Abstract
Vaccine development for Group A streptococcal (GAS) infection has been extensively focused on the N-terminal hypervariable or the C-terminal conserved regions of the M protein, a major virulence factor of GAS. We evaluated the immunogenicity and functional activity of the conserved C-terminal peptide vaccine candidate, J8, conjugated to CRM197, in two mouse strains: C3H (H2k) and Balb/c (H2d), and in rhesus macaques. Mice were immunized with J8-CRM197 formulated with Amorphous Aluminum Hydroxyphosphate Sulfate Adjuvant (AAHSA), and non-human primates were immunized with J8-CRM197 formulated with AAHSA, ISCOMATRIXTM adjuvant, or AAHSA/ISCOMATRIX adjuvant. J8-CRM197 was immunogenic in mice from both H2k and H2d backgrounds, and the antibodies generated bound to the surface of four different GAS serotypes and had functional bacterial opsonic activity. Mice immunized with J8-CRM197/AAHSA demonstrated varying degrees of protection from lethal challenge. We also demonstrated that J8-CRM197 is immunogenic in non-human primates. Our data confirm the utility of J8 as a potential GAS vaccine candidate and demonstrate that CRM197 is an acceptable protein carrier for this peptide.
Introduction
Group A Streptococcus (GAS) remains a leading cause of several diseases with global impact, ranging from mild (pharyngitis and impetigo) to invasive (cellulitis, bacteremia, pneumonia, necrotizing fasciitis and streptococcal toxic shock syndrome) infections, as well as non suppurative sequelae (acute rheumatic fever and post-streptococcal glomerulonephritis) which are prominent in developing countries.Citation1
M protein has been described as the major surface-expressed virulence factor and the determinant of naturally acquired immunity against GAS.Citation2,Citation3 Until recently, M protein has been the major focus of vaccine design, and two strategies targeting this antigen have been extensively studied. The most advanced work to date has focused on the highly immunogenic but hypervariable N-terminal region of M protein as the basis for a multi-epitopic vaccine approach. A 26-valent vaccine has been evaluated in human clinical trials and demonstrated to induce opsonic antibodies to several representative GAS serotypes.Citation4 However, the utility of such an approach has been questioned since serotype distribution of strains causing disease differs based upon geographical region studied and is complex and poorly understood in many parts of the world where vaccines would be of the greatest benefit; however, inclusion of representative N-terminal epitopes for specific geographical locations may be one approach to overcome these restrictions.Citation5-Citation8 A second approach is based on the more highly conserved C-terminal region of the M protein.Citation9-Citation11 This approach is expected to be advantageous since sequence conservation would imply broader strain coverage with fewer vaccine components; although, safety concerns exist since this region of the protein has been shown to induce T cell responses that are cross reactive with human cardiac myosin.Citation12 Good et al. defined a peptide sequence (p145) located in this region, which was recognized by antibodies in the sera of most adults living in areas of endemic streptococcal disease.Citation11,Citation13 A minimal B cell epitope and non-host reactive peptide derived from p145, termed J8, has been shown to be immunogenic and protective in mice, either administered as a peptide alone formulated with Freund's adjuvant in B10.BR (H2k) miceCitation14 or conjugated to Diphtheria Toxoid (DT) and formulated on an aluminum phosphate adjuvant.Citation15,Citation16
Here we describe conjugation of J8 to an alternate protein carrier, CRM197, a non-toxic analog of DT which has been used as the basis for multiple bacterial polysaccharide conjugate vaccines.Citation17 We show that J8-CRM197 is immunogenic in inbred mice from two genetic backgrounds, H2k and H2d, and, for the first time, demonstrate immunogenicity of this conjugate vaccine in non-human primates. Our results indicate that J8-CRM197 performs similarly to the previously reported J8-DT conjugate in mice, confirming that CRM197 can serve as an alternative carrier protein to DT for J8 conjugate vaccines.
Results
J8- CRM197 conjugate vaccine is immunogenic in Balb/c and C3H mice
Initial experiments were designed to assess the immunogenicity of J8-CRM197 formulated with AAHSA at a single concentration of 12.5 μg in inbred Balb/c (H2d) and C3H (H2k) mice. Immunogenicity was comparable in both mouse strains with total IgG titers of ~10,000 elicited after a single dose of vaccine and a subsequent 10-fold increase in titer following the second dose. No further increase in titer was observed after the third immunization (). No significant difference in response to the vaccine was observed between these genetically distinct inbred mice. (p = 0.8). We next performed a dose-ranging study by immunizing a smaller cohort of Balb/c mice (n = 3) with 12.5, 5, 1 or 0.1 μg of J8 peptide conjugated to CRM197. We observed no differences in J8-specific antibody titers across all four doses (), although there was a trend toward lower antibody responses at the 0.1 μg dose. No J8-specific antibodies were detected in control animals immunized with CRM197/AAHSA or Saline/AAHSA (data not shown).
Figure 1. Serum IgG antibody responses to J8 in Balb/c and C3H mice over time. Titers from C3H and Balb/c mice immunized with J8 -CRM197/AAHSA (n = 10) at 12.5μg/dose based on specific peptide content (A). Dose titration of J8-CRM197/AAHSA in Balb/c mice (n = 3), mice were immunized with 12.5, 5, 1 and 0.1 μg /dose, based on J8 peptide content (B).
J8-specific antibodies bind to M-protein on the GAS surface
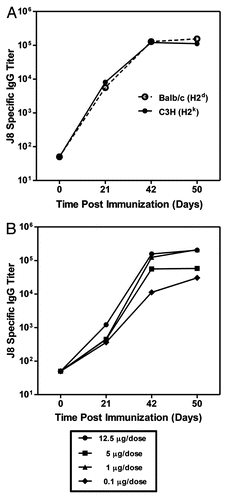
We examined the ability of antibodies elicited by immunization of mice with J8-CRM197 to recognize the corresponding M protein epitope on intact bacterial cells. We evaluated binding of J8-CRM197/AAHSA, CRM197/AAHSA and AAHSA immune serum to four different GAS serotypes (M1, M3, M6 and M97) and found that of the groups tested, the only serum that contained antibodies which bound to the surface of GAS was from mice immunized with J8-CRM197/AAHSA. One representative experiment using serotype M3, is shown in . Similar results were obtained with other GAS serotypes (data not shown).
Figure 2. Photomicrographs (representative of the slide) of immunofluorescent (fluorescein isothiocyanate) - stained Group A streptococci (GAS, serotype M3) viewed under the same conditions, using a microscope with a 100X objective. Bacteria were stained with pre-immune mouse sera (A), CRM197 immune serum (B), J8-CRM197 immune serum (C) and type M3 immune serum as positive control (D). All sera were taken after the last boost and diluted 1:50.
J8-CRM197 mouse antibodies have opsonophagocytic activity
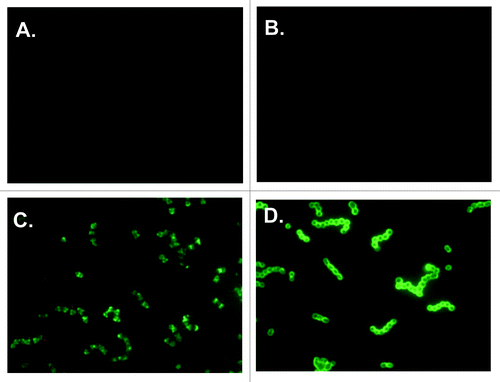
The functional activity of antibodies raised by immunization of mice with J8-CRM197 was evaluated in an opsonophagocytic assay using GAS 88/30 M97. Naïve mouse serum was tested in the same experiments to serve as a non-immune control for determination of percent opsonization. Results from four different immunization experiments are shown in ; two using Balb/c mice (Experiment-1 and Experiment-2), and two using C3H mice (Experiment-3 and Experiment-4). From each of the four experiments only J8-CRM197/AAHSA immune serum promoted killing of the bacteria (activity ranging from 57–93%) that was statistically significant compared with the negative controls [CRM197/AAHSA or AAHSA immune serum (p < 0.001)] (). Serum from mice immunized with GAS M97 was used as a positive control and in all experiments caused between 90–100% killing (data not shown).
Figure 3. Average percent opsonization of GAS M97 strain by sera taken one week after the last boost (Day 50). Filled symbols (●▲) represent two independent experiments using mouse sera taken from C3H immunized mice and open symbols (○∆) represent two independent experiments using sera taken from Balb/c mice. In all four experiments only J8-CRM197/AAHSA immune serum promoted killing of the bacteria (activity ranging from 57–93%) which was statistically significant compared with the negative control (CRM197/AAHSA or AAHSA) immune serum p < 0.001.
Immunization with J8-CRM197 induces limited protection in mice following systemic and intranasal challenge
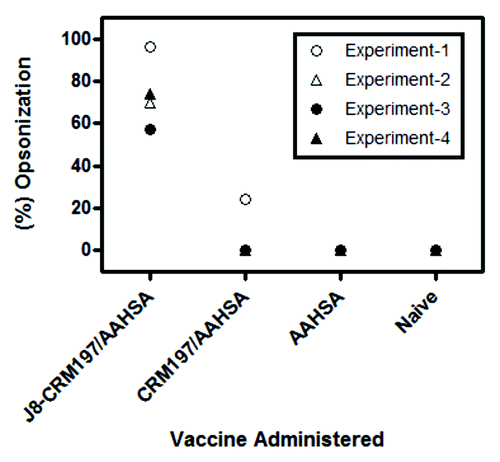
Groups of Balb/c and C3H mice were immunized with J8-CRM197/AAHSA, CRM197/AAHSA, AAHSA, heat-killed GAS M1 or recombinant M protein (strain M3). Balb/c mice were challenged (i.p.) with GAS pM1 SR in mucin. Ninety percent of the J8-CRM197/AAHSA immunized group and 100% of the heat-killed M1 immunized groups (positive control) survived challenge (p = 0.0001 relative to CRM197/AAHSA and AAHSA groups) (). In order to demonstrate protection in a second model system, C3H mice were challenged (i.n.) with a GAS M3 SR strain. Seventy percent of the J8-CRM197/AAHSA immunized group and 100% of the recombinant M protein immunized group (positive control) were protected from challenge, while 60% of the CRM197/MAA group also survived challenge (p = 0.001 relative to the AAHSA group) ().
Figure 4. Survival of C3H and Balb/c mice after challenge. Balb/c mice challenged intraperitoneally (i.p.) with GAS pM1 SR in mucin (A) 90% of the J8-CRM197/AAHSA immunized group and 100% of the heat-killed M1 immunized group (positive control) survived challenge p < 0.0001 relative to CRM197/AAHSA and AAHSA groups. C3H mice challenged intranasally (i.n.) with GAS M3 SR (B) 70% percent of the J8-CRM197/AAHSA immunized group and 100% of the recombinant M protein immunized group (positive control) were protected from challenge, while 60% of the CRM197/MAA group also survived challenge p < 0.001 relative to the AAHSA group.
J8-CRM197 is immunogenic in non-human primates
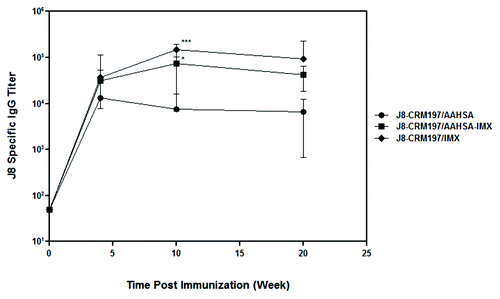
We evaluated immunogenicity of J8-CRM197 formulated with three adjuvant combinations, AAHSA, ISCOMATRIX adjuvant and AAHSA + ISCOMATRIX adjuvant, in rhesus macaques. shows the time course of the antibody responses to J8 as determined by ELISA. Pre-immune sera were tested as pooled samples while post-immunization titers were assessed from individual animals. IgG titers were lowest overall in animals immunized with J8-CRM197/AAHSA with titers of only ~6,000 and ~9,000 following one and two doses of conjugate, respectively. In contrast, J8-CRM197 formulated in ISCOMATRIX adjuvant alone or with an AAHSA/ISCOMATRIX adjuvant mixture induced IgG titers of > 20,000 after a single dose and > 60,000 following a second dose. Moreover, the post-dose two titers were sustained after a third dose. J8-specific IgG titers were statistically significantly different (p < 0.05) between groups immunized with J8-CRM197/AAHSA-ISCOMATRIX adjuvant or J8-CRM197/ISCOMATRIX adjuvant vs. the group immunized with J8-CRM197/AAHSA at both post-dose two and post-dose three time points. We attempted to evaluate sera from NHPs in both the OPK and IFA assays; unfortunately the pre-immune sera contained high levels of background antibodies that led to both high killing activity in the OPK as well as background fluorescence in the IFA which made the data very difficult to interpret.
Figure 5. J8-specific serum IgG titers in rhesus macaques immunized with J8-CRM197 formulated with three different adjuvants. Antibody titers were evaluated by ELISA. Note that after the second immunization, animals that received J8-CRM197/ISCOMATRIX adjuvant (IMX) had higher anti-J8 IgG titers statistically significant compared with the group that only received J8-CRM197/AAHSAA p = 0.0331*, and the group that received J8-CRM197/ISCOMATRIX adjuvant (IMX) p = 0.0001***.
Discussion
An efficacious vaccine for prevention of Group A streptococcal infection would be an important tool for reducing rheumatic fever and rheumatic heart disease in the developing world as well as for reducing recurrent pharyngitis in both the developing and developed world.Citation18 Recent reports detailing an increase in global expansion of macrolide resistant strains of Streptococcus pyogenes adds further impetus to the desire to develop such a vaccine.Citation19,Citation20 Although S. pyogenes is susceptible to penicillin, macrolides are the antibiotics of choice for individuals who are allergic to β-lactams.Citation21
M-protein, the major surface protein of GAS, is a coiled-coil protein composed of a highly variable N-terminal region, which is the focus of serotyping (M typing) and genotyping (emm typing), A-repeat and B-repeat domains which do not induce opsonic antibodies,Citation22 and a C-repeat domain, which is the most conserved of the three repeat regions and contains the J8 peptide. Previously described vaccine approaches utilizing the M protein conserved C-repeat region include: (1) a recombinant protein domain encompassing the C-terminal region of strain M6Citation9; (2) selected B and T cell epitopes from strain M5 presented as either synthetic peptides or recombinant proteinCitation23; and (3) the 12-amino acid minimal B cell epitope J8 presented as a synthetic peptide.Citation15 Murine studies evaluating J8 conjugated to DT and formulated with aluminum hydroxide established that this potential vaccine candidate induced opsonic antibodies which were protective in a lethal challenge model.Citation24,Citation25
The objectives of the current study were, first to assess the immunogenicity and protective efficacy in mice of a vaccine candidate consisting of the J8 peptide covalently conjugated to the non-toxic DT analog, CRM197, and second to demonstrate that immunogenicity of this vaccine candidate was extendable to non-human primates. The use of CRM197 as an alternate carrier protein to Diphtheria toxoid offers several potential advantages in terms of vaccine manufacturability and safety. CRM197 is a component of several licensed vaccines including PREVNAR7®, PREVNAR13® and HibTITER®.Citation17 As such, protocols for cGMP supply and release of the protein have been established. Importantly, a substantial safety profile for use of this protein as a carrier for bacterial polysaccharides has been established in humans, including infants. It is reasonable to expect that this safety profile would extend to its use as a carrier for peptide vaccine antigens as well. CRM197 has been evaluated pre-clinically for additional investigational conjugate vaccines including studies with GAS polysaccharides.Citation26
J8-CRM197 formulated with AAHSA was shown to be highly immunogenic in Balb/c (H2d) and C3H (H2k) mice at a peptide dose as low as 0.1 μg, however we choose the 12.5µg dose since opsonophagocytic (OPK) activity was reduced at lower doses (data not shown). Antibodies elicited by immunization with J8-CRM197/AAHSA bound to the surface of four different GAS strains (M1, M3, M6 and M97), confirming that the J8 epitope is conserved across these strains as well as demonstrating that the conjugation chemistry employed here does not compromise presentation of the J8 epitope. Furthermore, we demonstrated that J8-CRM197/AAHSA induces functional antibodies, which mediate opsonophagocytosis of GAS 88/30 M97 in vitro and lead to killing of bacteria by human phagocytic cells, a major pathway for bacterial clearance. Unfortunately, we were unable to expand upon this opsonophagocytosis data with additional serotypes of GAS as suitable human blood donors for phagocytic cells could not be identified for these types. This is likely due to the fact that most adults have been exposed to multiple GAS serotypes throughout their life and therefore have pre-existing M-protein based immunity. The opsonic activity of J8-CRM197 immune serum in four different mouse immunization experiments (two using Balb/c mice and two using C3H mice), ranged from 53% to 97%, but no opsonic activity was observed in the sera of control animals immunized with CRM197 or adjuvant alone.
J8-CRM197/AAHSA induced protective immunity in mice against two serotypes of GAS in two separate challenge models suggesting that protection mediated by this vaccine is not restricted to a single M-type, but rather may be broadly effective. In a systemic challenge study performed with GAS pM1 SR in Balb/c mice, the bacteria were mixed with mucin in order to decrease the bacterial dose required for lethality in the model as observed in experiments with other bacterial pathogens,Citation27 90% of J8-CRM197/AAHSA immunized animals were protected, while CRM197/AAHSA and AAHSA immunized mice did not survive. This study was repeated a total of three times, and J8-CRM197/AAHSA immunized mice were protected in two out of three experiments. In an intranasal challenge study performed with GAS M3 SR in C3H mice, 70% of J8-CRM197/AAHSA immunized mice were protected, in contrast to 10% survival observed in mice immunized with AAHSA alone. Unexpectedly, 60% of animals immunized with CRM197/AAHSA also survived. It has been previously reported that DT formulated with adjuvant also induced non-specific protection.Citation15 We attempted to overcome this issue by increasing the time between the last immunization and subsequent bacterial challenge; however some experiments continued to show non-specific protection by the CRM197 conjugate control (data not shown). This study was repeated a total of five times, and J8-CRM197/AAHSA immunized mice were protected in three of the experiments. However, since significant protection was not obtained in 100% of either the i.p. or i.n. challenge studies, the results of the mouse challenge models must be interpreted with caution. Since mouse models of infection for many bacterial pathogens can be quite variable, and GAS is not a natural mouse pathogen, non-human primates may serve as a more relevant model system for pre-clinical GAS vaccine evaluation.Citation28
It has been previously suggested that antibodies generated by immunization with the C-terminal region of GAS M protein exhibit cross-reactive binding to human serum albumin (HSA), and, therefore, are not expected to exhibit any specific functional activity.Citation29 Our data showing opsonophagocytic activity of J8-directed antibodies appears to contradict this assumption. Additionally, we performed ELISA assays to directly assess binding of sera from J8-immunized animals to HSA, and we failed to detect any binding even at antigen coating concentrations as high as 10 μg/ml (data not shown). This suggests that the J8 epitope is located outside of the HSA binding site localized to the C-terminal portion of M protein. Alternatively, J8 may form a portion of the HSA binding site, but binding to HSA is insufficient to abolish its specific opsonophagocytic activity.
To our knowledge, the current study describes for the first time immunogenicity of the J8 peptide conjugated to CRM197 in mice and non human primates. J8-CRM197 formulated on any of the three adjuvants tested in rhesus macaques was well-tolerated and no adverse effects were observed in any vaccinated animals throughout the time course of the study. All vaccinees sero-converted following a single immunization with 10 μg of J8-CRM197 conjugate, achieving serum IgG titers of 6,000 to 20,000. Following a second administration, titers rose to > 65,000 in animals immunized with J8-CRM197/ISCOMATRIX adjuvant or J8-CRM197/AAHSA/ISCOMATRIX adjuvant. Interestingly, animals immunized with J8-CRM197/AAHSA elicited lower IgG titers of ~10,000 after the second dose. Statistical comparisons indicated that the saponin-based ISCOMATRIX adjuvant is significantly more potent than AAHSA in non-human primates. No additional enhancement in IgG levels was observed in any group following a third immunization, suggesting that further enhancement will require additional evaluation of adjuvant, dose and immunization regimens. Our data demonstrating robust immunogenicity of J8-CRM197 formulated with ISCOMATRIX adjuvant in non-human primates establishes this immunogen as a viable vaccine candidate for further optimization and testing by itself or combined with other GAS antigens. We were unable to evaluate the functional activity of these sera due to high pre-existing antibodies to GAS in non-human primates. This may be due to previous exposure of the animals to this organism as has been described.Citation30,Citation31 In conclusion, we report that J8 conjugated to CRM197 is immunogenic and confers variable degrees of protection in mouse models. We also demonstrated that J8-CRM197 formulated with the human compatible adjuvants AAHSA, AAHSA + ISCOMATRIX adjuvant and ISCOMATRIX adjuvant is safe and immunogenic in non human primates which further supports pre-clinical development of this promising GAS vaccine candidate.
Methods
Ethics statement
All animal work was performed in strict accordance with the recommendations in the Guide for Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC), Merck Research Labs, West Point, PA
Synthesis of J8 peptide
The peptide (Ac-Gln-Ala-Glu-Asp-Lys-Val-Lys-Gln-Ser-Arg-Glu-Ala-Lys-Lys-Gln-Val-Glu-Lys-Ala-Leu-Lys-Gln-Leu-Glu-Asp-Lys-Val-Gln-Aha-Cys-NH2, Aha = 6-aminohexanoic acid, MW = 3541.1) was prepared by standard solid-phase peptide synthesis with Fmoc/t-Bu chemistry. The N-terminus was acetylated and the C-terminus was amidated. Briefly, the peptide was synthesized by solid phase chemistry on a NovaPEG Rink Amide resin (0.62 mmeq/g, EMD Biosciences) using a Symphony Synthesizer (Protein Technologies, Inc.). Acylations were performed with double couplings for 30 min with a 5-fold excess of amino acids activated with equimolar amounts of HBTU [2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate] and HOBt (N-hydroxybenzotriazole), and a 2-fold molar excess of DIEA (N,N-diisopropylethylamine). At the end of the assembly, the peptide was acetylated by treatment with a 10-fold molar excess of acetic anhydride. The dry peptide resin was then treated with cleavage mixture (82.5% trifluoroacetic acid, 5% phenol, 5% thioanisole, 5% water, 2.5% ethanedithiol) for 2h at room temperature. The filtered peptide solution was precipitated with cold diethyl ether, isolated by centrifugation, washed twice with cold diethyl ether, dried, resuspended and lyophilized. The peptide was purified by Reverse Phase HPLC on a C18 Jupiter Column (Phenomenex Jupiter 10μ, C18, 300A) and eluted with an appropriate linear gradient of an increasing concentration of acetonitrile containing 0.1% TFA. The purified peptide was characterized by electrospray mass spectrometry.
Synthesis of biotinylated J8 peptide
The peptide (Ac-Gln-Ala-Glu-Asp-Lys-Val-Lys-Gln-Ser-Arg-Glu-Ala-Lys-Lys-Gln-Val-Glu-Lys-Ala-Leu-Lys-Gln-Leu-Glu-Asp-Lys-Val-Gln-Aha-Cys (PEG-Biotin)-NH2, Aha = 6-aminohexanoic acid, PEG = polyethylene glycol, MW = 4066.8) was prepared by a reaction of purified J8 peptide with a 2-fold molar excess of EZ-Link maleimide-PEG2-Biotin (Thermo Fisher Scientific, Rockford, IL) in 0.1M ammonium acetate buffer, pH 7.0 for 1 h. The peptide was purified by Reverse Phase HPLC on a C18 Jupiter Column (Phenomenex Jupiter 10μ, C18, 300A) and eluted with an appropriate linear gradient of an increasing concentration of acetonitrile containing 0.1% TFA. The purified peptide was characterized by electrospray mass spectrometry.
Preparation of J8-CRM197 conjugate
Purified CRM197 carrier protein was obtained from Merck Research Laboratories (West Point, PA) and activated for conjugation by addition of maleimide groups to surface-accessible lysine residues using the heterobifunctional cross-linking reagent succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC, Thermo Fisher Scientific, Rockford, IL).Citation32 Briefly, a concentrated stock of CRM197 in phosphate buffer was diluted in 20mM HEPES, 0.15M NaCl, 0.005M EDTA, pH 7.3 to a total protein concentration of 1 mg/ml. Nine mg of SMCC (10-fold molar ratio to theoretical CRM197 lysine content) was dissolved in 1 ml of dry dimethyl sulfoxide (DMSO) and added to the CRM197 solution while it was being gently vortexed. The reaction mixture was incubated in the dark at room temperature for 3 h. The sample was then reduced in volume to 5 ml using an Amicon Ultra 4 centrifugal concentrator and desalted over a Hi Prep 26/10 desalting column (GE Healthcare Life Sciences) equilibrated in 20mM HEPES, 0.15M NaCl, 5mM EDTA, pH 7.3. The amount of maleimide incorporated into the CRM197 was determined using a 5, 5′-dithiobis-(2-nitrobenzoic acid) (DTNB) -based assay which relies upon the consumption of a fixed amount of N-acetyl cysteine from a standard solution. CRM197 total protein was determined by the bicinchoninic acid (BCA) protein assay. Conjugation of thiolated J8 peptide to maleimide-activated CRM197 was performed at a 2 to 1 molar ratio of peptide thiol to CRM197 maleimide. The required amount of thiolated J8 peptide was weighed out and placed in a 20 ml glass vial to which the activated CRM197 solution was added. The solution was mixed to ensure that the peptide was completely dissolved and then incubated overnight at room temperature. The conjugate was purified of un-reacted peptide by size exclusion chromatography over a 2.5 by 45 cm Superdex 30 column equilibrated and run in 20mM HEPES, 0.15M NaCl, pH 7.3. The molar loading ratio of peptide to carrier was determined by quantitative amino acid analysis using least squares linear regression analysis and quantitation of unique amino acid residues generated during the conjugation reaction.Citation33
The adjuvants used for formulation were amorphous aluminum hydroxyphosphate sulfate (AAHSA) and ISCOMATRIX adjuvant. AAHSA was obtained from the Merck Manufacturing Division, West Point, PA., and ISCOMATRIX adjuvant was obtained from CSL Biotherapies Inc., King of Prussia, PA.
Immunization of mice and non-human primates
Mouse experiments were approved by the Institutional Animal Care and Use Committee at Merck and Co., Inc. Five to seven week old female C3H or Balb/c mice were purchased from Taconic Farms (Hudson, NY) and housed in microisolator cages in the animal facility at Merck Research Laboratories (MRL), West Point, PA. Mice were intramuscularly (i.m.) immunized with J8-CRM197 (doses ranged from 0.1 to 12.5 μg based on peptide content), formulated with amorphous aluminum hydroxyphosphate sulfate adjuvant (AAHSA). Negative control animals received CRM197/AAHSA or AAHSA, while positive control animals received 10 μg of recombinant, full length M3 protein (rM3)Citation28 or 109 CFU of heat-killed GAS, serotype M1. For generation of high-titered immune sera for use as a positive control in OPK assays, mice were immunized intraperitoneally with 107 CFU of heat-killed GAS 88/30 strain (serotype M97).Citation34 All immunizations were performed on days 0, 21 and 42. Blood samples were collected on days -1, 20, 41 and 50. Samples from all dates were tested in ELISA and samples from day 50 were tested in immunofluorescence and bactericidal assays. The aluminum concentration in each of the vaccine formulations was 45 μg/dose.
Non-human primate experiments were approved by the Institutional Animal Care and Use Committee at both Merck and Co., Inc. and the University of Louisiana, New Iberia Research Center (NIRC, Lafayette, LA). A group of 12 rhesus macaques were included in the study. Selected animals were matched by age, sex and weight, housed in social settings and randomly assigned to three experimental groups of four individuals each that received different vaccine formulations. Four monkeys per group were immunized i.m with J8-CRM197 with various adjuvant formulations. Monkeys received a vaccine dose on days 0, 56 and 112. Vaccine antigen/adjuvant concentrations were: 20 μg/ml of J8 peptide formulated with AAHSA (450 μg/ml) and/or ISCOMATRIX™ (120 μg/ml). A vaccine dose of 0.5 ml was injected per animal. Serum samples were collected on days 0 (pre-immunization), 28, 56, 70, 84, 126 and 140. Only samples collected on days 0, and weeks 4, 10 and 20 were tested in ELISA.
Bacterial strains and culture conditions
For bacterial designations, M type and origin see . All the previously mentioned strains and a GAS serotype M6–12348 obtained from ATCC were grown in Todd Hewitt broth (THB) (Becton Dickinson) overnight and processed for use in immunofluorescence or bactericidal assays.
Table 1. Group A streptococcal strains used in these studies
ELISA assays
Immulon 2 HB (Thermo Labsystems, Catalog # 3455) 96 well plates coated with J8 peptide were used for testing mouse sera samples and Neutravidin (Pierce, Cat # 15129) 96-well plates coated with biotinylated J8 were used to test non-human primate sera samples. In both experiments the plates were coated with 0.25 μg/well of J8 peptide in coating solution (KPL, Cat # 50–84–00) overnight at 4°C. Wells were washed with PBS containing 0.05% Tween 20 (PBS-T), and unbound sites blocked with Blocking/Dilution Solution (KPL, Cat # 50–61–00) by incubation for 2 h at room temperature. Mouse or non-human primate sera were 5-fold serially diluted on the plates and incubated at room temperature for 1 h. Plates were washed with PBS-T and incubated with goat anti-mouse (Jackson Immuno Research, Cat # 115–035–071) or anti-rhesus (Rockland Immunochemicals Inc., Cat # 617–103–012) IgG HRP secondary antibody for 1 h at room temperature. Following an additional wash, ABTS (2,2'-Azinobis [3-ethylbenzothiazoline-6-sulfonic acid]-diammonium salt) substrate (KPL, Cat # 50–62–00) was added and after 30 min, the reaction was stopped with ABTS Stop Solution (KPL, Cat # 50–85–01) and absorbance measured at 405nm.
Immunofluorescence assay
Antibodies elicited after three immunizations with J8-CRM197 were tested in indirect immunofluorescence assays (IFA) against four different GAS strains.Citation35 Strains 88/30 M97, M3, M6 and M1 were grown overnight, centrifuged at 3000 rpm and the pellet resuspended in PBS to an O.D600 ~0.1. Ten μl of bacterial suspension per well was applied to 12-well glass microscope slides, dried overnight at room temperature, fixed with 3% formaldehyde, rinsed with PBS and stored at -20°C until used. Slides were brought to room temperature, washed in PBS and blocked with 5% skim milk in PBS-T in a humidified chamber at 37°C for 30 min, rinsed with PBS and incubated with a 1:200 dilution of purified non-specific human IgG (Serotec, Cat # PHP-001) for 1h at 37°C followed by incubation with J8 immune mouse sera (1:50 dilution). A rabbit serum with specificity for GAS (Statens Serum Institut, Cat # 22433) or serum from mice immunized with individual serotypes M1 or M3 or M97 heat killed GAS were included as a positive control. Slides were then washed and incubated with FITC-conjugated F(ab)2 goat anti mouse IgG (Invitrogen, Cat # 62–6311) diluted 1:50 in Evans blue (MP Biomedicals) and incubated for 45 min in the dark. Goat anti-rabbit IgG (H+L) secondary antibody if the rabbit positive serum was used (Invitrogen, Cat # A10526) was used for detection of the positive control antiserum. Wells were washed three times with PBS for 10 min. Mounting solution was added and stained bacteria were visualized at 100 × magnification using a fluorescence microscope.
Whole blood bactericidal assay
Murine antibodies were assayed for opsonic activity against GAS M97 as previously described.Citation3,Citation25,Citation36 Briefly, GAS were incubated with end-over-end rotation at 37°C for 3h in the presence of non opsonic human blood and either mouse immune or control naïve mouse sera. Antisera to heat killed GAS strain M97 was used as a positive control. Bacteria were plated in triplicate on sheep blood agar plates (Remel, Cat # 01202) and colony-forming units (cfu) were counted after 24h incubation. The percentage of bacteria killed was determined using the following equation: [(mean cfu negative control)-(mean cfu test sample)/mean cfu negative control] × 100.
Challenge experiments
Mice were challenged 50–55 d after the primary immunization regimen. Both the pM1 and M3 strains used for challenge experiments had been serially passaged by isolating them from infected mouse spleen to enhance virulence and made streptomycin resistant (SR),Citation25 then labeled (pM1 SR) and (M3 SR).
For systemic challenge: the pM1 SR strain was grown overnight in 10 ml aliquots of Todd Hewitt broth supplemented with 0.1% neopeptone (THBN). The cultures were combined and bacteria were collected by centrifugation at 3000 rpm for 10 min, the pellet was resuspended in cold THBN and serially diluted, cfu were determined by plating on sheep blood agar and the undiluted culture was held at 4°C overnight. The following day, the culture was resuspended to 1 × 108 cfu/ml in THBN, then serially diluted in THBN plus 5% mucin type II (Sigma, St. Louis, MO). The 1 × 105 cfu/ml dilution was further diluted to the inoculum challenge dose of 7.5 × 103 cfu/ml in mucin and injected intraperitoneally (i.p.) into Balb/c mice at 0.4 ml/mouse. The mice were observed daily for survival for 15 d. Inoculum concentration was confirmed by plating on blood agar plates.
For intranasal (i.n.) challenge: 48 h before challenge, mice were given streptomycin (200 μg/ml) in their drinking water. This strain was grown as described previously.Citation37 Briefly, a frozen stock of GAS M3 SR was cultured overnight in THB supplemented with 0.5% yeast extract (THYE) containing 200 μg/ml streptomycin (Teknova, Cat # S6550) at 37°C in 5% CO2. The overnight culture was subcultured and grown for 4.5 h at 37°C with 5% CO2 to an A600 between 0.9 and 1.0. The bacteria were collected by centrifugation, washed and resuspended in THYE containing 25% glycerol (final concentration) and frozen at -70°C. The concentration of viable bacteria (~1.7 × 109 cfu/ml), was confirmed by plating on sheep blood agar plates containing streptomycin (Teknova, Cat # B0143). On the day of the challenge the frozen GAS M3 SR was thawed and used undiluted (~1.7 × 109 cfu/ml). The mice were sedated with 100mg/kg ketamine (Fort Dodge Animal Health) and 10mg/kg xylazine (AnaSed®) injected intraperitoneally. The bacterial suspension (15 μl/nare) was administered to the sedated mice. The mice were observed daily for survival for 15 d.
Statistical analysis
Antibody responses were log transformed and multiple comparisons were done using Student's t-test. Opsonophagocytosis analysis was performed using Anova followed by Bonferroni's multiple comparison test. A log-rank (Mantel-Cox) test was used to determine the differences in protection between vaccine immunized and negative control groups.
| Abbreviations: | ||
| AAHSA | = | amorphous aluminum hydroxyphosphate sulfate adjuvant |
| BSA | = | bovine serum albumin |
| GAS | = | Group A streptococcus |
| IFA | = | immunofluorescence assay |
| OPK | = | opsonophagocytic killing |
| SR | = | streptomycin resistant |
Acknowledgments
The authors would like to specially thank: David Thiriot, Pat Ahl, Christopher Farrell and Jayme Cannon for preparation of peptide-conjugate formulations; Jodie Field, Robin Kaufhold, Jose Aste-Amezaga, Adam Finnefrock and the Merck WP Laboratory Animal Resources department for technical assistance and Michael Citron and Dan Freed for assistance in carrying out the non-human primate study. We also thank the New Iberia Research Center technical staff, veterinarian and study coordinators for their support with the monkey study.
Disclosure of Potential Conflicts of Interest
I.C.-A., E.O., R.W.H., D.D.N., C.W., J.G.J., J.H.H and J.M.S. are either current or former employees of Merck and Co., Inc. and may potentially own stock and/or hold stock options in the company.
Related Research Data
References
- Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis 2005; 5:685 - 94; http://dx.doi.org/10.1016/S1473-3099(05)70267-X; PMID: 16253886
- Fischetti VA, Hodges WM, Hruby DE. Protection against streptococcal pharyngeal colonization with a vaccinia: M protein recombinant. Science 1989; 244:1487 - 90; http://dx.doi.org/10.1126/science.2660266; PMID: 2660266
- Lancefield RC, Perlmann GE. Preparation and properties of type-specific M antigen isolated from a group A, type 1 hemolytic streptococcus. J Exp Med 1952; 96:71 - 82; http://dx.doi.org/10.1084/jem.96.1.71; PMID: 14946330
- McNeil SA, Halperin SA, Langley JM, Smith B, Warren A, Sharratt GP, et al. Safety and immunogenicity of 26-valent group a streptococcus vaccine in healthy adult volunteers. Clin Infect Dis 2005; 41:1114 - 22; http://dx.doi.org/10.1086/444458; PMID: 16163629
- Kaplan EL, Wotton JT, Johnson DR. Dynamic epidemiology of group A streptococcal serotypes associated with pharyngitis. Lancet 2001; 358:1334 - 7; http://dx.doi.org/10.1016/S0140-6736(01)06415-7; PMID: 11684215
- Tewodros W, Kronvall G. M protein gene (emm type) analysis of group A beta-hemolytic streptococci from Ethiopia reveals unique patterns. J Clin Microbiol 2005; 43:4369 - 76; http://dx.doi.org/10.1128/JCM.43.9.4369-4376.2005; PMID: 16145079
- Dale JB, Penfound TA, Chiang EY, Walton WJ. New 30-valent M protein-based vaccine evokes cross-opsonic antibodies against non-vaccine serotypes of group A streptococci. Vaccine 2011; 29:8175 - 8; http://dx.doi.org/10.1016/j.vaccine.2011.09.005; PMID: 21920403
- Steer AC, Law I, Matatolu L, Beall BW, Carapetis JR. Global emm type distribution of group A streptococci: systematic review and implications for vaccine development. Lancet Infect Dis 2009; 9:611 - 6; http://dx.doi.org/10.1016/S1473-3099(09)70178-1; PMID: 19778763
- Bessen D, Fischetti VA. Synthetic peptide vaccine against mucosal colonization by group A streptococci. I. Protection against a heterologous M serotype with shared C repeat region epitopes. J Immunol 1990; 145:1251 - 6; PMID: 1696296
- Guilherme L, Faé KC, Higa F, Chaves L, Oshiro SE, Freschi de Barros S, et al. Towards a vaccine against rheumatic fever. Clin Dev Immunol 2006; 13:125 - 32; http://dx.doi.org/10.1080/17402520600877026; PMID: 17162355
- Pruksakorn S, Currie B, Brandt E, Martin D, Galbraith A, Phornphutkul C, et al. Towards a vaccine for rheumatic fever: identification of a conserved target epitope on M protein of group A streptococci. Lancet 1994; 344:639 - 42; http://dx.doi.org/10.1016/S0140-6736(94)92083-4; PMID: 7520963
- Pruksakorn S, Galbraith A, Houghten RA, Good MF. Conserved T and B cell epitopes on the M protein of group A streptococci. Induction of bactericidal antibodies. J Immunol 1992; 149:2729 - 35; PMID: 1383324
- Relf WA, Cooper J, Brandt ER, Hayman WA, Anders RF, Pruksakorn S, et al. Mapping a conserved conformational epitope from the M protein of group A streptococci. Pept Res 1996; 9:12 - 20; PMID: 8727479
- Hayman WA, Brandt ER, Relf WA, Cooper J, Saul A, Good MF. Mapping the minimal murine T cell and B cell epitopes within a peptide vaccine candidate from the conserved region of the M protein of group A streptococcus. Int Immunol 1997; 9:1723 - 33; http://dx.doi.org/10.1093/intimm/9.11.1723; PMID: 9418133
- Batzloff MR, Hayman WA, Davies MR, Zeng M, Pruksakorn S, Brandt ER, et al. Protection against group A streptococcus by immunization with J8-diphtheria toxoid: contribution of J8- and diphtheria toxoid-specific antibodies to protection. J Infect Dis 2003; 187:1598 - 608; http://dx.doi.org/10.1086/374800; PMID: 12721940
- Brandt ER, Sriprakash KS, Hobb RI, Hayman WA, Zeng W, Batzloff MR, et al. New multi-determinant strategy for a group A streptococcal vaccine designed for the Australian Aboriginal population. Nat Med 2000; 6:455 - 9; http://dx.doi.org/10.1038/74719; PMID: 10742155
- Shinefield HR. Overview of the development and current use of CRM(197) conjugate vaccines for pediatric use. Vaccine 2010; 28:4335 - 9; http://dx.doi.org/10.1016/j.vaccine.2010.04.072; PMID: 20452430
- Bisno AL, Rubin FA, Cleary PP, Dale JB, National Institute of Allergy and Infectious Diseases. Prospects for a group A streptococcal vaccine: rationale, feasibility, and obstacles--report of a National Institute of Allergy and Infectious Diseases workshop. Clin Infect Dis 2005; 41:1150 - 6; http://dx.doi.org/10.1086/444505; PMID: 16163634
- Martin JM, Green M, Barbadora KA, Wald ER. Erythromycin-resistant group A streptococci in schoolchildren in Pittsburgh. N Engl J Med 2002; 346:1200 - 6; http://dx.doi.org/10.1056/NEJMoa013169; PMID: 11961148
- Richter SS, Heilmann KP, Dohrn CL, Beekmann SE, Riahi F, Garcia-de-Lomas J, et al. Increasing telithromycin resistance among Streptococcus pyogenes in Europe. J Antimicrob Chemother 2008; 61:603 - 11; http://dx.doi.org/10.1093/jac/dkm525; PMID: 18218647
- Bisno AL, Gerber MA, Gwaltney JM Jr., Kaplan EL, Schwartz RH, Infectious Diseases Society of America. Practice guidelines for the diagnosis and management of group A streptococcal pharyngitis. Clin Infect Dis 2002; 35:113 - 25; http://dx.doi.org/10.1086/340949; PMID: 12087516
- Fischetti VA, Windels M. Mapping the immunodeterminants of the complete streptococcal M6 protein molecule. Identification of an immunodominant region. J Immunol 1988; 141:3592 - 9; PMID: 2460540
- Guilherme L, Postol E, Freschi de Barros S, Higa F, Alencar R, Lastre M, et al. A vaccine against S. pyogenes: design and experimental immune response. Methods 2009; 49:316 - 21; http://dx.doi.org/10.1016/j.ymeth.2009.03.024; PMID: 19409999
- Batzloff MYH, Yan H, Davies M, Hartas J, Good M. Preclinical evaluation of a vaccine based on conserved region of M protein that prevents group A streptococcal infection. Indian J Med Res 2004; 119:Suppl 104 - 7; PMID: 15232173
- Olive C, Clair T, Yarwood P, Good MF. Protection of mice from group A streptococcal infection by intranasal immunisation with a peptide vaccine that contains a conserved M protein B cell epitope and lacks a T cell autoepitope. Vaccine 2002; 20:2816 - 25; http://dx.doi.org/10.1016/S0264-410X(02)00205-0; PMID: 12034109
- Kabanova A, Margarit I, Berti F, Romano MR, Grandi G, Bensi G, et al. Evaluation of a Group A Streptococcus synthetic oligosaccharide as vaccine candidate. Vaccine 2010; 29:104 - 14; http://dx.doi.org/10.1016/j.vaccine.2010.09.018; PMID: 20870056
- Knudsen JD, Frimodt-Møller N, Espersen F. Experimental Streptococcus pneumoniae infection in mice for studying correlation of in vitro and in vivo activities of penicillin against pneumococci with various susceptibilities to penicillin. Antimicrob Agents Chemother 1995; 39:1253 - 8; http://dx.doi.org/10.1128/AAC.39.6.1253; PMID: 7574511
- Skinner JM, Caro-Aguilar IC, Payne AM, Indrawati L, Fontenot J, Heinrichs JH. Comparison of rhesus and cynomolgus macaques in a Streptococcus pyogenes infection model for vaccine evaluation. Microb Pathog 2011; 50:39 - 47; http://dx.doi.org/10.1016/j.micpath.2010.10.004; PMID: 21035535
- Sandin C, Carlsson F, Lindahl G. Binding of human plasma proteins to Streptococcus pyogenes M protein determines the location of opsonic and non-opsonic epitopes. Mol Microbiol 2006; 59:20 - 30; http://dx.doi.org/10.1111/j.1365-2958.2005.04913.x; PMID: 16359315
- García A, Paul K, Beall B, McClure H. Toxic shock due to Streptococcus pyogenes in a rhesus monkey (Macaca mulatta). J Am Assoc Lab Anim Sci 2006; 45:79 - 82; PMID: 16995651
- Seegal B, Heller G, Jablonowitz J. Incidence of Hemolytic Streptococci and Pneumococci in the Pharyngeal Flora of Normal Rhesus Monkeys. Proc Soc Exp Biol Med 1936; 34:812 - 6
- Hermanson GT. Bioconjugate Techniques. London: Academic Press, 2008.
- Nahas DD, Palladino JS, Joyce JG, Hepler RW. Amino acid analysis of peptide loading ratios in conjugate vaccines: a comparison of direct electrochemical detection and 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate pre-column derivatization methods. Bioconjug Chem 2008; 19:322 - 6; http://dx.doi.org/10.1021/bc700232z; PMID: 18072716
- Siegert J, Sastalla I, Chhatwal GS, Medina E. Vaccination equally enables both genetically susceptible and resistant mice to control infection with group A streptococci. Microbes Infect 2006; 8:347 - 53; http://dx.doi.org/10.1016/j.micinf.2005.06.024; PMID: 16213175
- Olive C, Ho MF, Dyer J, Lincoln D, Barozzi N, Toth I, et al. Immunization with a tetraepitopic lipid core peptide vaccine construct induces broadly protective immune responses against group A streptococcus. J Infect Dis 2006; 193:1666 - 76; http://dx.doi.org/10.1086/504266; PMID: 16703510
- Olive C, Batzloff MR, Horváth A, Wong A, Clair T, Yarwood P, et al. A lipid core peptide construct containing a conserved region determinant of the group A streptococcal M protein elicits heterologous opsonic antibodies. Infect Immun 2002; 70:2734 - 8; http://dx.doi.org/10.1128/IAI.70.5.2734-2738.2002; PMID: 11953422
- Sumby P, Tart AH, Musser JM. A Non-Human Primate Model of Acute Group A Streptococcus Pharyngitis. In: Otto FDaM, ed. Methods in Molecular Biology. Totowa, NJ: Humana Press, 2006:255-67.