Abstract
Background: Patients with gastric cancer benefit from perioperative chemotherapy, however, treatment is toxic and many patients will relapse. The trifunctional antibody catumaxomab targets EpCAM on tumor cells, CD3 on T cells, and the Fcγ-receptor of antigen-presenting cells. While in Europe catumaxomab is approved for treating malignant ascites, it has not been investigated in the perioperative setting and its exact immunological mode of action is unclear.
Methods: In our study, gastric cancer patients received neoadjuvant platinum-based chemotherapy, one intraoperative application of catumaxomab, and 4 postoperative doses of intraperitoneal catumaxomab. Immunomonitoring was performed in 6 patients before surgery, after completion of catumaxomab treatment, and one month later.
Results: Intraperitoneal application of catumaxomab caused an increased expression of activation markers on the patients’ T cells. This was accompanied by a transient decrease in numbers of CXCR3+ effector T cells with a T-helper (Th)-1 phenotype in the peripheral blood. All patients evidenced pre-existing EpCAM-specific CD4+ and/or CD8+ T cells. While these cells transiently disappeared from the blood stream after intraperitoneal application of catumaxomab, we detected increased numbers of peripheral EpCAM-specific cells and a modified EpCAM-specific T-cell repertoire 4 weeks after completion of treatment. Finally, catumaxomab also amplified humoral immunity to tumor antigens other than EpCAM.
Conclusions: Our findings suggest that catumaxomab exerts its clinical effects by (1) activating peripheral T cells, (2) redistributing effector T cells from the blood into peripheral tissues, (3) expanding and shaping of the pre-existing EpCAM-specific T-cell repertoire, and (4) spreading of anti-tumor immunity to different tumor antigens.
Introduction
Gastric cancer is the fourth most common cancer and the second leading cause of cancer-related death worldwide with 5-year survival rates of less than 20%.Citation1 When diagnosed at an early stage, surgery can be performed with a curative intent, however, 20–50% of patients will eventually relapse.Citation2–Citation7 It has long been proposed that adjuvant chemotherapy might improve the perspective of patients with operable gastric cancer and a number of phase III trials have been conducted, however, definitive evidence of the efficacy of adjuvant chemotherapy is still missing.Citation8 Randomized trials have demonstrated that in patients with operable gastric or lower esophageal adenocarcinomas, the application of perioperative chemotherapy leads to a decrease in tumor size and stage and improves survival. However, chemotherapy causes significant toxicity and, accordingly, in the pivotal MAGIC study only ~40% of patients were capable of completing postoperative chemotherapy consisting of epirubicin/cisplatin/fluorouracil.Citation3 Therefore, the integration of adjuvant/perioperative approaches which are effective, more specific and less toxic seems desirable for improving the multimodal therapy of gastric cancer.
It has repeatedly been shown that an infiltration of the tumor tissue by lymphocytes,Citation9 in particular by memory T cells,Citation10,Citation11 has a beneficial effect on the survival of patients with esophageal adenocarcinoma or gastric cancer after potentially curative surgery. Unfortunately, immunotherapeutic approaches actively promoting the immune control of gastric cancer have not been available. The trifunctional monoclonal antibody catumaxomab binds to three different structures: its mouse IgG2a chain binds to the human tumor antigen EpCAM, its rat IgG2 chain binds to the human pan-T-cell antigen CD3 and a third functional section within the hybrid Fc region binds to Fcγ receptor type I and type III-expressing accessory cells. Animal models and in vitro experiments have suggested that the effect of catumaxomab is based on the simultaneous and direct activation of various components of the cellular immune system in the immediate tumor environment.Citation12–Citation18 However, catumaxomab’s exact mode of action is still unclear and it is unknown whether catumaxomab given intraperitoneally elicits a specific local or systemic immune response against its target antigen EpCAM.
Based on a randomized phase II/III trial, which showed a significantly prolonged puncture-free survival in the catumaxomab group (median 46 d) when compared with the group receiving paracentesis alone (median 11 d), catumaxomab was approved in Europe for the treatment of malignant ascites.Citation19 Applied as a peritoneal infusion, catumaxomab not only reduces malignant ascites in cancer patients but also decreases the number of tumor cells in the peritoneal cavity.Citation19–Citation22 The presence of free intraperitoneal malignant cells represents a negative prognostic factor in patients with gastric cancer undergoing a potentially curative resection.Citation23–Citation25 These findings suggest that single cells, exfoliated from the surface of the primary cancer, are responsible for peritoneal dissemination, which is a very frequent pattern of recurrence in gastric carcinoma patients.Citation2,Citation4–Citation7 In the perioperative or adjuvant settings, intraperitoneal chemotherapy has been applied to eradicate these remaining tumor cells,Citation26 however, results are far from being optimal. Therefore, we set up a clinical trial investigating if catumaxomab can be safely applied as part of a multimodal perioperative therapy and whether this might improve the prognosis of gastric cancer patients. The study is currently at two years of follow-up and clinical results will be demonstrated separately. We herein report the results of a detailed analysis of the immunological consequences of catumaxomab when applied intraperitoneally to gastric cancer patients in the adjuvant setting.
Results
Catumaxomab treatment induces a broad T-cell activation and redistribution of Th1-type memory T cells
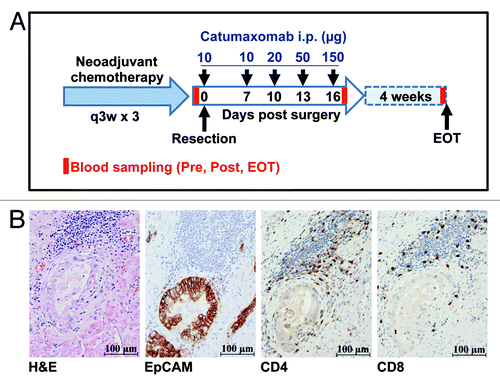
While EpCAM protein is absent from or only weakly expressed in normal gastric mucosa, up to 90% of patients with gastric cancer evidence an overexpression of EpCAM in their tumors as indicated by immunohistochemistry.Citation27 For the purpose of this study (), however, we specifically determined EpCAM expression in all 6 patients treated at our center by immunohistochemistry. Importantly, we detected strong expression of EpCAM protein in all cases (), with > 90% of tumor cells expressing the antigen, indicating that in our patients EpCAM indeed represented a meaningful target for catumaxomab treatment. Interestingly, even before the first administration of catumaxomab we observed in all 6 patients included an infiltration of the EpCAM-positive tumor tissues by varying numbers of CD4+ and CD8+ T cells (). This finding showed that basic numbers of this second cell type theoretically participating in the therapeutic effects of catumaxomab were already in close proximity to EpCAM-expressing tumor cells targeted by the antibody.
Figure 1. Tumors of gastric cancer patients treated with catumaxomab show strong expression of EpCAM and pre-therapeutic infiltration by CD4+ and CD8+ T cells. (A) Within the IP-CAT-GC-03 trial, a total of 6 patients were treated at our center with intraperitoneal (i.p.) catumaxomab as shown. Blood samples (Pre, Post, EOT) for immunomonitoring were collected at three time-points as indicated by red bars. (B) Resected tumors from all 6 gastric cancer patients were analyzed by immunohistochemistry (magnification × 400). Consecutive cuts are shown from the block of one representative subject (patient GC-3). In addition to a routine H&E stain, tumor samples were analyzed for the expression of EpCAM protein as well as for the presence of CD4+ and CD8+ T cells.
When we analyzed the effect of intraperitoneal catumaxomab application on the distribution of different lymphocyte subsets in the peripheral blood, we did not observe any convincing effect on percentages of CD4+ T cells, CD4+CD25+FOXP3+ T regulatory cells (Tregs), γ/δ T cells, and CD3+CD56+ natural killer T (NKT) cells when compared with baseline levels (data not shown). However, it seemed that catumaxomab induced a slight increase in the peripheral numbers of CD8+ T cells and a more pronounced rise in CD3-/CD56+ natural killer (NK) cells, in particular at later timepoints ().
Figure 2. Catumaxomab induces a mobilization of CD8+ T cells and NK cells and an activation of CD4+ and CD8+ T cells. (A) The catumaxomab-induced redistribution of lymphocytes subsets was analyzed in patients with gastric cancer (n = 6) shortly before surgery was performed (Pre), on day 17 after gastrectomy following infusion of the final dose of catumaxomab (Post), and 4 weeks after the last catumaxomab application (EOT) using four-color flow cytometry. Analysis was performed using a combination of a morphological lymphocyte gate and gates for CD8+ T cells (CD3+CD8+) and NK cells (CD3-CD56+), respectively. Logarithmically plotted values depict individual changes during treatment in comparison to results at baseline (Pre). (B) Fold changes in percentages of CD4+ and CD8+ T cells expressing activation markers CD69 and HLA-DR, respectively, were evaluated for the same patients and time points. Each patient (GC-01 to GC-06) is indicated by a separate symbol as given below the figure.
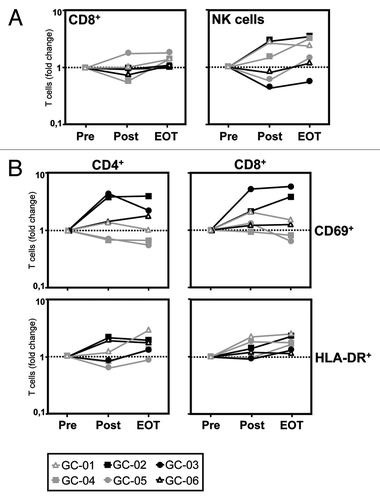
We next performed a serial analysis of the effect of catumaxomab treatment on the in vivo expression of different markers indicating the activation status and cytotoxic potential of the patients’ T cells. We did not observe any changes in CD4+ or CD8+ T cells expressing granzyme B, perforin, or CD107a (data not shown). However, we found a strong increase in CD4+ T cells (in 4/6 patients) and CD8+ T cells (in 5/6 patients), positive for the “early” activation marker CD69 after completion of catumaxomab treatment ( and ). In accordance with this finding, we observed a rise in CD4+ (in 5/6 patients) and CD8+ T cells (in 6/6 patients) expressing the “late” activation marker HLA-DR after another 4 weeks had passed ().
Table 1. Distribution of mechanistic elements across all patients (n = 6)
Catumaxomab causes CXCR3+ Th1-type T cells to egress from the peripheral blood
Chemokines are small proteins that direct the movement of circulating leukocytes to sites of inflammation or injury within peripheral tissues. Investigating whether catumaxomab treatment would specifically influence T cell subsets expressing certain chemokine receptors, we did not observe any convincing effect on the distribution of naïve/memory T-cell subpopulations as characterized by the expression of CD45RA and/or CCR7. We also did not detect a discriminating role of the expression of chemokine receptor CCR6 on the behavior of the patients’ T cells under the influence of catumaxomab (data not shown).
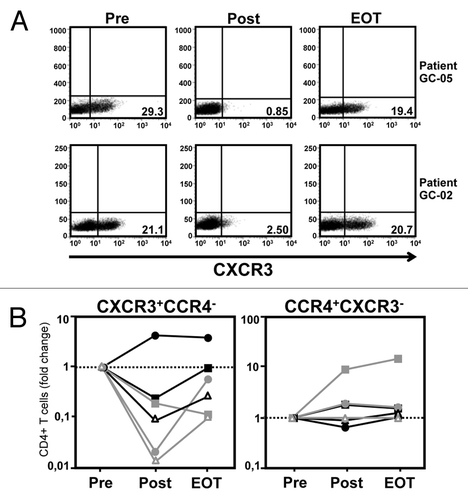
In contrast, we observed a dramatic effect of the intraperitoneal application of catumaxomab on the peripheral numbers of CD4+ T cells, and to a lesser extent also CD8+ T cells (), expressing chemokine receptor CXCR3 (). At baseline, 20–30% of all CD4+ T cells were usually positive for CXCR3, a chemokine receptor which is preferentially found on T cells with a T-helper type 1 (Th1) phenotype.Citation28 However, treatment with catumaxomab practically resulted in a depletion of CXCR3-expressing T cells from the peripheral blood of 5 out of 6 patients () and numbers had only returned to baseline levels 4 weeks after the last catumaxomab dose had been applied (). Interestingly, a comparable phenomenon was not observed for T cells expressing CCR4 (), a chemokine receptor which is predominantly found on T-helper type 2 (Th2) cells.Citation29
Figure 3. Application of catumaxomab causes CXCR3+ Th1-type T cells to leave the peripheral blood. (A) The catumaxomab-induced redistribution of lymphocytes as characterized by the expression of different chemokine receptors was analyzed in the peripheral blood of our patients (n = 6) shortly before surgery was performed (Pre), following infusion of the final dose of catumaxomab (Post), and 4 weeks after completion of catumaxomab treatment (EOT) using flow cytometry. Dot plots demonstrating the expression of chemokine receptor CXCR3 on CD4+ T cells at the different time points are shown for two representative patients. (B) Results for CD4+ T cells being either CXCR3+CCR4− (left) or CCR4+CXCR3− (right) are shown for all patients (see legend of regarding symbols for individual patients). Data are plotted logarithmically and depict fold changes in percentages of the given lymphocyte subset during treatment in comparison to results at baseline.
Application of catumaxomab leads to an amplification and shaping of pre-existing T-cell responses against EpCAM
Next, we asked the question whether the marked effects of catumaxomab on the distribution of effector-type T cells in our patients would also translate into a modulation of EpCAM-specific immunity. In order to amplify pre-existing antigen-specific memory T cells, we subjected our patients CD4+ and CD8+ T cells to one round of stimulation with a pool of overlapping peptides covering the complete amino acid sequence of EpCAM. Thereafter, we performed an IFN-γ ELISPOT using autologous targets pulsed with the same peptides combined in pools or as single peptides. Interestingly, we found that all 6 patients evidenced CD4+ and/or CD8+ T cells directed against EpCAM even before EpCAM-targeting catumaxomab was applied for the first time (). At baseline, CD4+ and CD8+ T-cell specificities were equally distributed between a number of different epitopes within the sequence of the full-length antigen ().
Figure 4. Application of catumaxomab results in an amplified and remodelled T-cell response against tumor antigen EpCAM. Patients with gastric cancer were analyzed before surgery was performed (Pre), following infusion of the final dose of catumaxomab (Post), and 4 weeks after completion of catumaxomab treatment (EOT). Frequencies of CD4+ and CD8+ T cells directed against 31 overlapping 20mer EpCAM peptides were determined in an ELISPOT assay following a single cycle of antigen-specific stimulation. (A) Black bars indicate numbers of patients evidencing CD4+ (upper lane) and/or CD8+ T-cell responses (lower lane) against 6 different pools of 5–6 peptides each at the given timepoints. (B) Exemplary IFN-γ ELISPOT results of patients GC-02 and GC-06 for CD4+ (two upper lanes) and CD8+ (two lower lanes) T-cell responses against single EpCAM peptides. Per well, 50 000 effector T cells were analyzed, background responses against irrelevant SSX2 peptide were usually < 10 spots/50 000 cells. (C) Frequencies of CD4+ and CD8+ T cells directed against individual EpCAM epitopes at the three timepoints (Pre, Post, EOT) are shown. Dots indicate spot numbers in ELISPOT assays with EpCAM peptide-pulsed target cells (T-APC) for 4 patients in whom the same individual EpCAM epitope was detectable at least at two of the three timepoints.
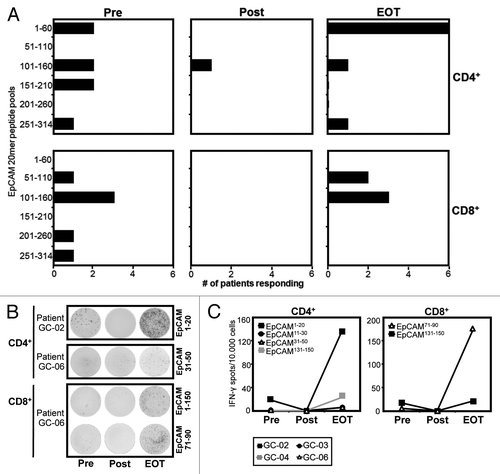
In accordance with our findings regarding CXCR3-positive Th1-type T cells, we found that detectable IFN-γ-secreting EpCAM-specific effector-type T cells had completely disappeared from the peripheral blood of all 6 patients () after the application of the final dose of catumaxomab (). At this time point, nearly all baseline responses against pools of overlapping EpCAM 20mer peptides had vanished () and we were not able to detect any T cells specific for single HLA class I- and II-restricted peptide epitopes we had previously identified ().
In marked contrast, CD4+ and CD8+ T-cell responses against EpCAM had reappeared 4 weeks after the last catumaxomab application (EOT) and in 4 out of 6 patients T-cell responses against pooled EpCAM peptides were even stronger than the ones recorded at baseline (). Accordingly, at EOT we observed enhanced responses against a number of single T cell epitopes we had identified when compared with baseline reactivity (). However, we did not only notice an amplification of pre-existing EpCAM-specific T-cell responses, it also seemed that catumaxomab treatment had resulted in a shaping of the tumor antigen-specific T-cell repertoire in the sense that responses against certain areas of the protein sequence had disappeared while others (i.e., CD4+ T-cell responses against amino acid region 1–60) were much more frequent than before the initiation of catumaxomab treatment ().
We next aimed at further describing the functional quality of the EpCAM-specific T-cell responses amplified and shaped by catumaxomab treatment. Among CD4+ cells, T regulatory cells (Tregs) represent a main obstacle of an effective anti-tumor T-cell responseCitation30 and might even be induced by immunotherapyCitation31 thus undermining a clinically relevant immune response. Performing intracellular staining for Th1-type cytokine IFN-γ, we detected a significant expansion of EpCAM-specific CD4+ T cells in the peripheral blood of patient GC-03 after completion of treatment with catumaxomab (). Importantly, these EpCAM-specific T cells, even after in vitro stimulation, did not co-express FOXP3, the most specific Treg marker to date,Citation32 suggesting that most of the EpCAM-targeting T cells amplified by catumaxomab indeed represented Th1-type memory effector cells.
Figure 5. Application of catumaxomab results in an expansion of effector-type T cells, and not Tregs, in the peripheral blood and in the tumor environment. (A) Intracellular cytokine staining followed by flow cytometry was performed using CD4+ T cells of patient GC-02 after one round of in vitro sensitization with pooled EpCAM peptides. Representative results of intracellular co-staining of IFN-γ and FOXP3 are shown for EpCAMCitation1–Citation20-specific CD4+ T cells obtained before initiation of catumaxomab therapy (Pre) and 4 weeks after completion of treatment (EOT). Dot plots show responses against the individual EpCAM peptide (upper row) and an SSX2 control peptide (lower row). (B) Bar graphs indicate mean numbers (+SEM) of CD4+ T cells specific for EpCAM amino acid region 151–210 (black bar) or control peptide SSX2 (white bar) within the malignant effusion of patient GC-02. Original ELISPOT data demonstrate spot numbers per well at 50 000 CD4+ T cells per well after presensitization with pooled EpCAM peptides.
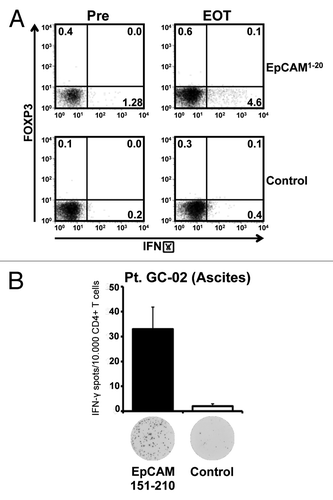
Interestingly, we could demonstrate that in the same patient EpCAM-specific effector-type T cells were also present in the immediate tumor environment. Patient GC-02 had developed a transient non-malignant ascites due to a local infection shortly after initiation of catumaxomab treatment. At this timepoint we detected significant numbers of CD4+ T cells specific for EpCAM amino acid region 151–210 in the patient’s benign effusion (), suggesting that EpCAM-specific T cells had indeed left the patient’s peripheral blood in order to travel into the immediate tumor environment.
Catumaxomab therapy also improves immune responses against tumor antigens other than EpCAM
Finally, we performed a broad analysis of autologous antibody responses against tumor antigens apart from EpCAM in order to answer the question whether the application of immunostimulating catumaxomab would lead to some form of antigen spreading. We analyzed our patients’ sera for immunoreactivity against a total number of 24 tumor antigens, most of which belonged to the strictly tumor-specific family of cancer-testis (CT) antigens (Table S1), a class of tumor-associated proteins, which are, apart from normal testis tissue, only expressed by malignant cells. Consequently, if a humoral response against these targets developed under catumaxomab treatment, this would mean that the agent indeed broadens tumor-specific immunity and does not simply cause, for example, unspecific autoimmune phenomena.
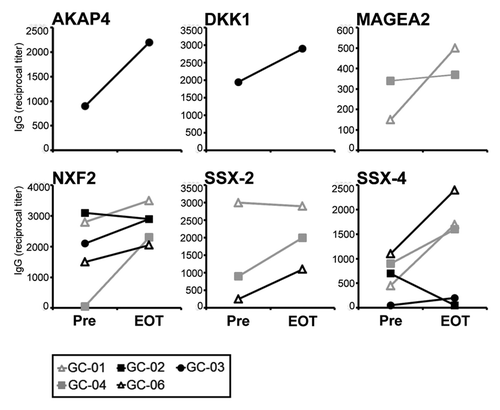
We did not at any of the three timepoints analyzed detect autologous IgG antibody responses against EpCAM (data not shown). This observation would be in agreement with a previous study suggesting that EpCAM only rarely induces spontaneous autoantibodies against native protein.Citation2 In contrast, in some of the patients we found baseline responses against a total number of 6 CT antigens, however, these responses were mostly low-titered (). Following application of catumaxomab, most of the pre-existing humoral responses increased in intensity and, in addition, in some patients, tumor-directed antibody responses even newly developed (). This observation suggested to us that catumaxomab is capable of amplifying pre-existing anti-tumor immune responses against a variety of targets and might even lead to spreading of immune responses from EpCAM to other tumor antigens.
Figure 6. Broad catumaxomab-induced modulation of humoral anti-tumor immunity. Lines indicate antibody responses against 6 different CT antigens in gastric cancer patients receiving catumaxomab in the adjuvant setting. Reciprocal IgG antibody titers against recombinant protein of the given CT antigen were measured by ELISA and are shown for two timepoints: before initiation of catumaxomab therapy (Pre) and 4 weeks after completion of treatment (EOT).
Discussion
In Europe, the trifunctional antibody catumaxomab has been approved for the treatment of malignant ascites caused by solid epithelial tumors and clinical studies are underway investigating its therapeutic efficacy in other indications. The exact immunological mode of action of catumaxomab, however, had remained unclear and we have here highlighted at least 4 different routes through which this immunotherapeutic agent seems to exert its effects on human tumors in vivo.
First, when applied as an adjuvant therapy, catumaxomab causes a prolonged activation of CD4+ and CD8+ T cells. It is well known that the process of T-cell activation is associated with de novo expression of surface molecules like CD69 and HLA-DR without requiring contact with the target antigen.Citation33 The two surface molecules show different kinetics with CD69 representing one of the earliest T-cell activation markers whereas HLA-DR expression indicates long-term activation. It has previously been indicated that catumaxomab treatment might result in an increased expression of CD69 on T cells.Citation22 We have shown here that in vivo, in addition to the expression of this short-term activation marker, catumaxomab also induces a prolonged activation of T cells as indicated by an enhanced expression of HLA-DR.
Unfortunately, in a number of clinical immunotherapy studies tumor progression occurred despite the induction of high numbers of tumor-specific T cells in the peripheral blood.Citation34,Citation35 A main reason for this failure of immunotherapeutic approaches seems to be that the effector T cells generated do not travel efficiently enough into the tumor tissue.Citation36,Citation37 On the other hand, it has repeatedly been suggested that the trifunctional antibody catumaxomab might enhance antitumor activity by redirecting T cells into the tumor,Citation38,Citation39 however, definitive experimental proof for this hypothesis has been lacking. We show here for the first time that treatment with catumaxomab specifically results in the depletion of CXCR3-expressing T cells from the peripheral blood of the patients followed by a return to baseline levels 4 weeks after the last catumaxomab dose had been applied. We believe that this phenomenon is based on the redistribution of this particular T-cell subtype from the peripheral blood into peripheral tissues, an idea that would be supported by a recent in vitro study indicating that catumaxomab promotes the transendothelial migration of activated T cells.Citation40
Chemokine receptor CXCR3 is preferentially expressed by effector T cells with a Th1 phenotypeCitation28 and traditionally T cells secreting Th1 cytokines (i.e., IFN-γ, TNF-α) have been considered critically important for the induction of cellular immunity and the generation of a relevant anti-tumor response in vivo.Citation41 It has also repeatedly been shown that the mobilization of Th1-type effector T cells into the tumor tissue and subsequent anti-tumor effects are mediated by chemokine receptor CXCR3 and its ligands CXCL9 and CXCL10Citation42–Citation47 and that, consequently, the presence of CXCR3+ T cells within the tumor has a positive influence on the patients’ outcome.Citation42,Citation44,Citation48 The antigens targeted by these prognostically relevant tumor-infiltrating T cells, however, have largely remained unidentified.
So far, no comprehensive analysis of spontaneous CD4+ and CD8+ T-cell responses against EpCAM has been performed. In our current study we detected T-cell responses comprising a large number of epitopes within the EpCAM sequence even before catumaxomab was applied for the first time. We observed two different effects of catumaxomab treatment on the EpCAM-specific T-cell repertoire. First, we found that EpCAM-specific CD4+ and CD8+ T cells, comparably to the finding with CXCR3-positive T cells, entirely disappeared from the patients’ peripheral blood after completion of catumaxomab treatment. Four weeks later, EpCAM-specific cells reappeared and in most cases the numbers of T cells targeting this tumor antigen were even higher than at baseline. Second, we found that after re-appearance in the peripheral blood the T-cell repertoire seemed more restricted to certain epitopes with the EpCAM sequence. We believe that this amplification and shaping of the EpCAM-specific memory T-cell repertoire is most likely due to a catumaxomab-induced re-exposure of pre-existing memory T cells to the target antigen.
It has recently been suggested that tumor antigen EpCAM might promote cancer immune evasion by favoring the development of Th2-type T cells.Citation49 Here, we have shown that the EpCAM-specific T cells, at least after catumaxomab-induced amplification and shaping of the responses, clearly express the phenotype of Th1-type effector T cells. This observation suggests that the EpCAM-specific cells represent one subgroup within the CXCR3-positive T cell compartment which might be guided into the tumor tissue following the application of catumaxomab—an idea that would further be supported by our observation of EpCAM-specific T cells infiltrating the peritoneal cavity after catumaxomab treatment.
It has been postulated that the activation of more than one class of immune effector cell is required for an optimal antitumor efficiency of catumaxomab treatment.Citation18 In vitro experiments have shown that catumaxomab through binding to their Fcγ receptor is capable of activating and maturing antigen-presenting cellsCitation18 Enhanced phagocytosis, processing, and presentation of tumor material by antigen-presenting cells might subsequently result in the induction of a polyclonal humoral and cellular catumaxomab-induced immune response against a variety of tumor antigens beyond EpCAM. Considering it possible that such an “antigen spreading” might in part be responsible for the therapeutic effect of catumaxomab, we analyzed our patients’ sera for immunoreactivity against a large number of tumor antigens apart from EpCAM, most of which belonged to the strictly tumor-specific CT antigen family. We found that following catumaxomab application, most of the pre-existing tumor-specific humoral responses increased in intensity and in some patients we even observed a de novo development of CT antigen-specific antibody responses. This observation suggested to us that catumaxomab is capable of amplifying pre-existing anti-tumor immune responses against a variety of targets and might even lead to spreading of immune responses from EpCAM to other tumor antigens.
Limitations of our current study include the small sample size and the lack of a control group, however, we would still suggest that catumaxomab, when applied as an adjuvant therapy, causes an activation of peripheral T cells, a redistribution of effector T cells from the blood into peripheral tissues, an expansion and shaping of the pre-existing tumor-specific T-cell repertoire, and spreading of humoral anti-tumor immunity to tumor antigens other than EpCAM. These findings highlight different routes through which catumaxomab might exert its effects in the adjuvant and maybe also in the metastasized clinical setting.
Methods
Patients and design of the clinical study
A total of 54 patients were included into this multicenter, open-label, single-arm, phase II trial (IP-CAT-GC-03). Out of this collective, 6 patients were enrolled at the University Medical Center Hamburg-Eppendorf and immunomonitoring was performed for this subgroup of patients. The clinical investigation was conducted according to the principles expressed in the Declaration of Helsinki and the trial had received approval from the ethics committee of the Ärztekammer Hamburg. All patients had provided written informed consent. Patients suffered from non-metastasized, nodal-positive or -negative gastric cancer with a T2–4 status according to the TNM classification. All patients received neoadjuvant platinum-based chemotherapy followed by en-bloc gastrectomy. Antibody treatment consisted of one intraoperative 10 µg bolus of catumaxomab (Removab®, Trion Pharma) applied into the peritoneum followed by 4 consecutive 3-h infusions of catumaxomab at escalating (10µg, 20µg, 50µg, 150µg) doses (). Adjuvant catumaxomab was applied intraperitoneally (i.p.) via an abdominal port on days 7, 10, 13, and 16 post surgery. Serum and peripheral blood were obtained from all patients treated at the University Medical Center Hamburg-Eppendorf shortly before surgery was performed (Pre), on day 17 after gastrectomy following infusion of the final dose of catumaxomab (Post), and 4 weeks after the last catumaxomab application (“end of treatment”; EOT).
Proteins and peptides
Recombinant full-length proteins of a total number of 22 cancer-testis antigens as well was of the tumor-associated carcinoembryonic antigen (CEA) and partial protein AKAP4 comprising amino acids 1–100 were obtained for our serological analyses (Table S1). Recombinant influenza nucleoprotein expressed in E. coli (Imgenex) served as a positive control in our ELISA assay. Full-length glutathione S-transferase (GST) either expressed in E. coli (Cell Systems) or in the wheat germ system (Abnova) was used as negative controls for the tumor antigens produced in the respective system. EpCAM 20mer peptides overlapping by 10 amino acids and covering the whole sequence of the protein were obtained from Iris Biotech. Single 20mer peptides derived from cancer-testis antigen SSX2 (Iris Biotech) were used as irrelevant controls in the read-out assays.
Immunohistochemistry
Immunohistochemistry was performed on formalin-fixed, paraffin-embedded tissue sections which had been obtained during gastrectomy for routine diagnostics. Briefly, consecutive cuts were deparaffinized and pretreated with 10 mmol/l citrate, pH 6.0 (Zymed) in a steam pressure cooker (Decloaking Chamber; BioCare Medical) followed by washing in distilled water. All further steps were performed at room temperature in a hydrated chamber. Slides were pretreated with peroxidase block (Dako) followed by blocking with goat serum diluted 1:5 in 50 mmol/l TRIS-HCl (pH 7.4) for 20 min. Staining was performed using murine monoclonal antibodies directed against EpCAM (clone VU-1D9; Novocastra), CD4 (clone 4B12; Dako), and CD8 (clone C8/144B; Dako). Slides were washed in 50 mmol/l TRIS-HCl and goat anti-mouse horseradish peroxidase-conjugated antibody (Dako) was applied for 30 min. After further washing, immunoperoxidase staining was developed using a diaminobenzidine chromogen kit (Dako), as per the manufacturer’s instructions.
Phenotypic analysis by flow cytometry
Peripheral blood mononuclear cells (PBMC) were prepared from heparinized blood or ascites using density gradient (Biochrom) centrifugation. PBMC were stained using the monoclonal antibodies listed in Table S2 and were analyzed by flow cytometry. Intracellular staining was performed after fixation and application of permeabilizing solution (BD Biosciences) according to the manufacturer’s instructions. Samples were measured using a FACSCalibur cytometer with BD Cell Quest TM Pro (Version 5.2.1) software (BD Biosciences) and analyzed using FlowJo Version 7.2.5 software (Tree Star).
Quantification of EpCAM-specific CD4+ and CD8+ T cells
Read-out-assays were performed following a single cycle of in vitro presensitization, as previously described.Citation50 Briefly, CD4+ and CD8+ T cells were sequentially purified from PBMC applying antibody-coated magnetic beads (Dynal). T cells were stimulated once with remaining irradiated CD8-CD4- cells pulsed with pools of 10–15 overlapping EpCAM peptides. After 10–20 d of culture in RPMI containing 10% SAB supplemented with glutamine, antibiotics, non-essential amino acids, IL-2 (10U/ml; Roche Diagnostics), and IL-7 (20ng/ml; R&D Systems), CD8+ and CD4+ T cells were harvested and were exposed to phytohemagglutinin (PHA; Roche Diagnostics)-stimulated CD4+ T cells (T-APC) pulsed over night with cognate or control peptides. In an ELISPOT assay, numbers of IFN-γ producing cells were determined applying a specific antibody kit (Mabtech) and resulting spots were counted using an AID EliSpot reader and EliSpot software version 3.2.3 (Autoimmun Diagnostika). The average of duplicates was calculated and a response was defined as positive if at least 10 spots per 10 000 cells were counted and EpCAM-induced responses exceeded background levels times three.
For the measurement of intracellular cytokines, pulsed T-APC were stained with 0.2 µM 5-(and-6) -carboxyfluorescein diacetate succinimidyl ester (CFSE; Molecular Probes) for 10 min at 37 °C. T-APC were then washed and incubated with presensitized effector T cells at a 1:2 ratio in 200 µl serum-free X-VIVO-15 medium (Lonza) at 37 °C for 7 h. Brefeldin-A (Sigma-Aldrich) at 10 μg/ml was added after the first two hours of culture. Cells were then fixed using FACS Lysing Solution (BD Biosciences) diluted 1:10, permeabilized using Permeabilizing Solution 2 (BD Biosciences), and stained with appropriate antibodies against CD4 and interferon (IFN)-γ (BD Biosciences). Co-staining of intracellular FOXP3 was performed applying the anti-FOXP3 mAb PCH101 (eBioscience). Cells were analyzed by flow cytometry gating on morphologically defined lymphocytes, CD4-positive and CFSE-negative cells.
Enzyme-linked-immunosorbent-assay (ELISA)
Immediately after patients’ blood sample was taken serum was frozen in 1 ml aliquots at −70 °C. 96-well-plates were coated over night at 4 °C with recombinant protein diluted in PBS at a final concentration of 1 μg/ml. Plates were blocked with PBS containing 3% milk powder for two hours at room temperature (RT). Sera diluted 1:100 in blocking buffer were added to the plates at 30 μl/well and incubated for two hours at RT. A secondary alkyline phosphatase (AP)-conjugated anti-human-IgG antibody (Southern Biotech) diluted at 1:3000 in blocking buffer was applied for one hour at RT. Detection reagent para-nitrophenyl phosphate (PNPP; Southern Biotech) was added to the plates and the phosphatase reaction took place at RT for 30 min before reaction arrest with 3N NaOH. Specific absorption was measured at 405 nm using a SunriseTM ELISA reader (Tecan).
In the screening part of the study, a sample was considered positive if the optical density (OD) measured exceed the autologous background signal measured with control protein GST by at least 50%. In the titration part of the study, serial serum dilutions were performed for antibody-positive samples and titers obtained with GST protein were used as reference values. For the calculation of titers, regression analyses were performed for the linear segment of the serum titration curves for the patient sample and pooled sera of five representative healthy donors. Titers were defined mathematically as the dilution at the intersection of both regression lines.
Statistics
Due to the relatively low number of patients included into this study, we decided to handle resulting data only descriptively without applying statistical tests.
Additional material
Download Zip (137.9 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study was supported by a grant from Fresenius Biotech.
Supplemental Material
Supplemental material may be found at www.landesbioscience.com/journals/vaccines/article/26065
Related Research Data
References
- Crew KD, Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol 2006; 12:354 - 62; PMID: 16489633
- Maehara Y, Hasuda S, Koga T, Tokunaga E, Kakeji Y, Sugimachi K. Postoperative outcome and sites of recurrence in patients following curative resection of gastric cancer. Br J Surg 2000; 87:353 - 7; http://dx.doi.org/10.1046/j.1365-2168.2000.01358.x; PMID: 10718807
- Cunningham D, Allum WH, Stenning SP, Thompson JN, Van de Velde CJ, Nicolson M, Scarffe JH, Lofts FJ, Falk SJ, Iveson TJ, et al, MAGIC Trial Participants. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med 2006; 355:11 - 20; http://dx.doi.org/10.1056/NEJMoa055531; PMID: 16822992
- Moriguchi S, Maehara Y, Korenaga D, Sugimachi K, Nose Y. Risk factors which predict pattern of recurrence after curative surgery for patients with advanced gastric cancer. Surg Oncol 1992; 1:341 - 6; http://dx.doi.org/10.1016/0960-7404(92)90034-I; PMID: 1341269
- Roviello F, Marrelli D, de Manzoni G, Morgagni P, Di Leo A, Saragoni L, De Stefano A, Italian Research Group for Gastric Cancer. Prospective study of peritoneal recurrence after curative surgery for gastric cancer. Br J Surg 2003; 90:1113 - 9; http://dx.doi.org/10.1002/bjs.4164; PMID: 12945079
- Wu CW, Lo SS, Shen KH, Hsieh MC, Chen JH, Chiang JH, Lin HJ, Li AF, Lui WY. Incidence and factors associated with recurrence patterns after intended curative surgery for gastric cancer. World J Surg 2003; 27:153 - 8; PMID: 12616428
- Yoo CH, Noh SH, Shin DW, Choi SH, Min JS. Recurrence following curative resection for gastric carcinoma. Br J Surg 2000; 87:236 - 42; http://dx.doi.org/10.1046/j.1365-2168.2000.01360.x; PMID: 10671934
- Okines AF, Cunningham D. Multimodality treatment for localized gastro-oesophageal cancer. Ann Oncol 2010; 21:Suppl 7 vii286 - 93; http://dx.doi.org/10.1093/annonc/mdq282; PMID: 20943630
- Setälä LP, Kosma VM, Marin S, Lipponen PK, Eskelinen MJ, Syrjänen KJ, Alhava EM. Prognostic factors in gastric cancer: the value of vascular invasion, mitotic rate and lymphoplasmacytic infiltration. Br J Cancer 1996; 74:766 - 72; http://dx.doi.org/10.1038/bjc.1996.434; PMID: 8795580
- Kamoi D, Ishii H, Takahashi H, Aoyama T, Toriyama T, Tanaka M, Kawamura Y, Kawashima K, Yoshikawa D, Amano T, et al. Sirolimus- vs. paclitaxel-eluting stent to coronary intervention in dialysis patients. Int J Cardiol 2013; 165:533 - 6; http://dx.doi.org/10.1016/j.ijcard.2011.09.078; PMID: 22000423
- Lee HE, Chae SW, Lee YJ, Kim MA, Lee HS, Lee BL, Kim WH. Prognostic implications of type and density of tumour-infiltrating lymphocytes in gastric cancer. Br J Cancer 2008; 99:1704 - 11; http://dx.doi.org/10.1038/sj.bjc.6604738; PMID: 18941457
- Gronau S, Schmitt M, Reinhardt P, Wiesneth M, Riechelmann H. [T-cells activated with a trifunctional bi-specific antibody in head and neck cancer]. Laryngorhinootologie 2005; 84:822 - 8; http://dx.doi.org/10.1055/s-2005-861448; PMID: 16358189
- Riesenberg R, Buchner A, Pohla H, Lindhofer H. Lysis of prostate carcinoma cells by trifunctional bispecific antibodies (alpha EpCAM x alpha CD3). J Histochem Cytochem 2001; 49:911 - 7; http://dx.doi.org/10.1177/002215540104900711; PMID: 11410615
- Gronau SS, Schmitt M, Thess B, Reinhardt P, Wiesneth M, Schmitt A, Riechelmann H. Trifunctional bispecific antibody-induced tumor cell lysis of squamous cell carcinomas of the upper aerodigestive tract. Head Neck 2005; 27:376 - 82; http://dx.doi.org/10.1002/hed.20170; PMID: 15818557
- Zeidler R, Reisbach G, Wollenberg B, Lang S, Chaubal S, Schmitt B, Lindhofer H. Simultaneous activation of T cells and accessory cells by a new class of intact bispecific antibody results in efficient tumor cell killing. J Immunol 1999; 163:1246 - 52; PMID: 10415020
- Ruf P, Lindhofer H. Induction of a long-lasting antitumor immunity by a trifunctional bispecific antibody. Blood 2001; 98:2526 - 34; http://dx.doi.org/10.1182/blood.V98.8.2526; PMID: 11588051
- Schmitt M, Schmitt A, Reinhardt P, Thess B, Manfras B, Lindhofer H, Riechelmann H, Wiesneth M, Gronau S. Opsonization with a trifunctional bispecific (alphaCD3 x alphaEpCAM) antibody results in efficient lysis in vitro and in vivo of EpCAM positive tumor cells by cytotoxic T lymphocytes. Int J Oncol 2004; 25:841 - 8; PMID: 15375531
- Zeidler R, Mysliwietz J, Csánady M, Walz A, Ziegler I, Schmitt B, Wollenberg B, Lindhofer H. The Fc-region of a new class of intact bispecific antibody mediates activation of accessory cells and NK cells and induces direct phagocytosis of tumour cells. Br J Cancer 2000; 83:261 - 6; PMID: 10901380
- Heiss MM, Murawa P, Koralewski P, Kutarska E, Kolesnik OO, Ivanchenko VV, Dudnichenko AS, Aleknaviciene B, Razbadauskas A, Gore M, et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. Int J Cancer 2010; 127:2209 - 21; http://dx.doi.org/10.1002/ijc.25423; PMID: 20473913
- Heiss MM, Ströhlein MA, Jäger M, Kimmig R, Burges A, Schoberth A, Jauch KW, Schildberg FW, Lindhofer H. Immunotherapy of malignant ascites with trifunctional antibodies. Int J Cancer 2005; 117:435 - 43; http://dx.doi.org/10.1002/ijc.21165; PMID: 15906359
- Burges A, Wimberger P, Kümper C, Gorbounova V, Sommer H, Schmalfeldt B, Pfisterer J, Lichinitser M, Makhson A, Moiseyenko V, et al. Effective relief of malignant ascites in patients with advanced ovarian cancer by a trifunctional anti-EpCAM x anti-CD3 antibody: a phase I/II study. Clin Cancer Res 2007; 13:3899 - 905; http://dx.doi.org/10.1158/1078-0432.CCR-06-2769; PMID: 17606723
- Jäger M, Schoberth A, Ruf P, Hess J, Hennig M, Schmalfeldt B, Wimberger P, Ströhlein M, Theissen B, Heiss MM, et al. Immunomonitoring results of a phase II/III study of malignant ascites patients treated with the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3). Cancer Res 2012; 72:24 - 32; http://dx.doi.org/10.1158/0008-5472.CAN-11-2235; PMID: 22044753
- Kodera Y, Yamamura Y, Shimizu Y, Torii A, Hirai T, Yasui K, Morimoto T, Kato T. Peritoneal washing cytology: prognostic value of positive findings in patients with gastric carcinoma undergoing a potentially curative resection. J Surg Oncol 1999; 72:60 - 4, discussion 64-5; http://dx.doi.org/10.1002/(SICI)1096-9098(199910)72:2<60::AID-JSO3>3.0.CO;2-1; PMID: 10518099
- Kodera Y, Nakanishi H, Ito S, Yamamura Y, Fujiwara M, Koike M, Hibi K, Ito K, Tatematsu M, Nakao A. Prognostic significance of intraperitoneal cancer cells in gastric carcinoma: detection of cytokeratin 20 mRNA in peritoneal washes, in addition to detection of carcinoembryonic antigen. Gastric Cancer 2005; 8:142 - 8; http://dx.doi.org/10.1007/s10120-005-0318-7; PMID: 16086116
- Kodera Y, Nakanishi H, Ito S, Mochizuki Y, Ohashi N, Yamamura Y, Fujiwara M, Koike M, Tatematsu M, Nakao A. Prognostic significance of intraperitoneal cancer cells in gastric carcinoma: analysis of real time reverse transcriptase-polymerase chain reaction after 5 years of followup. J Am Coll Surg 2006; 202:231 - 6; http://dx.doi.org/10.1016/j.jamcollsurg.2005.09.008; PMID: 16427547
- Shi C, Yang B, Chen Q, Yang J, Fan N. Retrospective analysis of adjuvant intraperitoneal chemotherapy effect prognosis of resectable gastric cancer. Oncology 2011; 80:289 - 95; http://dx.doi.org/10.1159/000329075; PMID: 21778768
- Went P, Vasei M, Bubendorf L, Terracciano L, Tornillo L, Riede U, Kononen J, Simon R, Sauter G, Baeuerle PA. Frequent high-level expression of the immunotherapeutic target Ep-CAM in colon, stomach, prostate and lung cancers. Br J Cancer 2006; 94:128 - 35; http://dx.doi.org/10.1038/sj.bjc.6602924; PMID: 16404366
- Kondo T, Ito F, Nakazawa H, Horita S, Osaka Y, Toma H. High expression of chemokine gene as a favorable prognostic factor in renal cell carcinoma. J Urol 2004; 171:2171 - 5; http://dx.doi.org/10.1097/01.ju.0000127726.25609.87; PMID: 15126779
- Bonecchi R, Bianchi G, Bordignon PP, D’Ambrosio D, Lang R, Borsatti A, Sozzani S, Allavena P, Gray PA, Mantovani A, et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med 1998; 187:129 - 34; http://dx.doi.org/10.1084/jem.187.1.129; PMID: 9419219
- Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol 2006; 6:295 - 307; http://dx.doi.org/10.1038/nri1806; PMID: 16557261
- Berntsen A, Brimnes MK, thor Straten P, Svane IM. Increase of circulating CD4+CD25highFoxp3+ regulatory T cells in patients with metastatic renal cell carcinoma during treatment with dendritic cell vaccination and low-dose interleukin-2. J Immunother 2010; 33:425 - 34; http://dx.doi.org/10.1097/CJI.0b013e3181cd870f; PMID: 20386464
- Yagi H, Nomura T, Nakamura K, Yamazaki S, Kitawaki T, Hori S, Maeda M, Onodera M, Uchiyama T, Fujii S, et al. Crucial role of FOXP3 in the development and function of human CD25+CD4+ regulatory T cells. Int Immunol 2004; 16:1643 - 56; http://dx.doi.org/10.1093/intimm/dxh165; PMID: 15466453
- Salgado FJ, Lojo J, Fernández-Alonso CM, Viñuela J, Cordero OJ, Nogueira M. Interleukin-dependent modulation of HLA-DR expression on CD4and CD8 activated T cells. Immunol Cell Biol 2002; 80:138 - 47; http://dx.doi.org/10.1046/j.1440-1711.2002.01055.x; PMID: 11940114
- Rosenberg SA, Sherry RM, Morton KE, Scharfman WJ, Yang JC, Topalian SL, Royal RE, Kammula U, Restifo NP, Hughes MS, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol 2005; 175:6169 - 76; PMID: 16237114
- Speiser DE, Liénard D, Rufer N, Rubio-Godoy V, Rimoldi D, Lejeune F, Krieg AM, Cerottini JC, Romero P. Rapid and strong human CD8+ T cell responses to vaccination with peptide, IFA, and CpG oligodeoxynucleotide 7909. J Clin Invest 2005; 115:739 - 46; PMID: 15696196
- Appay V, Jandus C, Voelter V, Reynard S, Coupland SE, Rimoldi D, Lienard D, Guillaume P, Krieg AM, Cerottini JC, et al. New generation vaccine induces effective melanoma-specific CD8+ T cells in the circulation but not in the tumor site. J Immunol 2006; 177:1670 - 8; PMID: 16849476
- Rezvani K, Yong AS, Mielke S, Jafarpour B, Savani BN, Le RQ, Eniafe R, Musse L, Boss C, Kurlander R, et al. Repeated PR1 and WT1 peptide vaccination in Montanide-adjuvant fails to induce sustained high-avidity, epitope-specific CD8+ T cells in myeloid malignancies. Haematologica 2011; 96:432 - 40; http://dx.doi.org/10.3324/haematol.2010.031674; PMID: 21134985
- Aoyama T, Yoshikawa T, Hayashi T, Kuwabara H, Mikayama Y, Ogata T, Cho H, Tsuburaya A, Rino Y, Masuda M. [S-1/krestin immunochemotherapy for patients with advanced gastric cancer]. Gan To Kagaku Ryoho 2011; 38:1921 - 3; PMID: 22202239
- Hirschhaeuser F, Walenta S, Mueller-Klieser W. Efficacy of catumaxomab in tumor spheroid killing is mediated by its trifunctional mode of action. Cancer Immunol Immunother 2010; 59:1675 - 84; http://dx.doi.org/10.1007/s00262-010-0894-1; PMID: 20652245
- Dettmar K, Seitz-Merwald I, Lindemann C, Schroeder P, Seimetz D, Atz J. Transient lymphocyte decrease due to adhesion and migration following catumaxomab (anti-EpCAM x anti-CD3) treatment in vivo. Clin Transl Oncol 2012; 14:376 - 81; http://dx.doi.org/10.1007/s12094-012-0811-5; PMID: 22551544
- Nishimura T, Nakui M, Sato M, Iwakabe K, Kitamura H, Sekimoto M, Ohta A, Koda T, Nishimura S. The critical role of Th1-dominant immunity in tumor immunology. Cancer Chemother Pharmacol 2000; 46:Suppl S52 - 61; http://dx.doi.org/10.1007/PL00014051; PMID: 10950149
- Berghuis D, Santos SJ, Baelde HJ, Taminiau AH, Egeler RM, Schilham MW, Hogendoorn PC, Lankester AC. Pro-inflammatory chemokine-chemokine receptor interactions within the Ewing sarcoma microenvironment determine CD8(+) T-lymphocyte infiltration and affect tumour progression. J Pathol 2011; 223:347 - 57; http://dx.doi.org/10.1002/path.2819; PMID: 21171080
- Chu Y, Yang X, Xu W, Wang Y, Guo Q, Xiong S. In situ expression of IFN-gamma-inducible T cell alpha chemoattractant in breast cancer mounts an enhanced specific anti-tumor immunity which leads to tumor regression. Cancer Immunol Immunother 2007; 56:1539 - 49; http://dx.doi.org/10.1007/s00262-007-0296-1; PMID: 17659370
- Hong M, Puaux AL, Huang C, Loumagne L, Tow C, Mackay C, Kato M, Prévost-Blondel A, Avril MF, Nardin A, et al. Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control. Cancer Res 2011; 71:6997 - 7009; http://dx.doi.org/10.1158/0008-5472.CAN-11-1466; PMID: 21948969
- Musha H, Ohtani H, Mizoi T, Kinouchi M, Nakayama T, Shiiba K, Miyagawa K, Nagura H, Yoshie O, Sasaki I. Selective infiltration of CCR5(+)CXCR3(+) T lymphocytes in human colorectal carcinoma. Int J Cancer 2005; 116:949 - 56; http://dx.doi.org/10.1002/ijc.21135; PMID: 15856455
- Pan J, Burdick MD, Belperio JA, Xue YY, Gerard C, Sharma S, Dubinett SM, Strieter RM. CXCR3/CXCR3 ligand biological axis impairs RENCA tumor growth by a mechanism of immunoangiostasis. J Immunol 2006; 176:1456 - 64; PMID: 16424173
- Pertl U, Luster AD, Varki NM, Homann D, Gaedicke G, Reisfeld RA, Lode HN. IFN-gamma-inducible protein-10 is essential for the generation of a protective tumor-specific CD8 T cell response induced by single-chain IL-12 gene therapy. J Immunol 2001; 166:6944 - 51; PMID: 11359856
- Mullins IM, Slingluff CL, Lee JK, Garbee CF, Shu J, Anderson SG, Mayer ME, Knaus WA, Mullins DW. CXC chemokine receptor 3 expression by activated CD8+ T cells is associated with survival in melanoma patients with stage III disease. Cancer Res 2004; 64:7697 - 701; http://dx.doi.org/10.1158/0008-5472.CAN-04-2059; PMID: 15520172
- Ziegler A, Heidenreich R, Braumüller H, Wolburg H, Weidemann S, Mocikat R, Röcken M. EpCAM, a human tumor-associated antigen promotes Th2 development and tumor immune evasion. Blood 2009; 113:3494 - 502; http://dx.doi.org/10.1182/blood-2008-08-175109; PMID: 19188665
- Atanackovic D, Matsuo M, Ritter E, Mazzara G, Ritter G, Jäger E, Knuth A, Old LJ, Gnjatic S. Monitoring CD4+ T cell responses against viral and tumor antigens using T cells as novel target APC. J Immunol Methods 2003; 278:57 - 66; http://dx.doi.org/10.1016/S0022-1759(03)00209-6; PMID: 12957396