
Abstract
Introduction: IC43 is a recombinant outer membrane protein-based vaccine against Pseudomonas aeruginosa (P. aeruginosa) consisting of OprF- and OprI- epitopes (Opr, outer membrane protein; OprF/I, OprF/OprI hybrid vaccine) with an N-terminal His 6 tag (Met-Ala-(His)6-OprF190–342-OprI21–83).
Objectives: The study aimed to confirm the optimal dose of IC43 in adults with regard to immunogenicity, safety, and tolerability after vaccination with three different dosages of IC43, compared with placebo, and to investigate a potential immune-enhancing effect of the adjuvant, aluminum hydroxide. Subjects were randomly allocated in a 1:1:1:1:1 ratio to one of five treatment groups: 50, 100, or 200 µg IC43 with adjuvant, 100 µg IC43 without adjuvant, or placebo (0.9% sodium chloride) and two intramuscular injections were given in the deltoid region 7 d apart.
Results: The primary immunogenicity analysis of OprF/I-specific IgG antibody titers on day 14 demonstrated statistically significant differences among treatment groups (P < 0.0001), with a significantly higher immune response detected in each IC43 treatment group compared with placebo. From day 0 to day 14, a ≥4-fold increase in OprF/I-specific immunoglobulin G (IgG) antibody titers were observed in >90% of subjects in all IC43 treatment groups in the per-protocol (PP) and intention-to-treat (ITT) populations; a ≥ 50-fold titer increase was observed in 42.6% subjects including all IC43 treatment groups. On day 90, OprF/I-specific IgGs started to decline in all IC43 treatment groups but remained significantly higher until 6 mo compared with placebo. Assessment of functional antibody induction by opsonophagocytic assay (OPA) followed a similar pattern compared with OprF/I-specific IgG kinetics. At day 14, a ≥2-fold increase in OPA titer was observed in 54.5% subjects within all IC43 treatment groups. An increase in antibody avidity index was observed after the second vaccination. At day 14, >96% of subjects in each IC43 treatment group had detectable OprF/I-specific IgG antibodies. Anti-histidine IgG antibody titers peaked on day 14 and were reduced on day 90 in all IC43 treatment groups. OprF/I-specific IgG secreted by antibody-secreting cell (ASC) was detected in all IC43 groups by B-cell ELIspot after the second vaccination and up to 6 mo. All vaccinations were safe and well tolerated up to the maximum cumulative dosage of 400 µg IC43.
Conclusion: IC43 doses equal to or greater than 50 µg were sufficient to induce a plateau of IgG antibody responses in healthy volunteers. Higher doses, whether adjuvanted or non-adjuvanted, were not more effective.
Methods: In this phase I, randomized, placebo-controlled, observer-blinded, multicenter clinical trial, 163 healthy volunteers (18−65 y) were randomly assigned to five treatment groups (1:1:1:1:1). Three groups received IC43 with adjuvant: 50 µg (n = 32), 100 µg (n = 33), or 200 µg (n = 33). One group received IC43 100 µg without adjuvant (n = 32), and one group received placebo (0.9% sodium chloride) (n = 33). Each subject received two intramuscular vaccinations, separated by a 7-d interval (days 0 and 7) (Fig. 1). Humoral immune response was assessed by measurement of outer membrane protein F/I (OprF/I)-specific antibodies determined by enzyme-linked immunosorbent assay (ELISA), anti-histidine antibodies determined by ELISA, and functional antibody activity determined by opsonophagocytic assay (OPA), up to 6 mo post-vaccination. Antibody avidity was measured on days 7 and 14 from samples that had detectable vaccine antibody-specific immunoglobulin G (IgG) antibody titers. At the Austrian site only, the B-cell ELIspot assay was used to determine specific ASC responses. Safety was assessed using adverse event monitoring and clinical laboratory tests. Local and systemic tolerability was recorded in a subject diary for 7 d after each vaccination and by investigators up to 6 mo post-vaccination.
Clinical trial registration number: NCT00778388
Figure 1. Study design. Subjects were randomly allocated in a 1:1:1:1:1 ratio to one of five treatment groups: 50, 100, or 200 µg IC43 with adjuvant, 100 µg IC43 without adjuvant, or placebo (0.9% sodium chloride) and two intramuscular injections were given in the deltoid region 7 d apart. Note: Vaccinations were given on day 0 and day 7. R, randomization; w/o adj, without aluminum hydroxide adjuvant. aPlacebo = 0.9% sodium chloride.
Introduction
Pseudomonas aeruginosa (P. aeruginosa), an aerobic gram-negative rod-shaped bacterium mostly found in water reservoirs and soil, is a classic opportunistic human nosocomial pathogen that causes under various host conditions acute or chronic infections.Citation1,Citation2
P. aeruginosa can become invasive when the first barrier of immune defense fails, for example, when the body surface becomes disrupted. High-risk groups for acquisition of P. aeruginosa include long-term hospitalized patients in intensive care units, burn victims, patients undergoing mechanical ventilation (ventilator-associated pneumonia), and immunocompromised individuals with diabetes, neutropenia, cancer, and post transplantation surgery.Citation2,Citation3P. aeruginosa is a leading cause of nosocomial infections and is responsible for 10% of hospital acquired infections.Citation4 Prevention of airway infections with P. aeruginosa is also a major goal in the treatment of cystic fibrosis patientsCitation5 and several studies indicate that colonization of the lower airways of COPD by P. aeruginosa is associated with enhanced airway inflammation, and probably has deleterious consequences on the course of the disease.Citation6
Serious infection due to strains of Pseudomonas aeruginosa that exhibit resistance to common antipseudomonal antimicrobials is an increasingly serious problem.Citation7 Fourteen percent of P. aeruginosa isolates are multi-drug resistant and some isolates from cystic fibrosis patients are resistant to all antibiotics approved by the United States Food and Drug Administration.Citation4 Therefore, other approaches for the prevention and cure of P. aeruginosa infections are required. Although the results of several attempts to develop a vaccine have been published, no P. aeruginosa vaccine is available.Citation5,Citation8-Citation13
In gram-negative bacteria lipopolysaccharides and outer membrane proteins are the major antigenic components of the bacterial envelope. Because lipopolysaccharide-based vaccines might cause systemic adverse reactions,Citation14 due to systemic inflammation after parenteral injection,Citation7 outer membrane protein-based vaccines with an acceptable toxicity profile are attractive clinical development candidates. P. aeruginosa-specific outer membrane proteins are structurally conserved among all known serotypes and still present after phenotypic conversion by overexpression of alginate to mucoid strains, a marker for the onset of chronic lung infection in CF patients.Citation15,Citation16
IC43, designed by Intercell AG, Austria, is a recombinant outer membaren protein-based vaccine against P. aeruginosa, consisting of epitopes of outer membrane proteins F (OprF) and I (OprI) with an N-terminal His6 tag. Published data from previous small studies investigating this vaccine in healthy volunteers and patients, showed that the OprF/OprI hybrid vaccine (OprF/I) meets most of the desired criteria such as immunogenicity and good tolerability.Citation17-Citation19 Because of considerable inter-individual differences in immune response against the IC43 vaccine, no definitive doses have yet been established.Citation18,Citation19 A possible enhancement of antibody induction using aluminum hydroxide adjuvanted as shown for other vaccinesCitation19 has not yet been tested for IC43 and data about the safety and tolerability of adjuvanted vs. not-adjuvanted IC43 do not exist .
We present the results of a phase I, randomized, placebo-controlled, observer-blinded clinical study to assess the safety and immunogenicity of three dose levels of adjuvanted IC43 and one dose level of IC43 without adjuvant. The main objective of the study was to confirm the optimal dose of IC43 using OprF/I-specific immunoglobulin G (IgG) antibody responses after vaccination with different doses of IC43 and to investigate the potential immune-enhancing effect of aluminum hydroxide adjuvant.
Results
Participants
A total of 202 subjects were screened, and 163 subjects were randomized and received at least one vaccination (32−33 subjects per treatment group). A total of 157 randomized subjects (96.3%) completed the study; 6 subjects (3.7%) were discontinued prematurely, none of them due to adverse events (AEs). Reasons for discontinuation were withdrawal of consent (2 subjects, 1.2%), loss to follow-up (3 subjects, 1.8%), and other reasons (1 subject, 0.6%).
Of the 157 randomized subjects, the median age was 36 y, the median weight was 76 kg, and the median body mass index was 24.7 kg/m2. There were slightly more female subjects (n = 85, 52.1%) than male (n = 78, 47.9%). Most subjects were Caucasian (n = 161, 98.8%). Demographic characteristics and medical history conditions were generally balanced across the treatment groups.
Major protocol deviations included: incorrect study treatment dosage (21 subjects [65.6%] in the IC43 50 μg group, and 2 subjects [6.1%] in the IC43 100 μg with adjuvant group); forbidden concomitant medication (1 subject [3.0%] in the IC43 100 μg with adjuvant group); treatment kit number issue (1 subject [3.1%] in the IC43 100 μg without adjuvant group); and second vaccination not given (2 subjects [6.1%] in the IC43 200 μg group, 1 subject [3.1%] in the IC43 50 μg group, and 1 subject [3.0%] in the placebo group). The 21 subjects in the 50 μg group who received the incorrect dose were given 100 µg in error by investigators, who injected the full volume of the vial instead of half the volume (i.e., 100 µl instead of 50 µl).
The numbers of subjects included in the PP population were 30, 31, 31, 11, and 32 subjects in the IC43 100 μg with adjuvant, IC43 100 μg without adjuvant, IC43 200 μg with adjuvant, IC43 50 μg with adjuvant, and placebo groups, respectively.
Immunogenicity results
Primary immunogenicity analysis
OprF/I-specific IgG antibodies (ELISA) at day 14 (7 d after second vaccination)
The primary immunogenicity analysis of OprF/I-specific IgG antibody titer on day 14 (7 d after the second vaccination) demonstrated statistically significant differences between treatment groups (P < 0.0001; ) with a significant study center effect (P = 0.0200) in the PP population and a non-significant study center effect (P = 0.0716) in the ITT population.
Figure 2. OprF/I-specific IgG antibody geometric mean titer by treatment group (per protocol population). The primary immunogenicity analysis of OprF/I-specific IgG antibody titer on day 14 (7 d after the second vaccination) demonstrated significant differences between treatment groups (P < 0.0001) with a significant study center effect (P = 0.0200) in the per protocol population and a non-significant study center effect (P = 0.0716) in the intention to treat population. Note: Statistically significant P values for pairwise comparisons of GMT ratio estimates are shown (analysis of variance with treatment group and study center as fixed factors, adjusted for multiple comparisons by Tukey’s Honestly Significant Difference test). D, day; GMT, geometric mean titer; IgG, immunoglobulin G; OprF/I, outer membrane protein OprF/I hybrid vaccine; w/o, without adjuvant.
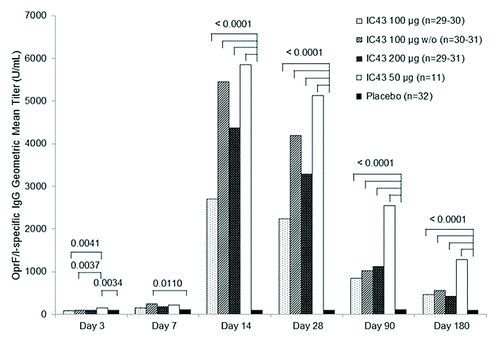
Each of the four IC43 groups produced a higher immune response compared with placebo (P < 0.0001), but no significant differences between the dose groups were observed.
Secondary immunogenicity analysis
OprF/I-specific IgG antibodies (ELISA) at days 3, 7, 28, 90, and 180
On day 3, the OprF/I-specific IgG antibody titer was low in all IC43 treatment groups. In the PP population, there was a statistically significant treatment effect (P = 0.0035) with some differences between individual treatment groups (), but in the ITT population there were no statistically significant differences.
On day 7, the OprF/I-specific IgG antibody titer remained low in all IC43 treatment groups with a statistically significant difference only observed between IC43 100 µg without adjuvant and placebo (P = 0.0110) in the PP population (). In the ITT population, statistically significant differences were observed between IC43 100 µg without adjuvant and placebo (P = 0.0111) and also between IC43 50 µg and placebo (P = 0.0493).
On days 28, 90, and 180, the OprF/I-specific IgG antibody titer in the IC43 treatment groups remained high compared with placebo, and pairwise comparisons between each IC43 treatment group and placebo were all statistically significant (PP and ITT populations) ().
From day 0 to day 14, a ≥4-fold increase in OprF/I-specific IgG antibody titer was observed in >90% of subjects in each IC43 treatment group (; PP population), and a 50-fold increase was observed in 42.6% of all subjects who received IC43 (range 36.4−53.3% across IC43 treatment groups). Similar results were seen in the ITT population.
Table 1. Number and percentage of subjects with a ≥4-fold increase in OprF/I-specific IgG antibody geometric mean titer from day 0 (first vaccination) to day 14 (7 d after second vaccination) (per-protocol-population)
On day 90, OprF/I-specific IgG antibody titer were reduced in all IC43 treatment groups but remained high until day 180; a ≥ 4-fold increase compared with baseline was observed in over 50% of subjects in each IC43 treatment group (). Similar results were seen in the ITT population.
Table 2. Number and percentage of subjects with a ≥4-fold increase in OprF/I-specific IgG antibody geometric mean titer from day 0 (first vaccination) to day 180 (6 mo after first vaccination) (per protocol population)
Functional antibodies (OPA)
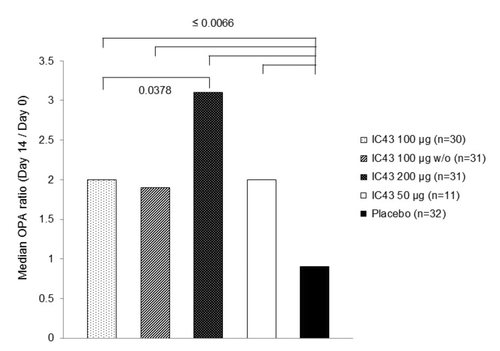
From day 0 to day 14, a ≥2-fold increase in OPA titer was observed in 54.5% of all subjects who received IC43 (range 46.7−63.3% across all IC43 treatment groups; PP population) and 1 subject (3.1%) in the placebo group. Median OPA ratios (changes in OPA titer from day 0 to day 14) are shown in ; all IC43 treatment groups showed statistically significant differences in OPA ratio compared with placebo, and there was a statistically significant difference between IC43 100 µg and IC43 200 µg (PP population). Similar results were seen in the ITT population.
Figure 3. Median OPA ratio (change in OPA titer from day 0 to day 14) by treatment group (per protocol population). From day 0 to day 14, a ≥2-fold increase in OPA titer was observed in 54.5% of all subjects who received IC43 (range 46.7%−63.3% across all IC43 treatment groups; PP population) and 1 subject (3.1%) in the placebo group. Note: P values from Wilcoxon test for unpaired observations on OPA ratio (day 14/day 0) are statistically significant for comparisons between each IC43 treatment group and placebo, and between IC43 100 μg and IC43 200 μg. OPA, opsonophagocytic assay; w/o, without adjuvant.
In a subset of responders (subjects with a ≥2-fold increase in OPA titer from day 0 to day 14), 40.0% subjects who received IC43 (range 14.3−63.2% across all IC43 treatment groups) also had a ≥2-fold increase in OPA titer from day 0 to day 180, and 21.8% subjects who received IC43 (range 0.0−26.3% across all IC43 treatment groups) had a ≥2-fold increase from day 0 to day 7 (PP population). The highest percentage was recorded in the 200 µg group at days 7 and 180. A higher percentage of subjects reached a 2-fold increase in OPA titer from day 0 to day 180 than from day 0 to day 7 in all IC43 treatment groups except for the IC43 100 µg without adjuvant group (14.3% from day 0 to day 180 vs. 21.4% from day 0 to day 7). Similar results were seen in the ITT population.
OprF/I-specific IgG titer and OPA ratio correlated significantly in all IC43 treatment groups (P ≤ 0.0007) except for IC43 50 µg (P = 0.4669) in the PP population; in the ITT population, there was a significant correlation for all IC43 treatment groups (P ≤ 0.0011).
Avidity of OprF/I-specific IgG antibodies
Avidity of OprF/I-specific IgG antibodies was measured in serum samples from subjects with detectable OprF/I-specific IgG titers (>350 U/mL), which was achieved at day 7 by 25.2% subjects who received IC43 (range 16.7%−35.5% across IC43 treatment groups) (PP population). By day 14, >96% of subjects in each IC43 treatment group had detectable OprF/I-specific IgG antibodies. This was not achieved by any subjects in the placebo group at any time.
The median avidity index of OprF/I-specific IgG antibodies in a subgroup of subjects who reached >350 U/mL of OprF/I-specific IgG antibodies at both days 7 and 14 is summarized in . Values were generally slightly higher for all active treatment groups at day 14 compared with day 7. On both day 7 and 14, the median avidity index was highest in the IC43 200 µg treatment group and lowest in the IC43 50 µg group. However, the median index values were generally in the lower half of the possible range of indices (0.1 to 4.5) for all groups, with the highest median affinity index at day 14 being 2.55 (IC43 200 μg group).
Table 3. Avidity index of OprF/I-specific IgG antibodies at day 7 and day 14 by treatment group (per protocol population; subgroup of subjects with detectable OprF/I-specific IgG antibodies at day 7 and day 14)
Results were similar in the ITT population.
Anti-histidine IgG antibodies
The median ratio compared with baseline of anti-histidine antibodies was greatest at day 14 (range 6.6−8.4 across IC43 treatment groups), decreasing at day 90 (range 2.2−4.0) and day 180 (1.6−2.5) (PP population). Results were similar for the ITT population.
From day 0 to day 14, a ≥4-fold increase in anti-histidine antibodies was observed in 54.5% of all subjects who received IC43 (PP population). The lowest proportion of subjects achieving at least a 4-fold increase was in the IC43 50 µg group (27.3%) and the highest was in the IC43 200 µg group (66.7%). Similar results were reported for the ITT population, although the percentage of subjects was higher in the IC43 50 µg group (51.6%).
Only a weak correlation between OprF/I-specific IgG antibody titer and anti-histidine antibodies was observed in the PP and ITT populations for all IC43 groups combined (Spearman coefficient of correlation 0.23 [P = 0.0193] and 0.25 [P = 0.0041], respectively).
PBMC analysis:
B cell ELIspot
The percentage of the OprF/I-specific IgG response compared with the total IgG response (% OprF/I-specific response) demonstrated that almost no ASC responses were measurable at baseline or following the first vaccination, with median percentage values on days 0 and 7 ranging between 0.00% and 0.01% for all IC43 treatment groups (PP population [n = 10−11 subjects] and ITT population [n = 11−13 subjects]).
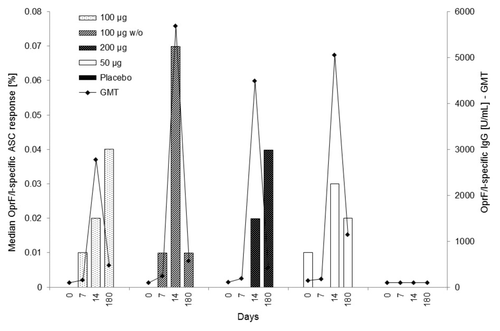
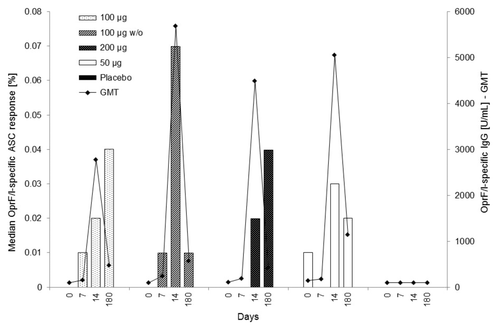
On day 14, all active IC43 treatment groups showed a measurable OprF/I-specific response (median percentage range 0.02%−0.07%, PP population), which further increased from day 14 to day 180 for the IC43 100 µg group in the PP population, and for both the IC43 100 µg and 200 µg groups in the ITT population. Responses remained measurable until day 180 for all active IC43 treatment groups (median percentage range 0.01%−0.04%, PP population). OprF/I-specific ASC responses were detected in 30% and 27% of subjects on day 14 and 180, respectively. At day 14, the highest median value was observed in the IC43 100 µg without adjuvant group (7.50%, median percentage 0.07%; PP population). On day 180, median values were higher than at day 14 for the IC43 100 µg group (4.50% compared with 1.75%). Results were similar in the ITT population.
The median OprF/I-specific ASC response (at Site 01 only) vs. the OprF/I-specific IgG titer (in PP population) at days 0, 7, 14, and 180 is illustrated in .
Figure 4. Median OprF/I-specific ASC response (%) vs. OprF/I-specific IgG geometric mean titer (U/mL) on days 0, 7, 14, and 180 (PP population). For median OprF/I-specific ASC response, the number of observations was n = 10 (100 μg), n = 9–10 (100 μg w/o), n = 9–11 (200 μg), n = 10–11 (50 μg), and n = 11–12 (placebo). For OprF/I-specific IgG geometric mean titer, the number of observations was n = 30 (100 μg), n = 30–31 (100 μg w/o), n = 30–31 (200 μg), n = 11 (50 μg), and n = 32 (placebo). OprF/I, outer membrane protein; OprF/I, hybrid vaccine; w/o, without adjuvant.
Safety results
All vaccinations were safe and well tolerated up to the maximum tested cumulative dose of 400 µg IC43.
There was no statistically significant difference in the percentage of subjects with at least one TEAE among the treatment groups (), but when all IC43 treatment groups were combined there was a significant difference compared with placebo (P = 0.0219, Fisher exact test). Also, there was a statistically significant difference among the treatment groups in the percentage of subjects with at least one treatment-related treatment-emergent adverse event (TEAE) (P < 0.0001, Fisher–Freeman–Halton test), with a higher percentage in all IC43 treatment groups compared with placebo (). The majority of events were mild or moderate; only a small percentage of subjects reported any severe AE, with no statistically significant difference between the combined IC43 groups and placebo (11 subjects [8.5%] in combined IC43 groups, 4 subjects [12.1%] in the placebo group; P = 0.5074, Fisher exact test). Individual TEAEs for which there was a statistically significant difference in the percentage of subjects reporting the event are included in .
Table 4. Overall incidence of TEAEs and treatment-related TEAEs, and TEAEs for which there was a statistically significant difference among treatment groupsa (safety population)
Only eight serious adverse events (SAEs) were experienced, by 6 subjects (2 subjects [6.1%] in the IC43 200 µg group: pregnancy and voluntarily induced abortion, knee distorted due to gonarthrosis; 4 subjects [12.5%] in the IC43 50 µg group: pregnancy and voluntarily induced abortion, acute coronary syndrome, muscle contusion due to work accident, gastroenteritis); none of the SAEs was considered to be treatment-related. There were no deaths during the study period. One subject (3.0%) in the IC43 200 µg group did not receive a second vaccination due to TEAEs of pyrexia, nausea, and vomiting, which occurred approximately 5 d after the first vaccination and were considered by the investigator to be probably related to study treatment, but they resolved spontaneously within 1–2 d. There were no discontinuations due to TEAEs.
There was no statistically significant difference in the percentage of subjects with at least one medically attended TEAE between the combined IC43 groups and placebo (51 subjects [39.2%] in combined IC43 groups, 8 subjects [24.2%] in the placebo group; P = 0.1550, Fisher exact test), and no significant differences for any individual medically attended TEAEs.
Local tolerability symptoms, as documented in the subject diary, were recorded on the majority of diary days. Local pain and tenderness were the most frequently reported tolerability symptoms in the 7-d periods following the first and second vaccinations, with statistically significant differences among the treatment groups (). For both pain and tenderness, there was a statistically significant difference between the combined IC43 treatment groups and placebo following both vaccinations (P ≤ 0.0004, Fisher exact test). These symptoms, mainly reported within the first 2 d after vaccination, were generally mild and occurred more frequently after vaccination with adjuvanted IC43 100 µg compared with IC43 100 µg without adjuvant. After the second vaccination, erythema/redness and induration also showed a statistically significant difference among treatments, with a higher incidence in the IC43 200 µg group ().
Table 5. Incidence of local and systemic tolerability symptoms recorded in the subject diary in the 7-d periods after the first and second vaccinations (safety population)
Systemic tolerability symptoms recorded by subjects in the 7-d periods following the two vaccinations were most frequently mild to moderate muscle pain, headache, excessive fatigue, and symptoms described as “other.” There was an overall statistically significant difference among the treatment groups for muscle pain after the first and second vaccinations (). Analysis of the combined IC43 treatment groups compared with placebo also showed a statistically significant difference for headache after the second vaccination (P = 0.0361, Fisher exact test).
Investigator-reported local and systemic tolerability results were similar to those reported in the subject diary.
Clinical laboratory data did not indicate any safety issues.
Discussion
This was a phase I study to confirm the optimal dose of IC43 vaccine against P. aeruginosa with regard to immunogenicity, safety, and tolerability in healthy volunteers. Subjects were vaccinated twice (day 0 and day 7) with adjuvanted doses of 50 µg, 100 µg, or 200 µg IC43, 100 µg IC43 without adjuvant, or placebo. Follow up for safety and immunogenicity continued until 180 d (approximately 6 mo) after the first vaccination. The major finding was that all doses of IC43 produced comparably high immune responses after 14 d.
An accelerated schedule (day 0 and 7 vaccinations) was used and the early immune response was tested, because it was hypothesized there would be a rapid rise in antibodies if some background immunity exists against this opportunistic pathogen.
Seven days after the first vaccination, OprF/I-specific IgG antibody responses remained low (GMT ≤ 250 U/mL), showing statistically significant differences only between IC43 100 µg without adjuvant and placebo in the PP and ITT populations.
These OprF/I IgG antibody kinetics indicate that no substantial IgG response was induced 7 d after a single vaccination. Whether a 2-dose vaccination regimen with an interval of only 7 d between vaccinations is required for a significant OprF/I IgG induction or if it simply takes 14 d until significant OprF/I IgG induction is reached after a single vaccinationCitation17 cannot be answered with the current study.
There are limited data on how quickly antibody titers can rise in response to modern vaccines. An OMP vaccine increased IgG levels measured by ELISA in healthy volunteers by only 10–20% 7 d after the first dose,Citation20 whereas a significant rise occurred only after 14 d. Another study with a rabies vaccine, showed that adequate levels of antibody of at least 0.5 IU/mL were developed by day 14 after 3 to 4 doses.Citation16 Seroconversion by day 7 was uncommon—a fact also seen in our trial. So it seems that even a rapid immunization schedule with 3 to 4 vaccinations in the first week, and even when combined with passive immunization for rabies post-exposure prophylaxis, takes about 14 d to achieve an adequate immune response.Citation21-Citation24
Looking at the kinetics of immune responses from a different angle, antibody responses to influenza A H5N1 virus infection in fatal cases and survivors showed that high neutralizing antibody titers were achieved only 2 to 3 weeks after symptom onset in the majority of H5N1-infected individuals tested.Citation25 Further, only one out of five patients had a significant antibody response within 5 d after the onset of disease. Infection should be an even stronger antigen challenge than any vaccination, so these data demonstrate that clinically relevant antibody responses are unlikely to occur within the first 7 d.
OprF/I-specific IgG antibody titers were significantly higher in all IC43 groups compared with placebo 7 d after the second vaccination (day 14). However, a dose dependency of the IgG antibody response upon vaccination with the applied doses was not observed, indicating that the plateau of IgG response is already reached with the 50 µg IC43 dose in the healthy volunteer population. Analysis of the ITT population confirmed results from the PP population, but the low sample size of the 50 µg group within the PP population has to be considered. These data differ from the findings of Mansouri et al.Citation17 where, after one vaccination, a maximal response was only observed for groups that received the 100- or 500-μg dose and no significant further increase of antibody titers was measured in these groups after the second or third dose. In contrast, individuals in the lower dose groups (20 or 50 μg) needed re-vaccination for significant antibody responses.Citation17 Differences in the vaccine preparation, differences in the vaccination schedule studied by Mansouri et al., who used a 4-weeks interval between the vaccinations, might be explanations for the described differences observed between our study and that of Mansouri et al. In addition sample size in the study by Mansouri et al. were low (8 subjects by group), therefore we believe our results are more representative
Immune responses might be enhanced by immunological adjuvants like aluminum salts.Citation26 In our trial, an immune-enhancing effect of aluminum hydroxide adjuvant was not observed.
A relatively rapid decline in OprF/I IgG titer was observed from day 90 in all IC43 treatment arms. However, IgG titers remained significantly higher compared with placebo until 180 d (6 mo) after vaccination. A three-dose vaccination regimen might be beneficial for a prolongation of the IgG peak response. Indeed, Mansouri et al. demonstrated the boosting ability of the OprF/I hybrid protein with a booster vaccination 6 mo after primary immunization.Citation17
Even though a dose of 50 µg IC43 without adjuvant might suffice for healthy subjects to induce a plateau of IgG response, the target populations for vaccination against P. aeruginosa are usually immunocompromised patients, in whom immune responses to vaccines are expected to differ to those in healthy volunteers.
While healthy volunteers needed only one vaccination for seroconversion in a previous study,Citation12 burn patients needed two vaccinations for a similar effect.Citation27 The mean antibody titers were about 85% of those of the healthy population and might be even less in more immunocompromised patients.Citation27 Thus, for patients with severe immunosuppression due to a variety of conditions, higher doses or more frequent boosters might be required.Citation28 Dose-finding and investigation of the necessity of an adjuvant to enhance the immune response still needs to be investigated for the target population.
Antibody avidity was measured twice (days 7 and 14) from OprF/I-specific IgG antibody titer detectable samples. An increase in the avidity index after the second vaccination and over time might be indicative of an affinity maturation of OprF/I-specific IgG antibodies in responders (subjects with detectable OprF/I-specific IgG titers at both day 7 and day 14).
IC43-induced functional IgG antibodies with opsonophagocytic activity were measured by OPA. Kinetics of OPA response followed a similar pattern to OprF/I-specific IgG kinetics (measured by ELISA) in responders. An increase in OPA titer from day 0 to day 14 of at least 2-fold was observed in all active treatment groups (≥46% of subjects in each IC43 treatment group) compared with 3% of subjects in the placebo group. All IC43 treatment groups showed significant differences in OPA ratio (day 14/day 0) compared with placebo.
Since IC43 is produced as a His-tagged protein, the induction of anti-histidine IgG was investigated and showed similar kinetics to that of OprF/I-specific IgG, with a peak response at day 14 and declining at day 90 for all IC43 treatment arms. To our knowledge, this appears to be the first report on anti-histidine antibodies.
Although their relevance is not clear yet, evaluation of potential toxic effects mediated by binding of vaccine-induced antibodies to normal human tissues (i.e., autoreactive antibodies) in preclinical toxicity studies did not indicate the induction of any anti-human tissue responses with the His-tagged OprF/I protein (unpublished results).
The lack of clinical relevance is also supported by the good safety profile of IC43, which was well tolerated up to doses of 200 µg per vaccination, and both systemic and local tolerability did not point to any safety issues with IC43.
OprF/I-specific IgG secreted by antibody-secreting cells was detectable in all IC43 groups by B-cell ELIspot after the second vaccination and up to day 180. However, no pre-existing OprF/I-specific memory B-cell responses (as precursor cells for ASC) were detected at day 0, which is consistent with the absence of a booster effect of the first vaccination on OprF/I-specific IgG response.
Potential limitations include that we have not assessed mucosal immune responses which might be important in chronic lung diseases. It would be interesting to determine mucosal antibodies in future trials. Also further studies should potentially assess the efficacy of a third vaccination which might lead to a prolonged response.
The strength, however, of the study is, that the sample size in the current study is reasonable for a Phase 1 to obtain a good indication on the immunogenicity and tolerability of the vaccine.
Conclusions
IC43 with and without aluminum hydroxide adjuvant was well tolerated in all treatment arms and raised no safety concerns. In healthy volunteers, both adjuvanted and non-adjuvanted IC43 was highly immunogenic in terms of OprF/I-specific IgG antibody induction 7 d after the second vaccination. The addition of adjuvant was not beneficial for the kinetics or strength of the response. IC43 doses equal to or greater than 50 µg were sufficient for healthy adults to reach an antibody response plateau. The antibody kinetics indicate that a two-vaccination regimen and an interval of at least 7 d are required for the induction of a significant IgG response in healthy volunteers Data are now required for evaluation of the IC43 vaccine in the target populations, which are patients at risk for acquiring Pseudomonas aeruginosa infections, such as intensive care patients, burn patients, CF patients or chronic lung disease patients.
Materials and Methods
Study design
This study was designed according to the note for guidance on clinical evaluation of new vaccines.Citation29
This was a phase I, randomized, placebo-controlled, observer-blinded, multicenter trial to assess the safety and immunogenicity of IC43 vaccination against P. aeruginosa in healthy adult volunteers. The study was conducted between August 2008 and April 2009 at 2 sites in Germany and 1 site in Austria. The study protocol was approved by the sites’ independent ethics committees, and the trial was conducted according to the Declaration of Helsinki 2008. Study participants provided written informed consent before any study-specific procedures were performed.
No formal sample size calculation was performed. A sample size of 32 subjects per group (160 total subjects) allowed for the detection of an effect size of 0.73 with a two-sided significance level of 5% (pairwise), a power of 80% and an estimated drop-out rate of 5% (t-test for independent observations).
After a screening period of up to 2 weeks (day−14 to day−1), during which inclusion and exclusion criteria were evaluated, subjects were randomly allocated in a 1:1:1:1:1 ratio to one of five treatment groups: 50, 100, or 200 µg IC43 with adjuvant, 100 µg IC43 without adjuvant, or placebo (0.9% sodium chloride). The randomization list was created using nQuery Advisor by a statistician at a contract research organization (Assign Data Management and Biostatistics GmbH). A hard copy and the electronic files were stored in a safe until the study was unblinded. The randomization was stratified by study center. Varying block sizes were used: 2 per group of 50 µg (with adjuvant), 100 µg (with adjuvant), 100 µg (without adjuvant), 200 µg (with adjuvant), and placebo in Blocks 1−5; 1 per group in Block 6; 2 per group in Blocks 7−16. Each subject was assigned the next free medication kit (randomization) number for the corresponding study center in ascending order.
Two intramuscular injections were given in the deltoid region (left arm or non-dominant arm) 7 d apart (days 0 and 7), using a conventional 25 gauge needle, approximately 5/8 in long. The subjects attended the study center for three follow-up visits (days 14, 28, and 90), and a final clinical evaluation was scheduled after 6 mo of follow-up (day 180). For the five different study groups and the overall study design see .
The trial was blinded for the subjects and not blinded for the investigator applying the vaccination, but blinded for a second independent investigator assessing local and systemic tolerability.
Serum was collected to assess humoral immunity at days 0, 3, 7, 14, 28, 90, and 180. Additional blood was taken at one site for peripheral blood mononuclear cell (PBMC) isolation and subsequent analysis of antibody-secreting cells (ASC). Adverse events (AEs) and clinical laboratory tests were assessed at each visit (day 0 through day 180). Systemic and local tolerability symptoms were recorded in a subject diary for the 7 d following each vaccination (days 0 to 7 and days 7 to 14) and by the investigator at each study visit.
Study population
Male and female subjects of good general health, aged 18−65 y, were enrolled in this study. Female participants with childbearing potential had to have a negative serum pregnancy test during screening, prior to any vaccination, and a willingness not to become pregnant during the entire study period by practicing reliable methods of contraception.
Exclusion criteria consisted of: a history of autoimmune diseases or malignancies; a history of severe hypersensitivity reactions or anaphylaxis; known hypersensitivity or allergic reactions to one of the components of the vaccine; use of any other investigational or non-registered drug or vaccine within 30 d before the first study vaccination or during the study; active or passive vaccination within 4 weeks before the first study vaccination or during the study period; immunodeficiency due to immunosuppressive therapy; administration of chronic (>14 d) immunosuppressants or immune-modifying drugs within 6 mo before vaccination; a family history of congenital or hereditary immunodeficiency; clinically significant diseases as judged by the investigator; any febrile acute infections within 4 weeks prior to randomization and during the study; intake of non-steroidal anti-inflammatory drugs or paracetamol within 3 d before the first study vaccination; body temperature >37.8 °C before the first study vaccination; infection with human immunodeficiency virus, hepatitis B, or hepatitis C; drug addiction within 6 mo prior to enrollment; and inability or unwillingness to avoid more than the usual intake of alcohol during the 48 h after vaccination.
Study vaccine
The P. aeruginosa vaccine IC43 consists of the C-terminal part of the outer membrane protein OprF and the entire mature outer membrane protein OprI with six histidine residues fused to the N-terminus of the recombinant protein (Met-Ala-[His]6-OprF190–342-OprI21–83). IC43 was produced in Escherichia coli. The purified protein was stored in phosphate buffered saline and diluted in 0.9% sodium chloride. Finally, the protein was either absorbed to aluminum hydroxide as adjuvant (400 µg protein per mL aluminum hydroxide) or directly filled in glass vials. The final protein concentration in both formulations was 100 µg/mL. Production and release were performed in accordance with good manufacturing practice. The drug substance (purified OprF/I fusion protein) was manufactured and released by Eurogentec S.A. Biologics, Liege Science Park-4102. The drug product was produced by SynCo Biopartners B.V. The volume injected was 0.5, 1, and 2 mL to achieve IC43 doses of 50, 100, and 200 μg, respectively. A volume of 1 mL 0.9% sodium chloride was injected for placebo doses.
IC43 with adjuvant (50, 100, and 200 µg doses) had batch number IC43-A-01/08, IC43 without adjuvant (100 µg dose only) had batch number IC43-B-02/08, and placebo (0.9% sodium chloride) had batch number 8054C18.
IC43 with adjuvant was stored at 2 °C to 8 °C. IC43 without adjuvant was stored at −20 °C (+/− 5 °C). All IC43 doses were shaken before use. Placebo saline solution was stored at room temperature (18 °C to 25 °C).
Study endpoints
The primary immunogenicity endpoint, OprF/I-specific IgG antibody titer at day 14, was measured by enzyme-linked immunosorbent assay (ELISA). Secondary immunogenicity endpoints included: OprI/F-specific antibody titer (ELISA) measured on days 3, 7, 28, 90, and 180; anti-histidine antibodies (ELISA) on days 0, 7, 14, 90, and 180; functional antibody induction measured by opsonophagocytic assay (OPA) on day 14 (and days 7 and 180 in a subset of responders, selected according to OPA activity on day 14); and antibody avidity measured on days 7 and 14 from samples that had detectable vaccine antigen-specific IgG antibody titers. PBMC analyses (B-cell ELIspot) were performed as supportive endpoints at one site only, on days 0, 7, 14, and 180.
Safety and tolerability endpoints were also considered primary. These included: adverse events (AEs) and serious adverse events (SAEs); clinical laboratory tests (hematology, clinical chemistry, and urinalysis); local tolerability symptoms (presence and severity of pain, tenderness, erythema/redness, edema, itching, and induration); and systemic tolerability symptoms (presence of headache, muscle pain, fever, flu-like symptoms, nausea, vomiting, rash, excessive fatigue, and other symptoms). AEs were recorded and clinical laboratory tests were performed at each study visit. AEs and concomitant diseases were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 11.1. Local and systemic tolerability was recorded by the subject in a diary for 7 d after each vaccination. Local tolerability was also evaluated before and up to 60 min after each vaccination by the investigator and at each study visit the injection site was inspected and evaluated by an independent investigator blinded for the treatment. Systemic tolerability was also evaluated at each visit and events were recorded as AEs.
Immunogenicity assays
The OprF/I protein used for the in-vitro assays was manufactured at Eurogentec S.A. according to the same process as the study vaccine.
ELISA
Human serum obtained at each study visit was analyzed for OprF/I-specific IgG by a validated ELISA. Microtiter plates were coated with 1 µg/mL OprF/I, stored at 2–8 °C for 12–72h, washed with PBS 0.05% Tween 20, and blocked using PBS 0.05% Tween 20 with 2% bovine serum albumin (BSA, fraction V, Biomol). Eight serial 4-fold dilutions of each serum sample were applied in duplicate starting at 1:20 dilution. Control wells with no sample, a quality control sample and a reference standard, both prepared from sera generated during a phase I clinical trialCitation17 were run on each plate. Plates were incubated at ambient temperature for 1–2 h, washed and rabbit anti-human IgG horseradish peroxidase conjugate (Dako) diluted in PBS - 0.05% Tween 20 with 2% BSA was added for 1–2 h. The presence of OprF/I-specific IgGs was detected by the addition of a substrate (Substrate solution I/II, Bender MedSystems). Quantification of specific IgGs was performed on the basis of the reference substance curve by four-parameter logistic fit and parallel line analysis using SoftMax Pro v5.2 Software (Molecular Devices). Responses below the limit of quantitation of the ELISA (350 U/mL) were replaced with 100 U/mL.
Opsonophagocytic assay (OPA)
OPA measured the uptake of OprF/I-coated fluorescent beads by phagocytes in the presence of functional antibodies. For the evaluation of functional antibodies, sera prepared at days 7, 14, and day 180 were analyzed with OPA. Carboxylate-modified fluorescent FluoSpheres® 1 µm (Molecular ProbesTM, Invitrogen) were coupled with OprF/I. Briefly, after washing in MES Buffer (pH 6.0, Sigma) FluoSpheres® were suspended in 200 µL MES buffer, mixed with 50 µL EDAC (100 mg/mL, 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide, Molecular Probes), and incubated in the dark at room temperature with gentle rotation (150 rpm) for 30 min. An amount of 250 µL OprF/I protein stock solution (1 mg/mL) was added and the mixture was incubated in the dark at room temperature over night with gentle rotation (150 rpm). After incubation, 50 µL glycine (100 mM in MES buffer) were added and incubation was allowed to proceed for another 30 min in the dark to block remaining active sites on the beads. Beads were washed with 50 mM PBS + 0.9%NaCl + 1% BSA and suspended in 50 mM PBS + 0.9% NaCl + 1% BSA+ 0.02% NaN3 and kept at 4 °C.
HL-60 human promyelocytic leukemia cells (CCL-240, American Type culture Collection) were differentiated into phagocytes by cultivation in growth medium (RPMI-1640 + 20% fetal bovine serum [FBS]) supplemented with 0.8% DMF (N,N-Dimethylformamide, Sigma) for 3 to 5 d. Six 5-fold dilutions of heat inactivated serum samples (30 min, 56 °C) were prepared in OPA buffer (Hanks buffered salt solution containing Ca2+ and Mg2+ [Gibco] supplemented with 2% BSA) starting at 1:5 dilution. Serum dilutions of 10 µL and 20 µl of OprF/I bead dilution (beads were pre-diluted to reach a final bead to cell ratio of approximately 4:1) were incubated in 96 well round bottom plates 30 min at 37 °C with shaking (150 rpm). An amount of 20 µL baby rabbit complement (Pel-freeze Biologicals) diluted in OPA buffer was added as external complement source (appropriate dilution was tested for each complement lot) and incubated 20 min at 37 °C, 150 rpm. To each well 105 differentiated HL-60 cells were added in 30 µL OPA buffer. Bead uptake by phagocytes was stopped after 30 min incubation at 37 °C by addition of 120 µL cold OPA buffer supplemented with 6.8 mM EDTA (EDTA, Fluka). Microtiter plates were kept on ice until analysis by flow cytometry. A Cytomics FC 500 MPL (multiplate loader) with MXP acquisition software (Beckman Coulter) was used for sample analysis. Each sample dilution was analyzed for the percentage of fluorescent HL-60 cells (phagocytes which have taken up fluorescent beads). The OPA titer (corresponding to serum dilution which induces half maximum uptake [EC50]) for each serum sample was determined by applying a four parameter logistic curve fit. OPA titer values below the lowest serum dilution tested (1:5) were replaced by 5.
Avidity
Measurement of antibody avidity was performed on days 7 and 14 from samples that had detectable vaccine antigen-specific IgG antibody titers (>350 U/mL, limit of quantitation of the IgG ELISA) using an OprF/I antigen-specific human serum IgG avidity ELISA. Avidity ELISA was performed according to a procedure modified fromCitation30). Plates were coated with OprF/I and blocked according to the procedure described for the IgG ELISA. Three individual dilutions were prepared for each sample to target an OD of 0.8 to 1.2. After 1 h of incubation plates were washed and four different concentrations of ammonium thiocyanate (0.5 M, 1 M, 2 M, 4 M NH4SCN) and PBS (control for 100% binding) were added for 15 min. According to their individual avidity, antigen-bound antibodies were dissociated by increasing concentrations of ammonium thiocyanate. Residual bound antibodies were then quantified by a rabbit anti-human IgG horseradish peroxidase conjugate/substrate reaction according to the procedure described for the OprF/I specific IgG ELISA. Results were calculated using analysis software SoftMax Pro v5.2 (Molecular Devices), Sample curves (log value of the percentage of eluted antibody against molarity of NH4SCN) were created by applying a four parameter logistic curve-fit. Avidity index was defined as the NH4SCN concentration resulting in 50% dissociation of antibody-antigen complex. Data presentations imputed 0.1 M for low values (defined as >50% stripping with a concentration of 0.5 M ammonium thiocyanate) and 4.5 M for high values (<50% stripping with 4 M ammonium thiocyanate).
Anti-6-histidine-IgG
In order to monitor possible responses and kinetics of responses to the 6-histidine-tag on OprF/I, an additional validated ELISA system was used for the detection of anti-6-histidine specific IgG antibodies in serum samples obtained on days 0, 7, 14, 90, and 180. Serum binding to an unrelated protein with a 6-histidine-tag (BSA coupled with a peptide containing the 6-histidine sequence AHHHHHHAPAC, piCHEM) was determined and compared with the binding to the variant without a 6-histidine tag. Serum samples were diluted at 1:20 and added in triplicates to microtiter plates, coated one half with BSA and one half with His-BSA. Plates were incubated at ambient temperature for 1–2 h, after washing rabbit anti-human IgG horseradish peroxidase conjugate (Dako) diluted in PBS - 0.05% Tween 20 with 2% bovine serum albumin was added for 1–2 h. Anti 6-histidine specific IgG antibodies are reported as ratio between mean ODs of three replicates obtained against His-BSA and the mean ODs of three replicates obtained against BSA. The relative change (fold increase) of the ratios from day 0 to 14 was calculated.
Cellular assay
B-cell ELIspot assay was used to determine specific antibody-secreting cell (ASC) responses, which represent memory B-cell responses, according to the published method.Citation31 Peripheral blood mononuclear cells (PBMCs) (5 × 105/mL) were pre-cultivated for 6 d at 37 °C, 5% CO2 in T25 cell culture flasks in medium supplemented with polyclonal activators: RPMI 1640 (Gibco), 10% FBS (PAA), Staphylococcus aureus Cowan (Sigma P7155, 1/10000), Phytolacca Americana pokeweed mitogen (Sigma, L8777, 0.1 µg/mL), 2-Mercapto-ethanol (50 µM, Gibco), and CpG-2006 oligonucleotide (6 µg/mL, Purimex). ELIspot plates (Multiscreen HTS plates, Millipore, MSIP N4510) were coated with recombinant OprF/I (5 µg/mL), anti-IgG capture antibody for determination of the total IgG response (5 µg/mL, goat anti-human Ig’s polyvalent, Caltag Medsystems), bovine serum albumin (2.5 µg/mL, fraction V, Biomol), and Agrippal (5 µg/mL, influenza vaccine, Novartis) as internal assay controls. Plates were washed and blocked with RPMI 1640 + 1% BSA for 2–3 h at 37 °C. After the polyclonal stimulation, PBMCs were washed and plated onto ELISpot plates at 4 different concentrations (8 × 105, 4 × 105, 2 × 105, 1 × 105 cells per well). For measurement of total IgG secreting B-cells a maximum of 2.5 × 104 cells were seeded per well. Cells were allowed to incubate on ELISpot plates for 6h at 37 °C and 5% CO2. Plates were washed and incubated with mouse anti-human IgG Fc PAN biotin-conjugate (Hybridoma Reagent Laboratory) over night at 2–8 °C. After further washing HRP-conjugated AvidinD (Vector Laboratories) was added for 1 h at ambient temperature and plates were developed with AEC substrate (3-amino-9-ethylcarbazole, Sigma). Plates were scanned and counted using an ImmunoSpot series 1 Analyzer (CTL) and Immunospot 2.08 Professional Analyzer Software. Counted spots per well were multiplied with respective dilution factor resulting in spot number/8 × 105 cells). Median spot number of total IgG response was set 100% in order to calculate the percentage of OprF/I specific response. Data represent the percentage of OprF/I-specific compared with total IgG secreting cells.
Statistical analysis
Statistical analyses were performed using SAS® version 9.2 (SAS Institute).
In the primary immunogenicity analysis, OprF/I-specific antibody geometric mean titers (GMTs) on day 14 were compared between the five treatment groups. GMTs and GMT ratios were estimated by applying an analysis of variance including the factors treatment group and study center. This was done using log 10-transformed data and taking the anti-log of the resulting point estimates for the least squares means, least squares means differences and the corresponding 95% confidence intervals (CIs). Tukey Honestly Significant Differences (HSD) test was applied for pairwise comparisons in the presence of a significant treatment effect. A similar analysis was performed for the secondary immunogenicity analysis of OprF/I-specific IgG antibody titers on days 3, 7, 28, 90, and 180. The number and percentage of subjects reaching 4- and 50-fold increases in OprF/I-specific IgG antibody titers from day 0 to day 14, and day 0 to day 180 were calculated and analyzed descriptively for each treatment group, including two-sided 95% CIs calculated according to Wilson's method recommended by Altman.Citation32
The number and percentage of subjects reaching a 2-fold increase in OPA titer from day 0 to day 14 were calculated and analyzed descriptively for each group, including two-sided 95% CIs.Citation33 A similar analysis of OPA titers, from day 0 to day 7 and day 0 to day 180, was performed for a subset of responders (subjects with OPA titer increase from day 0 to day 14 ≥ 2-fold). For all subjects, the OPA ratio (OPA titer day 14/OPA titer day 0) was compared between each treatment group using a Wilcoxon test for unpaired observations. A non-parametric correlation analysis (Spearman) for OprF/I-specific antibodies at day 14 vs. OPA titer ratio (day 14/day 0) was performed.
For antibody avidity evaluation, the number and percentage of subjects achieving detectable OprF/I-specific IgG antibodies (>350 U/mL) on days 7 and 14 were calculated. Descriptive statistics were calculated for a subgroup of subjects with detectable OprF/I-specific IgG antibodies (>350 U/mL) at both days 7 and 14. Data presentations imputed 0.1 M for low values (defined as >50% stripping with a concentration of 0.5 M ammonium thiocyanate) and 4.5 M for high values (<50% stripping with 4 M ammonium thiocyanate).
Summary statistics for anti-histidine IgG antibodies at days 7, 14, 90, and 180 were calculated. In a post-hoc analysis, the number and percentage of subjects reaching a 4-fold increase in anti-histidine antibodies from day 0 to day 14 were calculated and analyzed descriptively for each treatment group. In a further post-hoc analysis, a non-parametric correlation analysis (Spearman) for OprF/I-specific antibodies at day 14 vs. anti-histidine antibodies was performed.
An exploratory analysis of B-cell ELIspot data collected at 1 site was performed. The spot numbers determined for the total IgG response of ASCs and the OprF/I-specific response were summarized using descriptive statistics. Also the percentage of the OprF/I-specific IgG response compared with the total IgG response (% OprF/I-specific response) was assessed.
A treatment-emergent adverse event (TEAE) was defined as an AE for which the first onset, or worsening, was simultaneous with or after the first vaccination. TEAEs were tabulated with two-sided 95% CIs calculated according AltmanCitation19 and P values calculated to test for an overall treatment effect using the Fisher–Freeman–Halton test. Additionally, the IC43 treatment groups were combined and compared with the placebo group using Fisher exact test. AEs with incomplete dates and/or time of onset were considered TEAEs unless the incomplete date and/or time clearly proved that the onset was before the first vaccination. If severity or causality was missing, the worst case (i.e., severity = severe, causality = probable) was assumed. Local and systemic tolerability data were summarized by treatment group with 95% CIs and P values, as described for TEAEs. Clinical laboratory test parameters were summarized by treatment group using descriptive statistics.
The intention-to-treat (ITT) analysis population was defined as all randomized subjects who received at least one vaccination, and subjects were analyzed according to the treatment group to which they were randomized, not by the actual treatment they received. The per-protocol (PP) analysis population consisted of all subjects in the ITT analysis population with no major protocol deviations. The primary analyses for the immunogenicity data were to be based on the ITT population. However, 21 out of 32 subjects (65.6%) randomized to IC43 50 µg erroneously received 100 µg IC43 (on days 0 and 7), at 2 study centers. Therefore, immunogenicity results for the PP population are presented, and results for the ITT population are provided as supportive data.
The safety analysis population included all subjects who received at least one vaccination. This population was used for all demographic, safety and tolerability analyses.
| Abbreviations: | ||
| AE | = | adverse event |
| ASC | = | antibody-secreting cell |
| CI | = | confidence interval |
| ELISA | = | enzyme-linked immunosorbent assay |
| GMT | = | geometric mean titer |
| IgG | = | immunoglobulin G |
| ITT | = | intention-to-treat |
| MedDRA | = | Medical Dictionary for Regulatory Activities |
| OPA | = | opsonophagocytic assay |
| Opr | = | outer membrane protein |
| OprF/I | = | OprF/OprI hybrid vaccine |
| PBMC | = | peripheral blood mononuclear cell |
| PP | = | per protocol |
| P. aeruginosa | = | Pseudomonas aeruginosa |
| SAE | = | serious adverse event |
| TEAE | = | treatment-emergent adverse event |
Acknowledgments
We thank Erich Tauber, Astrid Kaltenböck, Nicole Haas, Gabriele Gartner-Wölfl, and Christoph Klade for their contributions to study design or study conduct.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Kerr KG, Snelling AM. Pseudomonas aeruginosa: a formidable and ever-present adversary. J Hosp Infect 2009; 73:338 - 44; http://dx.doi.org/10.1016/j.jhin.2009.04.020; PMID: 19699552
- Rello J, Ausina V, Ricart M, Puzo C, Quintana E, Net A, Prats G. Risk factors for infection by Pseudomonas aeruginosa in patients with ventilator-associated pneumonia. Intensive Care Med 1994; 20:193 - 8; http://dx.doi.org/10.1007/BF01704699; PMID: 8014285
- Parkins MD, Gregson DB, Pitout JD, Ross T, Laupland KB. Population-based study of the epidemiology and the risk factors for Pseudomonas aeruginosa bloodstream infection. Infection 2010; 38:25 - 32; http://dx.doi.org/10.1007/s15010-009-9145-9; PMID: 20012908
- National Nosocomial Infections Surveillance System. National Nosocomial Infections Surveillance (NNIS) system report, data summary from January 1992 through June 2004, issued October 2004. Am J Infect Control 2004; 32:470 - 85; http://dx.doi.org/10.1016/j.ajic.2004.10.001; PMID: 15573054
- Döring G, Meisner C, Stern M, Flagella Vaccine Trial Study Group. A double-blind randomized placebo-controlled phase III study of a Pseudomonas aeruginosa flagella vaccine in cystic fibrosis patients. Proc Natl Acad Sci U S A 2007; 104:11020 - 5; http://dx.doi.org/10.1073/pnas.0702403104; PMID: 17585011
- Parameswaran GI, Sethi S. Pseudomonas infection in chronic obstructive pulmonary disease. Future Microbiol 2012; 7:1129 - 32; http://dx.doi.org/10.2217/fmb.12.88; PMID: 23030418
- Linden PK, Kusne S, Coley K, et al. Use of parenteral colistin for the treatment of serious infection due to antimicrobialresistant Pseudomonas aeruginosa. Clin Infect Dis 2003; 37:154 - 60; http://dx.doi.org/10.1086/379611
- Pier G. Application of vaccine technology to prevention of Pseudomonas aeruginosa infections. Expert Rev Vaccines 2005; 4:645 - 56; http://dx.doi.org/10.1586/14760584.4.5.645; PMID: 16221066
- Bumann D, Behre C, Behre K, Herz S, Gewecke B, Gessner JE, von Specht BU, Baumann U. Systemic, nasal and oral live vaccines against Pseudomonas aeruginosa: a clinical trial of immunogenicity in lower airways of human volunteers. Vaccine 2010; 28:707 - 13; http://dx.doi.org/10.1016/j.vaccine.2009.10.080; PMID: 19887136
- von Specht BU, Lücking HC, Blum B, Schmitt A, Hungerer KD, Domdey H. Safety and immunogenicity of a Pseudomonas aeruginosa outer membrane protein I vaccine in human volunteers. Vaccine 1996; 14:1111 - 7; http://dx.doi.org/10.1016/0264-410X(96)00054-0; PMID: 8911006
- Jones RJ, Roe EA, Gupta JL. Controlled trials of a polyvalent pseudomonas vaccine in burns. Lancet 1979; 2:977 - 82; http://dx.doi.org/10.1016/S0140-6736(79)92559-5; PMID: 91774
- Kim DK, Kim JJ, Kim JH, Woo YM, Kim S, Yoon DW, Choi CS, Kim I, Park WJ, Lee N, et al. Comparison of two immunization schedules for a Pseudomonas aeruginosa outer membrane proteins vaccine in burn patients. Vaccine 2000; 19:1274 - 83; http://dx.doi.org/10.1016/S0264-410X(00)00235-8; PMID: 11137267
- Cripps AW, Peek K, Dunkley M, Vento K, Marjason JK, McIntyre ME, Sizer P, Croft D, Sedlak-Weinstein L. Safety and immunogenicity of an oral inactivated whole-cell pseudomonas aeruginosa vaccine administered to healthy human subjects. Infect Immun 2006; 74:968 - 74; http://dx.doi.org/10.1128/IAI.74.2.968-974.2006; PMID: 16428742
- Derhaschnig U, Bergmair D, Marsik C, Schlifke I, Wijdenes J, Jilma B. Effect of interleukin-6 blockade on tissue factor-induced coagulation in human endotoxemia. Crit Care Med 2004; 32:1136 - 40; http://dx.doi.org/10.1097/01.CCM.0000126265.08175.BE; PMID: 15190963
- Mutharia LM, Hancock RE. Surface localization of Pseudomonas aeruginosa outer membrane porin protein F by using monoclonal antibodies. Infect Immun 1983; 42:1027 - 33; PMID: 6315589
- Qiu D, Eisinger VM, Head NE, Pier GB, Yu HD. ClpXP proteases positively regulate alginate overexpression and mucoid conversion in Pseudomonas aeruginosa. Microbiology 2008; 154:2119 - 30; http://dx.doi.org/10.1099/mic.0.2008/017368-0; PMID: 18599839
- Mansouri E, Gabelsberger J, Knapp B, Hundt E, Lenz U, Hungerer KD, Gilleland HE Jr., Staczek J, Domdey H, von Specht BU. Safety and immunogenicity of a Pseudomonas aeruginosa hybrid outer membrane protein F-I vaccine in human volunteers. Infect Immun 1999; 67:1461 - 70; PMID: 10024596
- Larbig M, Mansouri E, Freihorst J, Tümmler B, Köhler G, Domdey H, Knapp B, Hungerer KD, Hundt E, Gabelsberger J, et al. Safety and immunogenicity of an intranasal Pseudomonas aeruginosa hybrid outer membrane protein F-I vaccine in human volunteers. Vaccine 2001; 19:2291 - 7; http://dx.doi.org/10.1016/S0264-410X(00)00550-8; PMID: 11257350
- Baumann U, Gocke K, Hagemann H, Freihorst J, von Specht BU. Mucosal immune response after nasal and systemic vaccination with a recombinant outer membrane protein F and I of Pseudomonas aeruginosa in healthy volunteers. [abstract] Pediatr Pulmonol Suppl 2002; 34:Suppl 24 275
- Jang IJ, Kim IS, Park WJ, Yoo KS, Yim DS, Kim HK, Shin SG, Chang WH, Lee NG, Jung SB, et al. Human immune response to a Pseudomonas aeruginosa outer membrane protein vaccine. Vaccine 1999; 17:158 - 68; http://dx.doi.org/10.1016/S0264-410X(98)00159-5; PMID: 9987150
- Keitel WA, Dekker CL, Mink C, Campbell JD, Edwards KM, Patel SM, Ho DY, Talbot HK, Guo K, Noah DL, et al. Safety and immunogenicity of inactivated, Vero cell culture-derived whole virus influenza A/H5N1 vaccine given alone or with aluminum hydroxide adjuvant in healthy adults. Vaccine 2009; 27:6642 - 8; http://dx.doi.org/10.1016/j.vaccine.2009.03.015; PMID: 19773098
- Rupprecht CE, Briggs D, Brown CM, Franka R, Katz SL, Kerr HD, Lett S, Levis R, Meltzer MI, Schaffner W, et al. Evidence for a 4-dose vaccine schedule for human rabies post-exposure prophylaxis in previously non-vaccinated individuals. Vaccine 2009; 27:7141 - 8; http://dx.doi.org/10.1016/j.vaccine.2009.09.029; PMID: 19925944
- Vodopija I, Baklaić Z, Vodopija R. Rabipur: a reliable vaccine for rabies protection. Vaccine 1999; 17:1739 - 41; http://dx.doi.org/10.1016/S0264-410X(98)00427-7; PMID: 10194832
- Wasi C, Chaiprasithikul P, Auewarakul P, Puthavathana P, Thongcharoen P, Trishnananda M. The abbreviated 2-1-1 schedule of purified chick embryo cell rabies vaccination for rabies postexposure treatment. Southeast Asian J Trop Med Public Health 1993; 24:461 - 6; PMID: 8160053
- Kitphati R, Pooruk P, Lerdsamran H, Poosuwan S, Louisirirotchanakul S, Auewarakul P, Chokphaibulkit K, Noisumdaeng P, Sawanpanyalert P, Puthavathana P. Kinetics and longevity of antibody response to influenza A H5N1 virus infection in humans. Clin Vaccine Immunol 2009; 16:978 - 81; http://dx.doi.org/10.1128/CVI.00062-09; PMID: 19458206
- Tagliabue A, Rappuoli R. Vaccine adjuvants: the dream becomes real. Hum Vaccin 2008; 4:347 - 9; http://dx.doi.org/10.4161/hv.4.5.6438; PMID: 18682690
- Mansouri E, Blome-Eberwein S, Gabelsberger J, Germann G, von Specht BU. Clinical study to assess the immunogenicity and safety of a recombinant Pseudomonas aeruginosa OprF-OprI vaccine in burn patients. FEMS Immunol Med Microbiol 2003; 37:161 - 6; http://dx.doi.org/10.1016/S0928-8244(03)00072-5; PMID: 12832120
- Centers for Disease Control and Prevention. Recommendations of the Advisory Committee on Immunization Practices (ACIP): use of vaccines and immune globulins for persons with altered immunocompetence. MMWR Recomm Rep 1993; 42:RR-4 1 - 18; PMID: 8474421
- Committee for Medicinal Products for Human Use (CHMP). Guidance on clinical evaluation of new vaccines. October 2006. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003870.pdf
- Goldblatt D. Simple solid phase assays of avidity. Immunochemistry 2, A Practical Approach, Oxford University Press 1997.
- Crotty S, Aubert RD, Glidewell J, Ahmed R. Tracking human antigen-specific memory B cells: a sensitive and generalized ELISPOT system. J Immunol Methods 2004; 286:111 - 22; http://dx.doi.org/10.1016/j.jim.2003.12.015; PMID: 15087226
- Altman D, Machin D, Bryant T, Gardner M. Statistics with confidence. 2nd ed. Bristol: BMJ Books; 2000.
- Jones RL, Froeschle JE, Atmar RL, Matthews JS, Sanders R, Pardalos J, Moeller L, Chin JE, Famula M, Briggs DJ, et al. Immunogenicity, safety and lot consistency in adults of a chromatographically purified Vero-cell rabies vaccine: a randomized, double-blind trial with human diploid cell rabies vaccine. Vaccine 2001; 19:4635 - 43; http://dx.doi.org/10.1016/S0264-410X(01)00238-9; PMID: 11535311