikka J, Smith L, Rolland A. Vaxfectin: a versatile adjuvant for plasmid DNA- and protein-based vaccines. Expert Opin Drug Deliv 2010; 7:1433 - 46; http://dx.doi.org/10.1517/17425247.2010.538047; PMID: 21118032
- Smith LR, Wloch MK, Ye M, Reyes LR, Boutsaboualoy S, Dunne CE, et al. Phase 1 clinical trials of the safety and immunogenicity of adjuvanted plasmid DNA vaccines encoding influenza A virus H5 hemagglutinin. Vaccine 2010; 28:2565 - 72; http://dx.doi.org/10.1016/j.vaccine.2010.01.029; PMID: 20117262
- Doukas J, Morrow J, Bellinger D, Hilgert T, Martin T, Jones D, et al. Nonclinical biodistribution, integration, and toxicology evaluations of an H5N1 pandemic influenza plasmid DNA vaccine formulated with Vaxfectin®. Vaccine 2011; 29:5443 - 52; http://dx.doi.org/10.1016/j.vaccine.2011.05.060; PMID: 21641955
- Schleiss MR. Could therapeutic vaccination of cytomegalovirus-seropositive persons prevent reinfection and congenital virus transmission?. J Infect Dis 2011; 203:1513 - 6; http://dx.doi.org/10.1093/infdis/jir144; PMID: 21592979
- Sabbaj S, Pass RF, Goepfert PA, Pichon S. Glycoprotein B vaccine is capable of boosting both antibody and CD4 T-cell responses to cytomegalovirus in chronically infected women. J Infect Dis 2011; 203:1534 - 41; http://dx.doi.org/10.1093/infdis/jir138; PMID: 21592981
- Heldwein EE, Krummenacher C. Entry of herpesviruses into mammalian cells. Cell Mol Life Sci 2008; 65:1653 - 68; http://dx.doi.org/10.1007/s00018-008-7570-z; PMID: 18351291
- Revello MG, Gerna G. Human cytomegalovirus tropism for endothelial/epithelial cells: scientific background and clinical implications. Rev Med Virol 2010; 20:136 - 55; http://dx.doi.org/10.1002/rmv.645; PMID: 20084641
- Schleiss MR, Lacayo JC, Belkaid Y, McGregor A, Stroup G, Rayner J, et al. Preconceptual administration of an alphavirus replicon UL83 (pp65 homolog) vaccine induces humoral and cellular immunity and improves pregnancy outcome in the guinea pig model of congenital cytomegalovirus infection. J Infect Dis 2007; 195:789 - 98; http://dx.doi.org/10.1086/511982; PMID: 17299708
- Lambert PH, Laurent PE. Intradermal vaccine delivery: will new delivery systems transform vaccine administration?. Vaccine 2008; 26:3197 - 208; http://dx.doi.org/10.1016/j.vaccine.2008.03.095; PMID: 18486285
- Hickling J, Jones R. Intradermal delivery of vaccines: a review of the literature and potential for development for use in low- and middle-income countries. Program for Appropriate Technology in Health (PATH) Report. Seattle, August 27, 2009: 94 pages. Available at: http://www.path.org/files/TS_opt_idd_review.pdf.
- Kenney RT, Frech SA, Muenz LR, Villar CP, Glenn GM. Dose sparing with intradermal injection of influenza vaccine. N Engl J Med 2004; 351:2295 - 301; http://dx.doi.org/10.1056/NEJMoa043540; PMID: 15525714
- Arnou R, Icardi G, De Decker M, Ambrozaitis A, Kazek MP, Weber F, et al. Intradermal influenza vaccine for older adults: a randomized controlled multicenter phase III study. Vaccine 2009; 27:7304 - 12; http://dx.doi.org/10.1016/j.vaccine.2009.10.033; PMID: 19849996
- Belshe RB, Newman FK, Wilkins K, Graham IL, Babusis E, Ewell M, et al. Comparative immunogenicity of trivalent influenza vaccine administered by intradermal or intramuscular route in healthy adults. Vaccine 2007; 25:6755 - 63; http://dx.doi.org/10.1016/j.vaccine.2007.06.066; PMID: 17692438
- Fynan EF, Webster RG, Fuller DH, Haynes JR, Santoro JC, Robinson HL. DNA vaccines: protective immunizations by parenteral, mucosal, and gene-gun inoculations. Proc Natl Acad Sci U S A 1993; 90:11478 - 82; http://dx.doi.org/10.1073/pnas.90.24.11478; PMID: 8265577
- McCluskie MJ, Brazolot Millan CL, Gramzinski RA, Robinson HL, Santoro JC, Fuller JT, et al. Route and method of delivery of DNA vaccine influence immune responses in mice and non-human primates. Mol Med 1999; 5:287 - 300; PMID: 10390545
- Arnou R, Eavis P, Pardo JR, Ambrozaitis A, Kazek MP, Weber F. Immunogenicity, large scale safety and lot consistency of an intradermal influenza vaccine in adults aged 18-60 years: Randomized, controlled, phase III trial. Hum Vaccin 2010; 6:346 - 54; http://dx.doi.org/10.4161/hv.6.4.10961; PMID: 20372053
- Dean HJ, Haynes J, Schmaljohn C. The role of particle-mediated DNA vaccines in biodefense preparedness. Adv Drug Deliv Rev 2005; 57:1315 - 42; http://dx.doi.org/10.1016/j.addr.2005.01.012; PMID: 15935876
- Frech SA, Kenney RT, Spyr CA, Lazar H, Viret JF, Herzog C, et al. Improved immune responses to influenza vaccination in the elderly using an immunostimulant patch. Vaccine 2005; 23:946 - 50; http://dx.doi.org/10.1016/j.vaccine.2004.06.036; PMID: 15603897
- Prausnitz MR, Mikszta JA, Cormier M, Andrianov AK. Microneedle-based vaccines. Curr Top Microbiol Immunol 2009; 333:369 - 93; http://dx.doi.org/10.1007/978-3-540-92165-3_18; PMID: 19768415
- Aguiar JC, Hedstrom RC, Rogers WO, Charoenvit Y, Sacci JB Jr., Lanar DE, et al. Enhancement of the immune response in rabbits to a malaria DNA vaccine by immunization with a needle-free jet device. Vaccine 2001; 20:275 - 80; http://dx.doi.org/10.1016/S0264-410X(01)00273-0; PMID: 11567774
- Babiuk S, Baca-Estrada ME, Foldvari M, Baizer L, Stout R, Storms M, et al. Needle-free topical electroporation improves gene expression from plasmids administered in porcine skin. Mol Ther 2003; 8:992 - 8; http://dx.doi.org/10.1016/j.ymthe.2003.09.008; PMID: 14664802
- Manam S, Ledwith BJ, Barnum AB, Troilo PJ, Pauley CJ, Harper LB, et al. Plasmid DNA vaccines: tissue distribution and effects of DNA sequence, adjuvants and delivery method on integration into host DNA. Intervirology 2000; 43:273 - 81; http://dx.doi.org/10.1159/000053994; PMID: 11251382
- Ledgerwood JE, Graham BS. DNA vaccines: a safe and efficient platform technology for responding to emerging infectious diseases. Hum Vaccin 2009; 5:623 - 6; PMID: 19779298
- Wang R, Epstein J, Baraceros FM, Gorak EJ, Charoenvit Y, Carucci DJ, et al. Induction of CD4(+) T cell-dependent CD8(+) type 1 responses in humans by a malaria DNA vaccine. Proc Natl Acad Sci U S A 2001; 98:10817 - 22; http://dx.doi.org/10.1073/pnas.181123498; PMID: 11526203
- Bodles-Brakhop AM, Heller R, Draghia-Akli R. Electroporation for the delivery of DNA-based vaccines and immunotherapeutics: current clinical developments. Mol Ther 2009; 17:585 - 92; http://dx.doi.org/10.1038/mt.2009.5; PMID: 19223870
- Luxembourg A, Evans CF, Hannaman D. Electroporation-based DNA immunisation: translation to the clinic. Expert Opin Biol Ther 2007; 7:1647 - 64; http://dx.doi.org/10.1517/14712598.7.11.1647; PMID: 17961089
- Prud’homme GJ, Glinka Y, Khan AS, Draghia-Akli R. Electroporation-enhanced nonviral gene transfer for the prevention or treatment of immunological, endocrine and neoplastic diseases. Curr Gene Ther 2006; 6:243 - 73; http://dx.doi.org/10.2174/156652306776359504; PMID: 16611045
- Sardesai NY, Weiner DB. Electroporation delivery of DNA vaccines: prospects for success. Curr Opin Immunol 2011; 23:421 - 9; http://dx.doi.org/10.1016/j.coi.2011.03.008; PMID: 21530212
- Vilalta A, Shlapobersky M, Wei Q, Planchon R, Rolland A, Sullivan S. Analysis of biomarkers after intramuscular injection of Vaxfectin-formulated hCMV gB plasmid DNA. Vaccine 2009; 27:7409 - 17; http://dx.doi.org/10.1016/j.vaccine.2009.08.075; PMID: 19735757
- Shlapobersky M, Wei Q, Sullivan S, Vilalta A. Vaxfectin-adjuvanted seasonal influenza protein vaccine: correlation of systemic and local immunological markers with formulation parameters. Vaccine 2009; 27:6404 - 10; http://dx.doi.org/10.1016/j.vaccine.2009.06.087; PMID: 19607952
- Brice GT, Dobaño C, Sedegah M, Stefaniak M, Graber NL, Campo JJ, et al. Extended immunization intervals enhance the immunogenicity and protective efficacy of plasmid DNA vaccines. Microbes Infect 2007; 9:1439 - 46; http://dx.doi.org/10.1016/j.micinf.2007.07.009; PMID: 17913540
- Leitner WW, Seguin MC, Ballou WR, Seitz JP, Schultz AM, Sheehy MJ, et al. Immune responses induced by intramuscular or gene gun injection of protective deoxyribonucleic acid vaccines that express the circumsporozoite protein from Plasmodium berghei malaria parasites. J Immunol 1997; 159:6112 - 9; PMID: 9550412
- Fuller DH, Corb MM, Barnett S, Steimer K, Haynes JR. Enhancement of immunodeficiency virus-specific immune responses in DNA-immunized rhesus macaques. Vaccine 1997; 15:924 - 6; http://dx.doi.org/10.1016/S0264-410X(96)00271-X; PMID: 9234549
- Keitel WA, Atmar RL. Preparing for a possible pandemic: influenza A/H5N1 vaccine development. Curr Opin Pharmacol 2007; 7:484 - 90; http://dx.doi.org/10.1016/j.coph.2007.06.004; PMID: 17644429
- Schleiss MR. Comparison of vaccine strategies against congenital CMV infection in the guinea pig model. J Clin Virol 2008; 41:224 - 30; http://dx.doi.org/10.1016/j.jcv.2007.10.008; PMID: 18060834
- Schleiss MR, Bourne N, Bernstein DI. Preconception vaccination with a glycoprotein B (gB) DNA vaccine protects against cytomegalovirus (CMV) transmission in the guinea pig model of congenital CMV infection. J Infect Dis 2003; 188:1868 - 74; http://dx.doi.org/10.1086/379839; PMID: 14673766
- Schleiss MR, Bourne N, Stroup G, Bravo FJ, Jensen NJ, Bernstein DI. Protection against congenital cytomegalovirus infection and disease in guinea pigs, conferred by a purified recombinant glycoprotein B vaccine. J Infect Dis 2004; 189:1374 - 81; http://dx.doi.org/10.1086/382751; PMID: 15073673
- Jimenez GS, Planchon R, Wei Q, Rusalov D, Geall A, Enas J, et al. Vaxfectin-formulated influenza DNA vaccines encoding NP and M2 viral proteins protect mice against lethal viral challenge. Hum Vaccin 2007; 3:157 - 64; http://dx.doi.org/10.4161/hv.3.5.4175; PMID: 17637571
- Leamy VL, Martin T, Mahajan R, Vilalta A, Rusalov D, Hartikka J, et al. Comparison of rabbit and mouse models for persistence analysis of plasmid-based vaccines. Hum Vaccin 2006; 2:113 - 8; PMID: 17012905
- Hebel H, Attra H, Khan A, Draghia-Akli R. Successful parallel development and integration of a plasmid-based biologic, container/closure system and electrokinetic delivery device. Vaccine 2006; 24:4607 - 14; http://dx.doi.org/10.1016/j.vaccine.2005.08.049; PMID: 16150516
- Khan AS, Pope MA, Draghia-Akli R. Highly efficient constant-current electroporation increases in vivo plasmid expression. DNA Cell Biol 2005; 24:810 - 8; http://dx.doi.org/10.1089/dna.2005.24.810; PMID: 16332178