
Abstract
Pandemic influenza A(H1N1)pdm09 virus is a global health threat and between 2009–2011 it became the predominant influenza virus subtype circulating in the world. The research describes the MSSCP (Multitemperature Single Strand Conformation Polymorphism) analysis of the hemagglutinin (HA) region encompassing major neutralizing epitope in pandemic influenza isolates from Taiwan. Several genetically distinct changes appeared in isolates obtained in 2010 and 2011. The majority of changes in HA protein did not result in significant modifications, however three modifications were localized in epitope E of H1 and one was part of the interface binding antibodies BH151 and HC45 possibly making the current vaccine less effective.-Taking into account the possibility of the emergence of influenza A with antibody evading potential, the MSSCP method provides an alternative approach for detection of minor variants which escape detection by conventional Sanger sequencing.
Introduction
Influenza A is a serious disease causing recurring outbreaks with significant negative effects on the general health status globally as well as on the economy of affected populations. In April of 2009 a novel strain of (H1N1) swine-origin influenza A was reported in Mexico and the United States.Citation1-Citation3 The rapid spread of this virus to over 70 countries of the world urged the WHO to raise its pandemic alert level to the highest phase 6 by June 11, 2009. This new A(H1N1)pdm09 virus was found to be a triple reassortant containing a combination of gene segments of swine, human, and avian origin.Citation4-Citation6 The 2009 pandemic influenza A turned out to be relatively mild in its symptoms when compared with other strains responsible for previous pandemics. Most individuals infected with the 2009 pandemic H1N1 strain usually developed uncomplicated illness with full recovery within a week. However, this may be a transient situation as influenza viruses are constantly undergoing changes, caused both by antigenic drift (mutations due to the error-prone genomic RNA amplification) as well as by antigenic shift (exchange of genomic segments during co-infection by two different strains). The best example of these phenomena was the 1918 Spanish flu pandemic virus. The initial outbreak was relatively mild in terms of clinical impact but acquired higher virulence when it returned in following season.
When influenza A is taken under consideration, changes are in some cases gradual and more predictable. Nonetheless the rate of genome mutation is high enough to cause the replacement of circulating strains every 3–5 y with variants that undergo antigenic changes sufficient for substantial or complete evasion of existing antibody response. Therefore, it is crucial to monitor not only the mutations leading to resistance against antiviral drugs but also those modifying important viral epitopes. The latter lead to gradual weakening of the antigen-antibody interactions and therefore reduce the efficacy of existing vaccine.
The viral surface protein hemagglutinin (HA) is the primary target for neutralizing antibodies in natural infections.Citation7 The antigenic variation of HA is the main mechanism employed by the influenza virus to escape the response of the host immune system. The HA of the pandemic A(H1N1)pdm09 strain has changed relatively little in its genetic and antigenic characteristics since it emerged in 2009. In 2010 a genetically distinct variant containing several amino acid changes in hemagglutinin and neuraminidase emerged in Singapore, Australia, and New Zealand. It was a dominant strain in the second and third quarters of 2010 in those countries, although it did not represent a significant antigenic change of the virus.Citation8
The study presents an analysis of the HA region encompassing the main neutralizing epitope of HA from Taiwan isolates of flu seasons 2009–2011. Isolates from samples obtained in 2010 and 2011 show significant changes in HA amino acid composition. Three of the detected mutations are likely to affect the virus-antibody interaction.
Results
Nineteen clinical specimens (throat or nasal swabs) from outpatients with influenza-like illnesses, who developed severe complications, were collected in Taiwan during the 2009–2011 flu season (). To determine viral genotype partial nucleotide sequences (positions 393–1182) of the HA gene were first analyzed by RT-PCR using primers published by the WHO. All the isolates in this study were characterized as A/California/07/2009-like viruses. To verify whether the observed clinical diversity could be correlated with genetic alterations or the presence of minor genetic variants the isolates were further analyzed. Bearing in mind the crucial role of hemagglutinin mutations for influenza virus virulence, representative HA gene fragments encompassing nucleotides 125 to 302 were amplified from cDNAs. This region corresponds to a fragment of influenza virus HA1 polypeptide starting 25 amino acids (H1 numbering is used throughout this paper) after the N-terminal signal peptide of hemagglutinin. A crucial part of the influenza A/H1N1 epitope reacting with neutralizing antibodies is located within this region. To check for the presence of minor genetic variants of the A(H1N1)pdm09 pandemic strains within the obtained amplicones there was performed MSSCP (Multitemperature Single Strand Conformation Polymorphism) analysis.
Table 1. Sample information and clinical symptoms of flu infection among A(H1N1)pdm09 Taiwan patients
MSSCP is a native electrophoretic separation performed under sequentially changed gel temperature. This improves the sensitivity of mutation detection and reduces time of analysis. The temperature changes increase the probability for the PCR products to adopt different ssDNA conformations during the electrophoretic run if they contain nucleotide substitutions All the amplified HA fragments, including corresponding fragments of the reference seasonal (s) (A/Brisbane/59/2007) and pandemic (p) (A/Mexico/4486/09) strains, were denatured and the resulting ssDNA fragments were subjected to the native electrophoresis in optimal conditions for the MSSCP analysis (15–10–5 °C, 450 Vxh/per phase, 10% polyacrylamide gel). Results of this experiment (after visualization with silver stain) are shown in . According to the electrophoretic profiles () none of the samples contains fragments corresponding to the predominant influenza A seasonal strain (s) which excludes the possibility of co-infection with seasonal and pandemic strains. Samples designated as 2009–02626, 2009–00940, 2009–08542, 2010–00842, 2010–06031, 2011–02054, 2011–00623, 2009–06078, 2009–04909, 2009–00937, 2009–08575, 2011–04512, 2011–02068, and 2011–04611 exhibited MSSCP profiles identical to the reference pandemic strain while the electrophoretic profiles of five samples: 2010–03994, 2011–01219, 2010–01164, 2010–05270, and 2010–05347 were different from that of the pandemic reference strain. For further analysis, if profiles reflected distinct DNA sequences, ssDNA bands from the samples indicated by arrows in were extracted from the gel, re-amplified and the PCR products were Sanger sequenced. Additionally, the reference pandemic ssDNA bands were analyzed in the same manner.
Figure 1. New genetic variants among A(H1N1)pdm09 isolates collected at Taiwan between 2009–2011 detected by MSSCP genotyping. RT-PCR products of hemagglutinin gene obtained from pandemic Taiwan A(H1N1)pdm09 virus isolates, as well as reference seasonal (s) and pandemic (p) strains of influenza virus A(H1N1)pdm09 were denatured and ssDNA’s were separated on a 9% polyacrylamide gel using MSSCP method under optimum electrophoretic conditions. DNA bands were visualized with silver stain. Strains are indicated as follows: s – reference seasonal strain, p – reference pandemic strains. Taiwan isolates are described with symbols listed in . Note the presence of five distinct MSSCP electrophoretic profiles (arrows at the bottom of the figure) (samples number: 2010–03994, 2011–01219, 2010–01164, 2010–05270, 2010–05347) among samples, which based on RT-PCR assay, were classified as A(H1N1)pdm09 strain. Dividing lines indicate grouping of images from different parts of the same gel.
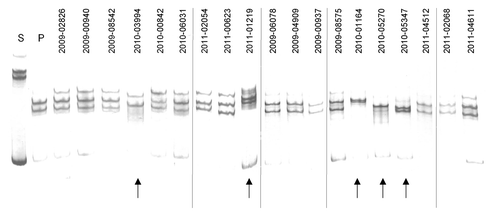
Sanger sequencing of the ssDNA bands confirmed that 14 out of the 19 analyzed samples were identical with the A(H1N1)pdm09 pandemic strain reference sequence (). For the five samples with electrophoretic profiles different from the reference strain, Sanger sequencing revealed the presence of many point mutations. Schematic representation of all detected mutations and their localization within analyzed HA amplicone are presented in . Sample 2010–03994 contained two point mutations, 2011–01219 – eight, 2010–01164 – three, 2010–05270 – seven, and 2010–05347 – five. Six mutations were present in more than one sample () and nine were unique to single isolates. It seems unlikely that mutations arose during the short passages of the original virus from swabs in MDCK cells.
Table 2. Genetic diversity of HA gene fragment in Taiwan A(H1N1)pdm09 isolates
Figure 2. Schematic representation of genetic diversity of hemagglutinin (HA) sequence in five Taiwan isolates of A(H1N1)v pan09 strain. Black arrows above and below A(H1N1)v pdm09 reference sequence indicate modified DNA codons. Red letters show nucleotide changes. Altered amino acids in HA protein sequence are also shown. Blue star over the description indicates amino acids localized in the epitope E of HA. Additionally, red arrows underline three point mutations, which are translated to amino acids in the epitope E of HA.
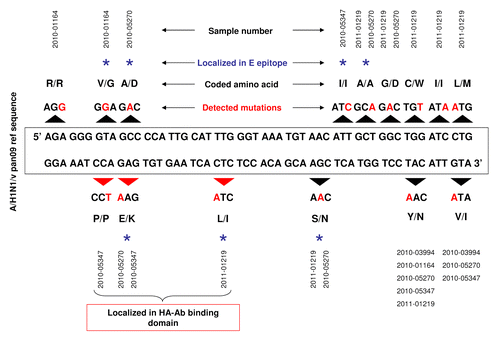
DNA codons containing detected point mutations were translated to amino acids and compared with the pandemic reference sequence. Furthermore, their physico-chemical properties and localization within HA protein structure were also determined. All detected point mutations as well as corresponding changes of amino acids in comparison to reference sequence are summarized in . Two mutations (TAC > AAC, GTA > ATA) close to the vicinity of 3′ end of the amplicones were not analyzed as they were localized within the primer binding region. Five mutations were synonymous substitutions () that did not affect the HA protein sequence. In three cases the mutations led to substitutions preserving the physico-chemical properties of the encoded amino acids. However, some of the non-synonymous substitutions affected the physico-chemical properties of amino acids (). Three point mutations (GTA > GGA, GCC > GAC, GAG > AAG) leading to amino acid changes (valine (V)›glycine (G), alanine (A)›aspartic acid (D), glutamic acid (E)›lysine (K) respectively), were localized in a region presumed to be part of epitope E of H1. To check if these changes could affect HA-Ab binding and modulate the potential immune response against A(H1N1)v we analyzed this region by molecular modeling and estimated the possible impact on vaccine effectiveness.
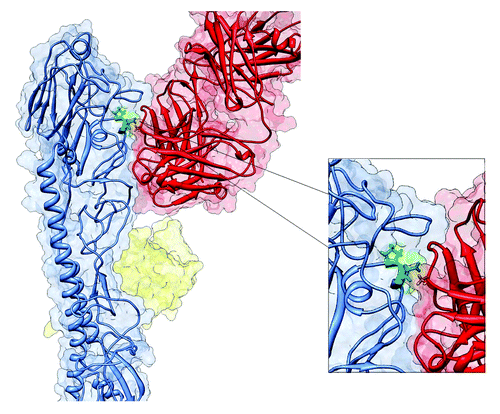
Moreover using sequence based structural alignment method we have matched the HA1 structure of H1 hemagglutinin (3ZTN) with the neutralizing antibody directed to H3 hemagglutinin from 1QFU, the 3-D structures were processed and aligned to obtain maximum resemblance of their HA-Ab binding interfaces. According to , one of the detected mutations - E66K was localized in an HA-Ab binding interface.
Figure 3. Structure of HA-Ab complex. Hemagglutinin protein monomer (blue color) interacting which neutralizing antibody (red color). Amino acid 66, localized on the HA-Ab interface is marked in green.
Substitutions in the amino acid sequence that distinguish a circulating strain from a vaccine strain, can be characterized by antigenic distance measured by values like P value or pepitopeCitation9 and then used for determination of how these substitutions may influence the vaccine effectiveness level predicted by a previously established mathematical model. Provided quantifications are based on five substitutions. The calculated pepitope value based on substituted amino acids located in epitope E shows that these substitutions can result in 17.2% decrease of the predicted vaccine effectiveness (52.7%)
pepitope: 5/34 = 0.147
Vaccine effectiveness: –1.19 x 0.147 + 0.53 = 0.355 (35.5%)
52.7% – 35.5% = 17.2%
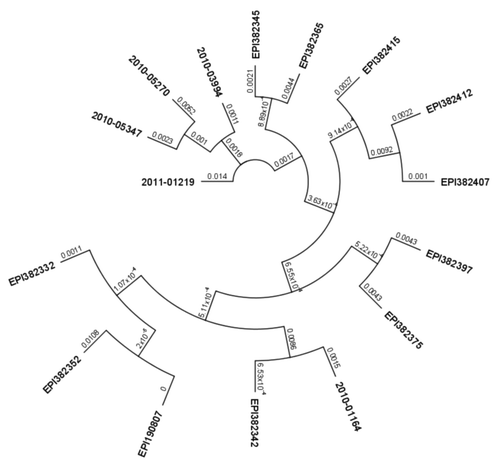
The mutated fragments listed in were compared with available HA sequences of Taiwan pandemic influenza isolates deposited in the NCBI and EpiFlu data banks. The phylogenetic tree constructed for the 935 nt HA gene fragment showed that the isolates may be divided into several clusters (). The isolates described in this work form a distinguishable cluster except isolate 2010–01164 which shares the V47G mutation with a group represented by isolate EPI382342. What’s more, identified A48D and E66K mutations were not found in any other group of isolates that were subjected to comparative analysis. They may therefore reveal novel changes in HA which appeared in the 2010/2011 flu season and suggest a short-term or geographically limited emergence of influenza A virus strains with a high antibody evading potential
Figure 4. Phylogenetic tree of nucleotide sequences of hemagglutinin gene of Taiwan isolates of influenza A(H1N1)pdm09 virus. All sequences included in phylogenetic analysis were 935 nucleotides long. All Taiwan isolates deposited from 2009 through 2011 were retrieved from the EpiFlu Database. Each isolate (EPI210250 HA, EPI382332 HA, EPI382342 HA, EPI382415 HA, EPI190807 HA, EPI382397 HA, EPI382345 HA, EPI382352 HA, EPI382407 HA, EPI382412 HA, EPI382365 HA, EPI382375 HA) represents a number of identical sequences used in alignment. Isolates studied in this paper are described in . Numbers on branches indicate number of nucleotide substitutions per site.
Discussion
Over four years have passed since the emergence of a novel H1N1 influenza A virus (A(H1N1)v) which caused the first global pandemic in the 21st century. The virus is still circulating in many countries, though at a much lower level than in its peak season of 2009–2010.Citation10 The impact of A(H1N1)v during the upcoming seasons will be directly correlated with the immunity of the population provided the virus does not undergo significant antigenic changes.Citation11 Therefore as long as the virus is genetically stable and the percentage of population resistant to infection is high (immunity being attained either by vaccinations or prior exposures to cross-reacting strains) it should not pose a major risk to public health. However, the influenza virus is a master of immune evasion and the possibility of an explosive emergence of new mutants of A(H1N1)v cannot be excluded.
The evasion of the host immune response by an influenza virus can be accomplished in at least three ways. First, by sequential changes in HA (and other influenza virus genes) owing to the error-prone copying of the RNA genome by the viral RNA polymerase; this phenomenon is called as antigenic drift. Second, by much more abrupt changes due to antigenic shift during co-infection with different HA subtypes; it can be described as importation by reassortment of a gene encoding a different HA subtype concurrently with or without other viral gene segments. The third mode is a special case of the antigenic shift involving intrasubtypic reassortment which may occur during co-infection with two different strains of the same serotype. The HA serotype in the progeny virus which undergo reaasortment remains the same, though the HA sequence may differ significantly from the HA sequences of the parental viruses.
The intrasubtypic reassorment may be particularly dangerous when two major strains of the same serotype co-circulate in an area, similarly to the case with the seasonal and pandemic A(H1N1) viruses. It was shown previously that among patients in Polish hospitals co-infection with these two strains was observed at a fairly high frequency.Citation12 In contrast, in the selected samples diagnosed as pandemic A(H1N1)pdm09 in Taiwan, mixed infections with seasonal and pandemic strains were not detected but instead some samples exhibited unique, specific electrophoretic profiles different from the A/Mexico/4486/09 reference pandemic strain. Five out of 19 samples contained point mutations. Notably, all samples isolated in 2009 were identical to the reference sequence even though originating from Mexico and the mutations were observed only in samples isolated in 2010 and 2011, suggesting geographically limited virus evolution.
The MSSCP method applied for these studies provided data only for a part of HA sequence. However in a single electrophoretic run, it was possible to obtain both analytical (screening) data, as well as to detect putative minor genetic variants in a particular sample. The additional advantage of the method was its relatively low cost in comparison to other analytical methods like e.g., next generation sequencing, thus making it feasible for wide-scale epidemiological investigation.
To assess the potential impact of the detected genetic changes on the virus fitness we analyzed them more closely. Conclusions were based on the epitope mapping done by Deem and PanCitation13 in which epitopes A-E of H3 hemagglutinin were aligned with the corresponding amino acids in H1 hemagglutinin. After sequence alignment similarity of the epitopes was verified by 3-D structure alignment of H1 and H3 hemagglutinins. Therefore we refer to this bioinformatically mapped regions of H1 as epitopes A-E of H1 hemagglutinin. Three amino acid substitutions affecting their physico-chemical properties: (GTA > GGA, GCC > GAC, and GAG > AAG), detected in three Taiwanese isolates localized to the HA region presumed to be a part of the epitope E of H1.Citation13 It is also worthy to point out that residue 66 (E) of H1 hemagglutinin is also a part of nine amino acid sequence (ILGNPECEL) listed among predicted epitopes for A2 supertype of MHC class I moleculeCitation14 by Influenza Research DatabaseCitation15
Hemagglutinin is a homo-trimer with each of the subunits comprising two polypeptides: HA1 and HA2. They fold into an α-helical stem and a globular domain, both of which can interact with the host cell membrane. Moreover neutralizing antibodies can bind to the both structures, as seen in crystallographic studies e.g, 3ZTN or 1EO8, but primarily they target the globular domain where the HA receptor binding site is also located.Citation16,Citation17 Based on the approach proposed by Deem and PanCitation13 we used data for H3 hemagglutinin from the 1QFU complex and matched its structure with that of H1 hemagglutinin (3ZTN) to predict their interaction interface. The HA-Ab interface with the BH151 antibody is formed by 14 residues (50–54, 65–70, and 81–85 of HA1). The E66K mutation is within this interface and could affect it as it changes both the charge and size of the amino acid. Single mutations within HA-AB interface or even those located in its close vicinity, have already been shown as the ones allowing viruses to escape neutralization.Citation18 Mutations of the interface residue D54 to N or to Y make the influenza virus immune to the HC45 and BH151 antibodies.Citation19,Citation20 This effect may be caused by introducing a new glycosylation site.Citation21 It is noteworthy that the mutation E66K may create a new glycosylation site and residue 66 is in close contact with D54 forming a hydrogen bond with its main chain in the crystal structure 1EO8.
On the other hand, although mutations at a protein-protein interface may be a strong indication of structural hindrances hampering complex formation, there are also studies showing the ability of antibodies to bind protein variants with one or a few amino acids changed.Citation20,Citation22 The close proximity to a known escape mutation residue, exposure, and interface location suggest the major role of residue 66 in modulating HA-Ab affinity. Nevertheless this hypothesis needs further studies.
It is also worth mentioning that the observed substitutions present in epitope E of H1 may affect vaccine effectiveness when they become widespread. The vaccine effectiveness undergoes annual changes partially due to antigenic distances between the recommended vaccine strain and the circulating strain.Citation13 One of factors to quantify such antigenic distance is peptope value which describes the highest fraction of substituted amino acids in all epitopes.Citation23,Citation24 Recent studies showed that vaccine effectiveness correlates with the pepitope value thus one can quantify the impact of substituted amino acids on the predicted level of vaccine effectiveness for influenza-like illnesses, which is 52.7%.Citation24 According to our pepitope and vaccine effectiveness calculations, the five substitutions found in epitope E of HA can decrease the predicted vaccine effectiveness from 52.7% to 35.2%. Obviously, there are many other factors that can alter the effectiveness of the vaccine. Although the presented calculations are only based on available epidemiological data, conclusions derived from such mathematical models may be useful in future understanding of influenza virus diversity.Citation24
Materials and Methods
Sample collection and virus propagation
Clinical specimens (throat or nasal swabs) from outpatients (southern, eastern, northern, and central Taiwan communities) with influenza-like illnesses and from hospitalized patients who developed severe complications, were collected and transported to the laboratories of the influenza surveillance network in Taiwan, coordinated by the Centers for Disease Control (Taiwan CDC), for influenza diagnosis using virus culture or/and real-time RT-PCR. All influenza isolates from positive cases were transported to the Taiwan CDC where they were shortly passaged in MDCK cells at 34 °C in serum free DMEM medium (Life Technologies, cat no. 21855–025) with TPCK-trypsin (Thermo Scientific, cat no. 20–233) and further characterized by analyzing the viral antigenicity and sequences of HA genes. Sample information with clinical features of A(H1N1)pdm09 infection is summarized in .
Total RNA extraction and cDNA synthesis
Viral RNA was extracted from cultured viral isolates using QIAamp Viral RNA Mini Kits (Qiagen, cat. no. 52904). In brief, 140 µl of a clinical specimen was mixed with AVL buffer, followed by 560 µl of ethanol. The resultant suspensions were applied on a QIAamp Mini column for RNA binding. After washing with wash buffers and eluting with TE buffer, the eluted viral RNA was used in the reverse transcription assay. Alternatively, automated extraction was conducted using a MagNa Pure LC extraction system (Roche). For cDNA preparation, BluePrint(TM) 1st Strand cDNA Synthesis Kit (Takara BIO Inc., cat. no. 6110) was used. Template viral RNA and random primers were mixed in a total volume of 10 µl. After incubating at 65 °C for 5 min followed by cooling on ice, reaction buffer, reverse transcriptase and water were added. The resultant mixture was incubated at 30 °C for 10 min, then at 42 °C for 30 min. The obtained cDNA was used in PCR assays. To determine the viral genotype, partial nucleotide sequences (position 393–1182) of the HA genes were first analyzed by RT-PCR using primers published by the WHO. Genotypes of the isolated viruses were then determined by blasting the sequences through the NCBI database. All of the A(H1N1)pdm09 isolates in this study were identified as A/California/07/2009-like viruses.
Hemagglutinin gene fragment amplification by PCR
The primers specific for a fragment of HA gene of both pandemic A(H1N1)v and seasonal A(H1N1)v strain were as follows: H1msscp1 (5′-AGTAACACAC TCTGT-3′) and H1msscp2 (5′-ACAATGTAGG ACCATGA-3′). The primers were synthesized by IBB PAN (Warsaw, Poland). PCR was performed in a 25 µl reaction volume with standard reagents, 0.4 µM each primer and 1 µl of cDNA solution. The assay was performed in a GeneAmp PCR System 2700 (Applied Biosystems Inc.) according to the following procedure: 7 min. at 94 °C for initial denaturation, then 45 cycles of denaturation at 94 °C for 10 s, annealing at 46 °C for 30 s and extension at 68 °C for 35 s. After the last cycle the reaction was completed by a final extension at 68 °C for 7 min.
MSSCP-based minor genetic variants enrichment procedure
The PCR products were analyzed by the MSSCP method at strictly controlled (± 0.2 °C) gel temperature in a dedicated apparatus: DNAPointer® System/BioVectis as described by Kaczanowski et al.Citation25 Briefly, the PCR products were heat denatured and ssDNA conformers were resolved on a 9% polyacrylamide gel in native conditions (TBE buffer), the gel temperature decreasing during the run from the initial 15 °C to 10 °C to 5 °C. Electrophoretic profiles of analyzed amplicones were compared with reference samples representing seasonal: A/Brisbane/59/2007 and pandemic: A/Mexico/4486/09 A(H1N1) strains. Subsequently, DNA bands were visualized by silver nitrate staining (SilverStain DNA Kit, BioVectis, cat. no. 200–101). Fragments of the MSSCP gel containing bands of interest (for samples with electrophoretic profile in accordance with pandemic reference sample: the first ssDNA band from the bottom of the gel; for samples with electrophoretic profile distinct from pandemic reference sample: the first ssDNA band from the top of the gel) were cut out, ssDNA was eluted and re-amplified using primers and PCR conditions as described above. For subsequent DNA Sanger sequencing (3730xl DNA Analyzer, Applied Biosystems) 1/10 vol. of obtained PCR products, purified with exonuclease I (Exo) and shrimp alkaline phosphatase (Sap) enzymes (Fermentas, cat. no. EN0581 and EF0511) was used.Citation26
Bioinformatic analysis of HA structures
HA structures were downloaded from the RCSB website http://www.rcsb.org (PDB accession codes: 3ZTN and 1QFU) and subjected to a short 200 step minimization procedure with the backbone atoms fixed. Minimization was performed with Tripos SybylX and AMBER force field to relax crystalographic constrains imposed especially on the surface exposed amino acid sidechains. Following structures where superimposed using the Matchmaker softwareCitation27 by: first creating a pairwise sequence alignment with the Needleman and Wunsch algorithm and BLOSUM-62 matrix, and then fitting the aligned residue pairs. RMSD between 165 aligned atom pairs was 1.111 A, yielding a highly matched structural alignment. The aligned crystallographic structures were analyzed with the emphasis on the interface between HA protein and neutralizing antibody chains: H and L chains from 1QFU. Volume analysis was conducted with VolSurf as implemented in Tripos SybylX (Tripos Inc.) packages. UCSF Chimera was used for hydrogen bond network analysis, contact/clash estimation and electrostatic surface generation.
Calculation of pepitope value and vaccine effectiveness
All calculations were based on data shown in Pan et al.Citation24 The P value which is used to measure antigenic distance between circulating and vaccine strainCitation21 was calculated for every epitope of H1 (A-E) using equation 8 in reference Citation24:
P value = Number of substitutions in the epitope / Number of amino acids in the epitope.
The calculation was based on the assumption that the only difference between the hypothetical strain and vaccine strain are the five amino acid substitutions present in epitope E of H1. The Pepitope value is the highest of all P values.Citation24 The P value for epitope E was taken as the pepitope value for further calculations. As shown in chart on in reference Citation24 based on epidemiological data for various circulating and vaccine strains from 1982 to 2008 taken from in reference Citation24, vaccine effectiveness is a decreasing linear function (R2 = 0.68) of pepitope described as:
Vaccine effectiveness = –1.19pepitope + 0.53.
The predicted value of vaccine effectiveness calculated by Pan et al.Citation24 was 52.7%, for pepitope = 0.
Phylogenetic analysis of A(H1N1)pdm09 isolates
Two hundred and twenty one Taiwan origin A(H1N1) hemagglutinin nucleotide sequences deposited from 2009 through 2011 were downloaded from EpiFlu database. The sequences were aligned in Geneious Alignment Tool and duplicated sequences were removed. The 12 unique sequences were then aligned with the five sequences from our study. After extraction, merged regions comprising nucleotides 183 - 291 and 364–1189 were used to create a phylogenetic tree based on Neighbor-joining method with Tamura Nei genetic distance model using Geneious Tree Builder.
| Abbreviations: | ||
| Ab | = | antibody CDC, Centers for Disease Control |
| HA | = | hemagglutinin |
| MSSCP | = | Multitemperature Single Strand Conformation Polymorphism |
| PCR | = | polymerase chain reaction |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We acknowledge the authors and laboratories originating and submitting the sequences from GISAID’s EpiFlu™ Database, on which this research is based (Tsao KC, Shih SR, Chen GW, Huang CG, Chang SC, Lin TY for sequences: EPI382375, EPI382365, EPI382412, EPI382407, EPI382352, EPI382345, EPI382397, EPI382415, EPI382342, EPI382332; CDC and WHO CCRRI for sequence EPI210250; Houng HSH, Gong H, Li T, Verratti K, Wu HS, Liu MT, Yang JR, Lyons AG, and Kuschner RA for sequence EPI210250). All submitters of data may be contacted directly via the GISAID website http://www.gisaid.org.
K.L. and B.S. were supported by a PBS grant “Influenza vaccine—Innovative methods for subunit antigens production” (National Center for Research and Development, Poland).
References
- Dawood FS, Jain S, Finelli L, Shaw MW, Lindstrom S, Garten RJ, Gubareva LV, Xu X, Bridges CB, Uyeki TM, Novel Swine-Origin Influenza A (H1N1) Virus Investigation Team. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N Engl J Med 2009; 360:2605 - 15; http://dx.doi.org/10.1056/NEJMoa0903810; PMID: 19423869
- Naffakh N, van der Werf S. April 2009: an outbreak of swine-origin influenza A(H1N1) virus with evidence for human-to-human transmission. Microbes Infect 2009; 11:725 - 8; http://dx.doi.org/10.1016/j.micinf.2009.05.002; PMID: 19442755
- Smith GJD, Vijaykrishna D, Bahl J, Lycett SJ, Worobey M, Pybus OG, Ma SK, Cheung CL, Raghwani J, Bhatt S, et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 2009; 459:1122 - 5; http://dx.doi.org/10.1038/nature08182; PMID: 19516283
- Garten RJ, Davis CT, Russell CA, Shu B, Lindstrom S, Balish A, Sessions WM, Xu X, Skepner E, Deyde V, et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 2009; 325:197 - 201; http://dx.doi.org/10.1126/science.1176225; PMID: 19465683
- Neumann G, Noda T, Kawaoka Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 2009; 459:931 - 9; http://dx.doi.org/10.1038/nature08157; PMID: 19525932
- Shinde V, Bridges CB, Uyeki TM, Shu B, Balish A, Xu X, Lindstrom S, Gubareva LV, Deyde V, Garten RJ, et al. Triple-reassortant swine influenza A (H1) in humans in the United States, 2005-2009. N Engl J Med 2009; 360:2616 - 25; http://dx.doi.org/10.1056/NEJMoa0903812; PMID: 19423871
- Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 2000; 69:531 - 69; http://dx.doi.org/10.1146/annurev.biochem.69.1.531; PMID: 10966468
- Barr IG, Cui L, Komadina N, Lee RT, Lin RT, Deng Y, Caldwell N, Shaw R, Maurer-Stroh S. A new pandemic influenza A(H1N1) genetic variant predominated in the winter 2010 influenza season in Australia, New Zealand and Singapore. Euro Surveill 2010; 15:19692; PMID: 21034722
- Gupta V, Earl DJ, Deem MW. Quantifying influenza vaccine efficacy and antigenic distance. Vaccine 2006; 24:3881 - 8; http://dx.doi.org/10.1016/j.vaccine.2006.01.010; PMID: 16460844
- http://www.nimr.mrc.ac.uk/who-influenza-centre/annual-and-interim-reports/
- Morens DM, Taubenberger JK, Fauci AS. The 2009 H1N1 pandemic influenza virus: what next?. MBio 2010; 1:e00211 - 10; http://dx.doi.org/10.1128/mBio.00211-10; PMID: 20877580
- Pajak B, Stefanska I, Łepek K, Donevski S, Romanowska M, Szeliga M, Brydak LB, Szewczyk B, Kucharczyk K. Rapid differentiation of mixed influenza A/H1N1 virus infections with seasonal and pandemic variants by multitemperature single-stranded conformational polymorphism analysis. J Clin Microbiol 2011; 49:2216 - 21; http://dx.doi.org/10.1128/JCM.02567-10; PMID: 21471335
- Deem MW, Pan K. The epitope regions of H1-subtype influenza A, with application to vaccine efficacy. Protein Eng Des Sel 2009; 22:543 - 6; http://dx.doi.org/10.1093/protein/gzp027; PMID: 19578121
- Sidney J, Peters B, Frahm N, Brander C, Sette A. HLA class I supertypes: a revised and updated classification. BMC Immunol 2008; 9:1 - 15; http://dx.doi.org/10.1186/1471-2172-9-1; PMID: 18211710
- Squires RB, Noronha J, Hunt V, García-Sastre A, Macken C, Baumgarth N, Suarez D, Pickett BE, Zhang Y, Larsen CN, et al. Influenza research database: an integrated bioinformatics resource for influenza research and surveillance. Influenza Other Respir Viruses 2012; 6:404 - 16; http://dx.doi.org/10.1111/j.1750-2659.2011.00331.x; PMID: 22260278
- Corti D, Voss J, Gamblin SJ, Codoni G, Macagno A, Jarrossay D, Vachieri SG, Pinna D, Minola A, Vanzetta F, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 2011; 333:850 - 6; http://dx.doi.org/10.1126/science.1205669; PMID: 21798894
- Fleury D, Daniels RS, Skehel JJ, Knossow M, Bizebard T. Structural evidence for recognition of a single epitope by two distinct antibodies. Proteins 2000; 40:572 - 8; http://dx.doi.org/10.1002/1097-0134(20000901)40:4<572::AID-PROT30>3.0.CO;2-N; PMID: 10899782
- Meng EC, Pettersen EF, Couch GS, Huang CC, Ferrin TE. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinformatics 2006; 7:339; http://dx.doi.org/10.1186/1471-2105-7-339; PMID: 16836757
- Gigant B, Fleury D, Bizebard T, Skehel JJ, Knossow M. Crystallization and preliminary X-ray diffraction studies of complexes between an influenza hemagglutinin and Fab fragments of two different monoclonal antibodies. Proteins 1995; 23:115 - 7; http://dx.doi.org/10.1002/prot.340230113; PMID: 8539243
- Knossow M, Skehel JJ. Variation and infectivity neutralization in influenza. Immunology 2006; 119:1 - 7; http://dx.doi.org/10.1111/j.1365-2567.2006.02421.x; PMID: 16925526
- Fleury D, Barrère B, Bizebard T, Daniels RS, Skehel JJ, Knossow M. A complex of influenza hemagglutinin with a neutralizing antibody that binds outside the virus receptor binding site. Nat Struct Biol 1999; 6:530 - 4; http://dx.doi.org/10.1038/9299; PMID: 10360354
- Fleury D, Wharton SA, Skehel JJ, Knossow M, Bizebard T. Antigen distortion allows influenza virus to escape neutralization. Nat Struct Biol 1998; 5:119 - 23; http://dx.doi.org/10.1038/nsb0298-119; PMID: 9461077
- Zhou H, Pophale R, Deem MW. Chapter 10: Computer-associated vaccine design. Influenza: Molecular Virology, Caister Academic Press 2009; 173-191.
- Pan K, Subieta KC, Deem MW. A novel sequence-based antigenic distance measure for H1N1, with application to vaccine effectiveness and the selection of vaccine strains. Protein Eng Des Sel 2011; 24:291 - 9; http://dx.doi.org/10.1002/1522-2683(200109)22:16<3539::AID-ELPS3539>3.0.CO;2-T; PMID: 11669539
- Kaczanowski R, Trzeciak L, Kucharczyk K. Multitemperature single-strand conformation polymorphism. Electrophoresis 2001; 22:3539 - 45; http://dx.doi.org/10.1002/1522-2683(200109)22:16<3539::AID-ELPS3539>3.0.CO;2-T; PMID: 11669539
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 1977; 74:5463 - 7; http://dx.doi.org/10.1073/pnas.74.12.5463; PMID: 271968
- Meng EC, Pettersen EF, Couch GS, Huang CC, Ferrin TE. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinformatics 2006; 7:339; http://dx.doi.org/10.1186/1471-2105-7-339; PMID: 16836757