Abstract
The activity and circulation of influenza viruses in Lombardy — Northern Italy — (a region with nearly 10 out of the 60 million inhabitants of Italy) were investigated during two consecutive seasons (2010–2011 and 2011–2012), as part of the Italian Influenza Surveillance Network. The molecular characteristics of the hemagglutinin (HA) sequence of circulating viruses were analyzed to investigate the emergence of influenza viral variants. In the surveyed area, the influenza activity of these two post-pandemic seasons was similar in terms of both time frame and impact. The timing of the influenza epidemics was similar to the timing seen prior to the emergence of the pandemic A(H1N1) virus in 2009. A(H1N1)pdm09 was the predominant virus circulating during the 2010–2011 post-pandemic season and then—unexpectedly—almost disappeared. The HA sequences of these A(H1N1)pdm09 viruses segregated in a different genetic group with respect to those identified during the 2009 pandemic, although they were still closely related to the vaccine viral strain A/California/07/2009. Influenza A(H3N2) viruses were the predominant viruses circulating during the 2011–2012 season, accounting for nearly 88% of influenza viruses identified. All HA sequences of the A(H3N2) viruses isolated in the 2011–2012 season fell into the A/Victoria/208/2009 genetic clade (although the A/Perth/16/2009 virus was the reference vaccine strain). B viruses presented with a mixed circulation of viral variants during these two seasons: viruses belonging to both B/Victoria and B/Yamagata lineages co-circulated in different proportions, with a notable rise in the proportion of B/Yamagata viruses (B/Wisconsin/1/2010-like) during the 2011–2012 epidemic. In conclusion, the continuous monitoring of the characteristics of circulating viruses is an essential tool for understanding the epidemiological and virological features of influenza viruses, for monitoring their matching with seasonal vaccine strains, and for tuning vaccination strategies.
Introduction
Seasonal influenza occurs every year — usually in winter — and kills nearly 500,000 people worldwide at the very least.Citation1 This unique epidemiological feature rests on the pronounced tendency of influenza viruses to change the genetic and antigenic characteristics of their surface antigens hemagglutinin (HA) and neuraminidase (NA) — the main targets for the human immune system.Citation2 Influenza viruses can undergo extensive genetic changes and are even able to jump species, sometimes evolving into a virus to which humans are highly vulnerable. This is the event underlying the occurrence of global influenza pandemics, such as those seen in 1918 (H1N1), 1957 (H2N2), 1968 (H3N2), and 2009 (H1N1).Citation1-Citation3
Every year more than 300 million doses of influenza vaccine are produced to control and prevent worldwide.Citation1 For over 50 y, the process by which an effective vaccine is developed and manufactured has relied on the international cooperation of a wide range of public health partners brought together under the coordination of the World Health Organization (WHO) in the Global Influenza Surveillance Network (GISN).Citation4 The main goals of this network are: establishing the epidemiological patterns of influenza, assessing the burden of influenza, updating the composition of seasonal influenza vaccines, assessing and monitoring the antiviral susceptibility of circulating influenza strains, detecting and identifying novel influenza viruses that have the potential to cause a pandemic.Citation4 Altogether, the surveillance activities performed by this network are critical to make the annual update of vaccine composition a reliable option rather than a risky bet to use for the control of such a global and persistent public health threat as influenza.
As part of the Italian Influenza Surveillance Network, the activity and circulation of influenza viruses in Northern Italy (Lombardy, the most populous Italian region: nearly 10 million of the 60 million inhabitants of Italy) were investigated during two consecutive seasons (2010–2011 and 2011–2012). The molecular characteristics of the HA sequence of circulating viruses were analyzed to investigate the emergence of influenza viruses variants.
Results
2010–2011 and 2011–2012 influenza activity in Northern Italy
In the surveyed area, the influenza activity of these two post-pandemic seasons was similar in terms of both time frame (both seasons lasted 11 weeks and peaked in week 5) and impact (with peak incidence of about 8‰ and cumulative incidence close to 70‰). Influenza activity peaked in February and declined by the end of March. The timing of the 2010–2011 and 2011–2012 influenza seasons was similar to the timing seen prior to the emergence of the pandemic A(H1N1) virus in 2009 (). In fact, these figures show that all returned to normalcy after the unusual event of the 2009 pandemic, characterized by a sharp and early peak in week 44 and very high morbidity rates (incidence of ILI at peak over 13‰). It is noteworthy that the cumulative incidences registered in the two analyzed seasons were about 2-fold higher than that reported during the 2009 pandemic, which accounted for a sharper wave than the broad ones observed in the last two seasons ().
Figure 1. Morbidity rates of influenza-like illness (ILI) ( × 1,000 inhabitants) reported in Lombardy during the 2009–2010 (pandemic), 2010–2011 and 2011–2012 seasons.
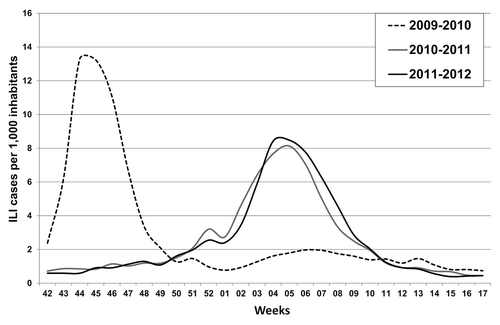
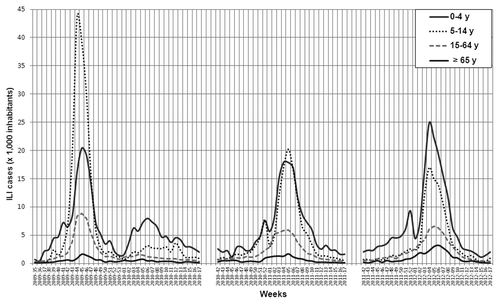
Whereas the 2010–2011 and the 2011–2012 epidemic waves almost overlapped in terms of the ILI incidence in the total population, several differences can be observed in terms of the pattern of ILI according to different age groups. shows the trend of ILI morbidity incidence by age group during the 2009 pandemic and the two post-pandemic influenza seasons analyzed. It is noteworthy that ILI incidence among the elderly (≥ 65 y) was 2-fold higher during the 2011–2012 season than in both the 2010–2011 epidemic and the 2009 pandemic.
Figure 2. Morbidity rates of influenza-like illness (ILI) ( × 1,000 inhabitants) by age group reported in Lombardy during the 2009–2010 (pandemic), 2010–2011 and 2011–2012 seasons.
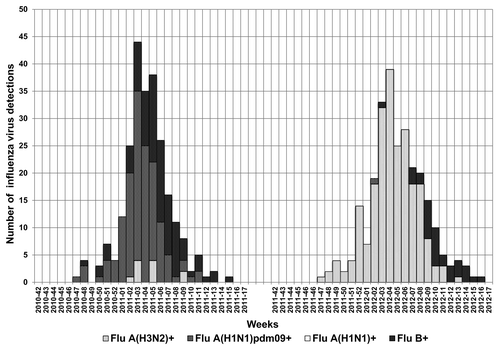
Results of virological surveillance showed that 43.3% (273/631) and 46.8% (266/568) of respiratory samples collected from outpatients with an ILI were positive for the detection of any influenza viruses (type A or B) during the 2010–2011 and the 2011–2012 epidemics, respectively. During the 2010–2011 season, influenza activity in Lombardy was related to influenza A viruses (accounting for 65.2% of total detections) and to influenza B viruses (accounting for 34.8% of total detections). Subtyping data revealed that 61.2% (167/273) of all influenza detections were influenza A(H1N1)pdm09 and 4% (11/273) A(H3N2). As expected, most influenza A viruses detected in the post-pandemic season belonged to pandemic A(H1N1)pdm09 subtype. This subtype almost disappeared in the following season. In fact, the 2011–2012 influenza activity in Lombardy was mainly associated with influenza A viruses: influenza A viruses detections (87.6%) predominated over type B (12.4%). Within type A, the H3 subtype (87.2%, 232/266) predominated over H1pdm09 viruses (0.4%, 1/266). The 2011–2012 epidemic was characterized by the almost exclusive circulation of A(H3N2) viruses. Influenza B detections were low in number, accounting for 12.4% (33/266) of total influenza detections compared with 34.8% in the previous season. After a peak in influenza A activity, influenza B activity followed in both seasons ().
Figure 3. Results of virological surveillance of influenza activity and circulation of influenza viruses in Lombardy during the 2010–2011 and 2011–2012 seasons.
Influenza virus analyses
A spatial-temporal representative number of influenza positive specimens collected during the two analyzed seasons were genetically characterized.
Influenza A(H1N1) viruses
Most of the influenza viruses detected in the surveyed area in the 2010–2011 season were A(H1N1). All H1N1 viruses were of the 2009 pandemic lineage. No former seasonal influenza viruses were detected. These viruses circulated almost exclusively during the 2010–2011 season: their circulation increased from week 51–2010, peaked in week 3–2011, and stopped in week 12–2011. Only one A(H1N1)pdm09 virus was detected during the 2011–2012 season.
shows a phylogenetic tree for the HA sequence of selected A(H1N1)pdm09 viruses. The HA sequences of the A(H1N1)pdm09 viruses detected in the 2010–2011 season fell into different phylogenetic groups, according to WHO classification.Citation5 Most (56.4%) HA sequences of the A(H1N1)pdm09 viruses fell into group 5 (A/Astrakhan/1/2011-like) and were characterized by amino acid mutations D97N, R205K, I216V, and V249L. 20.5% of HA sequences — characterized by amino acid variations D97N and S185T — belonged to group 6 (A/St Petersburg/27/2011-like), whereas the remaining sequences were dispersed among group 3 (A/Hong Kong/3934/2011-like; 5.1% of sequences), group 4 (A/Christchurch/16/2010-like; 12.8% of sequences), and group 7 (A/St Petersburg/100/2011-like; 5.1% of sequences). Most recent viruses fell into group 5, 6 or 7. Each genetic group can be defined by encoded amino acid substitutions, as reported in .
Figure 4. Phylogenetic tree based on A(H1N1)pdm09 viruses HA1 amino acid sequences. Only bootstrap values above 70% value are shown. The reference viruses are in bold. The vaccine A(H1N1) virus for the 2010–2011, 2011–2012, and 2012–2013 seasons (A/California/07/2009) is underlined.
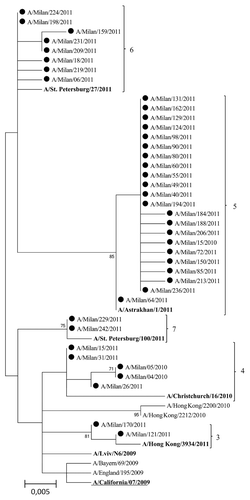
Table 1. Encoded HA amino acid substitutions defining the genetic groups within the A(H1N1)pdm09 viruses detected in the 2010–2011 season
Influenza A(H3N2) viruses
Influenza A(H3N2) viruses were identified only sporadically (4% of total detections) during the 2010–2011 post-pandemic season, whereas they were the predominating viruses in the 2011–2012 season in the surveyed area, accounting for 87.2% of all influenza viruses identified. The A(H3N2) virus activity started increasing from week 52–2011, peaked in week 4–2011, and then gradually decreased.
Phylogenetic analysis of the HA sequences of representative A(H3N2) viruses is shown in . The HA sequences of the A(H3N2) viruses isolated in the last two seasons fell into different phylogenetic groups, according to WHO classification.Citation5 Most (75%) of the HA sequences from A(H3N2) viruses detected during the 2010–2011 season — characterized by a low circulation of this viral subtype (H1pdm09: H3 ratio = 15) — fell into the A/Victoria/208/2009 genetic clade. These sequences were A/Alabama/04/2011-like and segregated into group 7. The remaining sequence (25%) from 2010–2011 season fell into the A/Perth/16/2009 genetic clade — the reference strain of both 2010–2011 and 2011–2012 vaccine composition.
Figure 5. Phylogenetic tree based on A(H3N2) viruses HA1 amino acid sequences. Only bootstrap values above 70% value are shown. Sequences from viruses detected during the 2010–2011 season are highlighted by a black dot; those from the 2011–2012 season by a gray dot. The reference viruses are in bold. The vaccine A(H3N2) virus for the 2010–2011 and 2011–2012 seasons (A/Perth/16/2009) is underlined. The vaccine A(H3N2) virus for the 2012–2013 season (A/Victoria/361/2011) is indicated by an arrow.
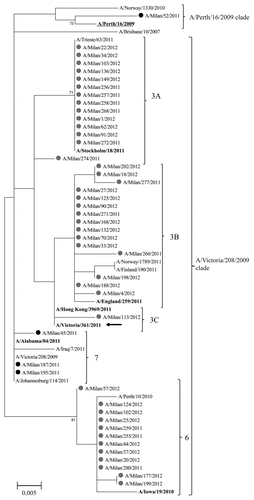
All HA sequences of the A(H3N2) viruses isolated in the 2011–2012 season fell into the A/Victoria/208/2009 genetic clade and predominantly into groups 3 (71.4% of sequences) or 6 (28.6% of sequences). Group 3 can be sub-divided into three groups: 3A (A/Stockholm/18/2011-like), 3B (A/England/259/2011-like), and 3C (A/Hong Kong/3969/2011-like), which included 33.3%, 35.7%, and 2.4% of study sequences, respectively. Group 6 included sequences that shared a high similarity with the A/Iowa/19/2010 strain. The amino acid characteristics of HA sequences of the genetic groups identified are listed in . It is significant that almost all mutations occurred in the epitopes.
Table 2. Encoded HA amino acid substitutions defining the genetic groups within the A/Victoria/208/2009(H3N2) clade
Influenza B viruses
B viruses presented with a mixed circulation of viral variants, since viruses belonging to both B/Victoria and B/Yamagata lineages co-circulated during these two seasons in different proportions: in the 2010–2011 season, influenza viruses of the B/Victoria-lineage predominated by a 1.5-fold margin over those of the B/Yamagata-lineage, while during the 2011–2012 season, viruses belonging to the B/Yamagata-lineage predominated by a 1.8-fold margin over those belonging to the B/Victoria one. This represents a considerable rise in the proportion of B/Yamagata viruses.
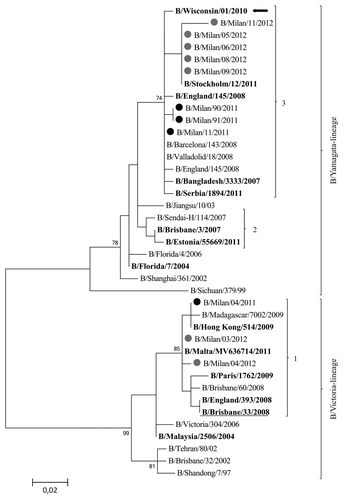
presents the phylogenetic analysis of the HA sequences of B viruses isolated during the two analyzed seasons. One-third of influenza B viruses characterized during these two seasons fell into the B/Victoria-lineage. The HA sequences of these viruses fell into clade 1, with only a small number of amino acid substitutions compared with the HA sequences of the B/Brisbane/60/2008 strain, the recommended vaccine virus included in both 2010–2011 and 2011–2012 vaccine composition. Two-thirds of B viruses molecularly characterized belonged to the B/Yamagata-lineage and fell into clade 3, represented by the reference virus B/Bangladesh/3333/2007 and the vaccine virus B/Wisconsin/1/2010. Within clade 3, the B viruses isolated during the 2011–2012 fell into group B/Stockholm/12/2011-like, characterized by the amino acid substitutions V29A and L172Q. It is noteworthy that all B viruses detected in individuals who underwent influenza vaccination in the 2010–2011 and 2011–2012 seasons (in both cases including the B/Brisbane/60/2008 strain belonging to the B/Victoria-lineage) belonged to the B/Yamagata-lineage.
Figure 6. Phylogenetic tree based on influenza B viruses HA1 protein sequences. Only bootstrap values above 70% value are shown. Sequences from viruses detected in the 2010–2011 season are highlighted by a black dot; those from the 2011–2012 season by a gray dot. The reference viruses are in bold. The vaccine B virus for the 2010–2011 and 2011–2012 seasons (B/Brisbane/60/2008) is underlined. The vaccine B virus for the 2012–2013 influenza season (B/Wisconsin/1/2010) is indicated by an arrow.
Discussion
As part of the Italian Influenza Surveillance Network, the activity and circulation of influenza viruses in Lombardy were investigated in the post-pandemic era during two consecutive seasons. Special attention was paid to the molecular characterization of the HA sequence of circulating viruses in order to assess the emergence of new HA variants or vaccine-escape viruses in the territory.
During the 2010–2011 winter season, influenza activity in Europe peaked in January and February 2011 and declined by the end of March. In many countries, after a peak in influenza A activity, influenza B activity followed. Cumulative influenza virus typing and subtyping showed that influenza A(H1N1)pdm09 predominated, with influenza B viruses representing fewer than 30% of the total viruses identified, while A(H3N2) viruses accounted for only 2% of identified viruses. Despite both being in the minority, influenza B and A(H3N2) activity increased as compared with A(H1N1)pdm09 activity in the previous season.Citation9
Regarding the 2011–2012 influenza season, in Europe activity started later than in the 2010–2011 season, but increased slowly over consecutive weeks after week 52–2011 and peaked sharply in weeks 8–2012 and 9–2012; this contrasts with the broad peak (from week 52–2010 to week 6–2011) seen in the 2010–2011 season. Influenza detections reported across the whole WHO Euro region were about half those reported in the 2010–2011 season,Citation9 but, again, type A detections (90.5%) predominated over type B (9.5%). In Europe the pattern of ILI was inconsistent, with some countries showing low levels of ILI (e.g., England, Romania, Switzerland, Russian Federation, Germany) and others reporting activity at levels similar to (e.g., Italy, Spain) or even greater than (e.g., Sweden) those of last year, with most countries reporting later starts, and subsequent peaks, in influenza activity than usual. Within type A, the H3 subtype (96%) predominated over H1pdm09 viruses (4%) by a 24-fold margin. Influenza B detections were low in number, accounting for 9.5% of total influenza detections compared with 31.5% in the previous season.Citation5
In Lombardy, the 2010–2011 and 2011–2012 seasons were characterized by medium influenza activity. Their epidemic waves almost overlapped in terms of both time frame and impact. This epidemiological picture differed from that observed in Europe,Citation5,Citation9 where the 2011–2012 influenza season seemed to occur late and have less impact than the previous one. However, the timing of the last influenza seasons was similar to the timing seen prior to the emergence of the pandemic A(H1N1) virus in 2009.
From an epidemiological point of view, the 2010–2011 influenza season was characterized by higher morbidity rates in school-age children (aged 5 to 14 y), whereas the main feature of the 2011–2012 season was higher morbidity rate (2-fold higher) among the elderly (≥ 65 y), one of the main targets of influenza vaccination in Italy. According to reports in the WHO Euro region,Citation5,Citation9 A(H1N1)pdm09 virus mainly circulated in the 2010–2011 season, whereas A(H3N2) was the predominant virus in the 2011–2012 epidemic.
As expected, the influenza A(H1N1)pdm09 viruses accounted for the majority of viruses detected in the 2010–2011 post-pandemic season in the surveyed area. The phylogenetic analysis of HA sequences of the A(H1N1)pdm09 viruses circulating in Lombardy showed that viruses isolated during the 2010–2011 season segregated in a different genetic group with respect to those identified during the 2009 pandemic. Even so, the post-pandemic A(H1N1)pdm09 viruses were still closely related to the vaccine viral strain A/California/07/2009. In a previous study,Citation10 the global and local phylogenetic analyses showed that all the HA sequences sampled in the post-pandemic season (2010–2011) grouped into at least four highly significant Italian clades, whereas those of the pandemic season (2009–2010) were interspersed with isolates from other countries at the tree root. The time of the most recent common ancestor of the strains circulating in the pandemic season in Italy was estimated to be between the spring and summer of 2009, whereas the Italian clades of the post-pandemic season originated in the spring of 2010 and showed radiation in the summer/autumn of the same year. The local phylogeography analysis showed that the first season of infection originated in Northern Italian high-density populated localities, whereas the second one involved less densely populated localities.
In the 2010–2011 season, influenza B viruses predominated along with influenza A(H1N1)pdm09 viruses representing nearly 40% of the total viruses identified. During this epidemic, influenza viruses of the B/Victoria-lineage predominated by a 1.5-fold margin over those of the B/Yamagata-lineage. In the surveyed area, B viruses circulated also during the 2011–2012 season, but were not so common (nearly 12% of the total influenza viruses identified). It is noteworthy that in the 2011–2012 season, viruses belonging to the B/Yamagata-lineage predominated by a 1.8-fold margin over those belonging to the B/Victoria one, thus representing a considerable rise in the proportion of B/Yamagata viruses. Overall, B viruses presented with a mixed circulation of viral variants, since viruses belonging to both B/Victoria and B/Yamagata lineages co-circulated during these two seasons in different proportions. As previously reported by other authors,Citation11-Citation13 this epidemiological feature — where a B virus lineage is unable to play a firm role in the epidemiological circulation — suggests that the inclusion in influenza vaccine of strains belonging to both B lineages may be advisable. However, licensed trivalent seasonal influenza vaccines contain antigens from only a single influenza B virus and thus provide limited immunity against circulating influenza B strains of the lineage not present in the vaccine. In recent years, predictions about which B lineage will predominate in an upcoming influenza season have been no better than chance alone, correct in only 5 of the 10 seasons from 2001 to 2011.Citation14 Seasonal influenza vaccines could be improved by inclusion of influenza B strains of both lineages. The resulting quadrivalent influenza vaccines would allow influenza vaccination campaigns to respond more effectively to current global influenza epidemiology.Citation15,Citation16
In the surveyed area, the 2011–2012 epidemic wave was sustained almost exclusively by influenza A(H3N2) viruses, which accounted for 87.2% of total influenza virus detections, similarly to what was observed across the whole WHO Euro region.Citation5 One of the main features of the 2011–2012 season was that the consultation rate for ILI in the age group ≥ 65 y was 2-fold higher than those registered during the 2009 pandemic and the 2010–2011 season. The impact of A(H3N2) virus infection was particularly important among the elderly. This piece of evidence provided an early warning about the sub-optimal immune protection elicited by vaccine against circulating viruses, since people aged over 65 y are one of the targeted groups for yearly influenza vaccination. Estimates from the I-MOVE European studyCitation17 suggest that, among the target groups for vaccination, the effectiveness of the 2011–2012 influenza vaccine was low to moderate against medically-attended ILI confirmed as influenza A(H3N2). This may be explained by a limited match identified between the circulating influenza A(H3N2) virus strains and the vaccine strain.Citation5,Citation18 In addition, in most European countries, excess mortality coincided with or followed reported increases in influenza activity. This pattern has been regularly observed in the pre-pandemic yearsCitation19-Citation21 and corroborates the association of influenza — particularly influenza A(H3N2) — with excess deaths in the elderly.Citation22 In addition, the excess deaths in the elderly may be further explained by the imperfect match of circulating influenza A(H3N2) viruses with the strain included in the 2011–2012 vaccine. Indeed, the phylogenetic analysis of A(H3N2) viruses circulating in Northern Italy in the 2011–2012 season showed that A(H3N2) viruses belonged to the A/Victoria/208/2009 genetic clade characterized by sequences all carrying substitutions in HA epitope sites with respect to viruses of the A/Perth/16/2009 genetic clade (strain included in 2011–2012 vaccine composition). Among analyzed H3 sequences from the 2011–2012 epidemic, about one-third fell into group 6 (A/Iowa/19/2010-like) whereas two-thirds fell into group 3, which can be sub-divided into three groups: 3A (A/Stockholm/18/2011-like), 3B (A/England/259/2011-like) and 3C (A/Hong Kong/3969/2011-like). As reported by WHO, the majority of viruses in each of the predominating genetic groups (groups 3A, 3B, 3C, 5 and 6) showed reduced reactivity with antisera raised against the vaccine virus A/Perth/16/2009 in both virus neutralisation and HI assays.Citation5 It is noteworthy that most HA sequences from A(H3N2) viruses detected during the 2010–2011 post-pandemic season — characterized by a low circulation of this viral subtype – fell into the A/Victoria/208/2009 genetic clade. This was an early warning of the circulation of the genetic drift HA variant during the previous epidemic season. In fact, as previously reported,Citation23 the time scale reconstruction of the phylogeny suggests that several A(H3N2) groups appeared independently between the summer and the winter 2011. However, the common origin of all the circulating A(H3N2) strains dated back to the 2009 pandemic period.
Conclusion
Epidemiological and virological surveillance of influenza is an essential informative tool for tuning vaccination strategies. The results of this study show that surveillance of genetic changes in the HA domain may reveal immune escape and provide early information on newly emerging strains with epidemiologic inference. This would greatly enhance the ability to monitor the antigenic drift and to detect rarer antigenic shifts of the virus virtually in real-time. The phylogenetic approach is of particular importance for the evaluation of the introduction and circulation of new variants in the area. Therefore, it should be implemented within the framework of influenza virological surveillance.
Materials and Methods
The Influenza Surveillance Network methodology and samples
The influenza epidemiological and virological surveillance network (InfluNet) consists of pediatricians and general practitioners, who survey approximately 2% of the general population, reporting weekly on the number of new cases of influenza-like illness (ILI), and collect respiratory samples for virological evaluation.Citation24 The standard case definition of ILI is abrupt onset of fever (> 38°C), one or more respiratory symptoms (non-productive cough, sore throat, or rhinitis), and one or more systemic symptoms (myalgia, headache, severe malaise).Citation25
Respiratory samples (nasal swabs) were collected from outpatients with a clinical presentation of ILI during the 2010–2011 and 2011–2012 seasons in Lombardy. Overall, 1,199 nasal swabs were collected: 631 during the 2010–2011 season and 568 during the 2011–2012 season.
Nucleic acids purification, amplification, sequencing and phylogenetic analyses
RNA was extracted from each specimen using the Invisorb® Spin Virus RNA Mini kit (STRATEC Molecular GmbH, Catalogue number: 1040300300). An one-step real-time RT-multiplex-PCR assay was performed to simultaneously detect and type A and B influenza viruses by using primer/probe sets for two different genome regions: the matrix region of influenza type A virus and the nucleoprotein region of influenza type B virus.Citation26 The subtyping of influenza A positive samples was performed by a one-step real-time RT-PCR assay using specific primer/probe sets for the HA gene.Citation26
The molecular characterization of influenza viruses was performed by sequence analysis of the globular head region of the HA protein (HA1 subunit) specific for A(H1N1) influenza virus (nt. 64–1,058), A(H3N2) influenza virus (nt. 174–1,056), and B (nt. 12–1,169) influenza virus.Citation11,Citation27,Citation28 The HA nucleotide sequences were obtained by automated DNA sequencing (ABI PRISM® 3100 Genetic Analyzer, Life Technologies). Multiple sequence alignment was conducted using ClustalX, version 2.0.10. Phylogenetic trees were constructed by means of the Maximum likelihood methodCitation29 and JTT + G or I modelCitation30 using MEGA package version 5.05.Citation31 Only bootstrap values above 70% were identified as distinct groups.Citation32 Sequences were deposited into GenBank National Center for Biotechnology Information (NCBI).Citation33
| Abbreviations: | ||
| WHO | = | World Health Organization |
| GISN | = | Global Influenza Surveillance Network |
| HA | = | Hemagglutinin |
| RT | = | retro transcription |
| PCR | = | polymerase chain reaction |
Acknowledgments
This work was supported by a grant from Direzione Generale Sanità, Regione Lombardia (grant no. 5988, 30/6/2011). Special thanks to Dr. Jennifer S. Hartwig for editorial assistance.
Submitted
10/19/12
Accepted
10/30/12
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Lambert LC, Fauci AS. Influenza vaccines for the future. N Engl J Med 2010; 363:2036 - 44; http://dx.doi.org/10.1056/NEJMra1002842; PMID: 21083388
- Nicholson KG, Wood JM, Zambon M. Influenza. Lancet 2003; 362:1733 - 45; http://dx.doi.org/10.1016/S0140-6736(03)14854-4; PMID: 14643124
- Fukuyama S, Kawaoka Y. The pathogenesis of influenza virus infections: the contributions of virus and host factors. Curr Opin Immunol 2011; 23:481 - 6; http://dx.doi.org/10.1016/j.coi.2011.07.016; PMID: 21840185
- World Health Organization (WHO) Global Influenza Surveillance Network. (GISN). Strengthening the WHO Global Influenza Surveillance Network (GISN). 2011. Available at: http://www.who.int/influenza/gisrs_laboratory/GISN_Meeting_Report_apr2011.pdf
- World Health Organization (WHO) Influenza Centre London. September 2012 interim report. Report prepared for the WHO annual consultation on the composition of influenza vaccine for the Southern Hemisphere 2013. 17th–19th September 2012. Available at: http://www.nimr.mrc.ac.uk/documents/about/Interim_Report_September_2012_2.pdf
- Nunthaboot N, Rungrotmongkol T, Malaisree M, Kaiyawet N, Decha P, Sompornpisut P, et al. Evolution of human receptor binding affinity of H1N1 hemagglutinins from 1918 to 2009 pandemic influenza A virus. J Chem Inf Model 2010; 50:1410 - 7; http://dx.doi.org/10.1021/ci100038g; PMID: 20726599
- Wiley DC, Wilson IA, Skehel JJ. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 1981; 289:373 - 8; http://dx.doi.org/10.1038/289373a0; PMID: 6162101
- Lees WD, Moss DS, Shepherd AJ. A computational analysis of the antigenic properties of haemagglutinin in influenza A H3N2. Bioinformatics 2010; 26:1403 - 8; http://dx.doi.org/10.1093/bioinformatics/btq160; PMID: 20388627
- World Health Organization (WHO) Influenza Centre London. September 2011 interim report. Report prepared for the WHO annual consultation on the composition of influenza vaccine for the Southern Hemisphere 26th–30th September 2011. Available at: http://www.nimr.mrc.ac.uk/documents/about/interim-report-sep-2011.pdf
- Zehender G, Pariani E, Piralla A, Lai A, Gabanelli E, Ranghiero A, et al. Reconstruction of the evolutionary dynamics of the A(H1N1)pdm09 influenza virus in Italy during the pandemic and post-pandemic phases. PLoS One 2012; 7:e47517; http://dx.doi.org/10.1371/journal.pone.0047517; PMID: 23152755
- Ansaldi F, D’Agaro P, De Florentiis D, Puzelli S, Lin YP, Gregory V, et al. Molecular characterization of influenza B viruses circulating in northern Italy during the 2001-2002 epidemic season. J Med Virol 2003; 70:463 - 9; http://dx.doi.org/10.1002/jmv.10418; PMID: 12767012
- Puzelli S, Frezza F, Fabiani C, Ansaldi F, Campitelli L, Lin YP, et al. Changes in the hemagglutinins and neuraminidases of human influenza B viruses isolated in Italy during the 2001-02, 2002-03, and 2003-04 seasons. J Med Virol 2004; 74:629 - 40; http://dx.doi.org/10.1002/jmv.20225; PMID: 15484280
- Chen GW, Shih SR, Hsiao MR, Chang SC, Lin SH, Sun CF, et al. Multiple genotypes of influenza B viruses cocirculated in Taiwan in 2004 and 2005. J Clin Microbiol 2007; 45:1515 - 22; http://dx.doi.org/10.1128/JCM.02189-06; PMID: 17329451
- Ambrose CS, Levin MJ. The rationale for quadrivalent influenza vaccines. Hum Vaccin Immunother 2012; 8:81 - 8; PMID: 22252006
- Belshe RB. The need for quadrivalent vaccine against seasonal influenza. Vaccine 2010; 28:Suppl 4 D45 - 53; http://dx.doi.org/10.1016/j.vaccine.2010.08.028; PMID: 20713260
- Reed C, Meltzer MI, Finelli L, Fiore A. Public health impact of including two lineages of influenza B in a quadrivalent seasonal influenza vaccine. Vaccine 2012; 30:1993 - 8; http://dx.doi.org/10.1016/j.vaccine.2011.12.098; PMID: 22226861
- Kissling E, Valenciano M, I-MOVE Case-Control Studies Team. Early estimates of seasonal influenza vaccine effectiveness in Europe among target groups for vaccination: results from the I-MOVE multicentre case-control study, 2011/12. Euro Surveill 2012; 17:pii: 20146; PMID: 22516046
- European Centre for Disease Prevention and Control (ECDC). 2012. Risk assessment. Seasonal influenza 2011-2012 in Europe (EU/EEA countries). 9 March 2012. Available at: http://ecdc.europa.eu/en/publications/Publications/120312-TER-Seasonal-influenza-risk-assessment.pdf
- Hardelid P, Pebody R, Andrews N. Mortality caused by influenza and respiratory syncytial virus by age group in England and Wales 1999-2010. Influenza Other Respi Viruses 2013; 7:35 - 45; http://dx.doi.org/10.1111/j.1750-2659.2012.00345.x; PMID: 22405488
- Nielsen J, Mazick A, Glismann S, Mølbak K. Excess mortality related to seasonal influenza and extreme temperatures in Denmark, 1994-2010. BMC Infect Dis 2011; 11:350; http://dx.doi.org/10.1186/1471-2334-11-350; PMID: 22176601
- Nunes B, Viboud C, Machado A, Ringholz C, Rebelo-de-Andrade H, Nogueira P, et al. Excess mortality associated with influenza epidemics in Portugal, 1980 to 2004. PLoS One 2011; 6:e20661; http://dx.doi.org/10.1371/journal.pone.0020661; PMID: 21713040
- Mazick A, Gergonne B, Nielsen J, Wuillaume F, Virtanen MJ, Fouillet A, et al. Excess mortality among the elderly in 12 European countries, February and March 2012. Euro Surveill 2012; 17:pii: 20138; PMID: 22516003
- Pariani E, Amendola A, Ebranati E, Ranghiero A, Lai A, Anselmi G, et al. Genetic drift influenza A(H3N2) virus hemagglutinin (HA) variants originated during the last pandemic turn out to be predominant in the 2011-2012 season in Northern Italy. Infect Genet Evol 2012; 13C:252 - 60; PMID: 23174527
- InfluNet website. Available at: http://www.iss.it/iflu/
- Ministero della Salute website. Available at: http://www.salute.gov.it/
- World Health Organization (WHO) Global Influenza Surveillance Network. Manual for the laboratory diagnosis and virological surveillance of influenza. 2011. Available at: http://whqlibdoc.who.int/publications/2011/9789241548090_eng.pdf
- Ellis JS, Alvarez-Aguero A, Gregory V, Lin YP, Hay A, Zambon MC. Influenza AH1N2 viruses, United Kingdom, 2001-02 influenza season. Emerg Infect Dis 2003; 9:304 - 10; PMID: 12643824
- Ellis JS, Chakraverty P, Clewley JP. Genetic and antigenic variation in the haemagglutinin of recently circulating human influenza A (H3N2) viruses in the United Kingdom. Arch Virol 1995; 140:1889 - 904; http://dx.doi.org/10.1007/BF01322680; PMID: 7503689
- Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 1981; 17:368 - 76; http://dx.doi.org/10.1007/BF01734359; PMID: 7288891
- Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 1992; 8:275 - 82; PMID: 1633570
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011; 28:2731 - 9; http://dx.doi.org/10.1093/molbev/msr121; PMID: 21546353
- Felsenstein J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985; 39:783 - 91; http://dx.doi.org/10.2307/2408678
- GenBank National Center for Biotechnology Information (NCBI). Available at: http://www.ncbi.nlm.nih.gov/