
Abstract
The importance of follicular T helper (TFH) cells and the germinal center (GC) reaction in the humoral immune response has become clear in recent years, however the role of TFH cells and the GC in an HIV vaccine strategy remains unclear. In this study, we primed mice with gp120-encoding DNA and boosted with gp120 protein, a regimen previously shown to induce high titers of high affinity and cross-reactive anti-gp120 Abs. Priming with gp120 DNA caused increased TFH cell differentiation, GC B cells, and antigen-specific antibody titers, compared with priming with gp120 protein. Priming with DNA also caused more activated CD4+ T cells to become TFH cells and more GC B cells to become memory cells. Deletion of BCL6 midway through the vaccine regimen resulted in loss of TFH cells and GCs, and, unexpectedly, increased anti-gp120 IgG titers and avidity. Our data suggests vaccination with gp120-encoding DNA elicits a stronger and more rapid TFH and GC response than gp120 protein. Furthermore, we demonstrate that the GC reaction may actually limit antigen-specific IgG secretion in the context of repeated immunizations.
Introduction
To make an effective vaccine, one of the common objectives has been the generation of high affinity, protective antibodies which, in the case of HIV, are cross-clade reactive and have neutralizing capacity.Citation1,Citation2 While much focus has been placed on serum antibody analysis in the development of HIV vaccines, little research has been done on evaluating the robustness of germinal center (GC) development with different vaccination approaches. The GC reaction or GC response represents the rapid clonal expansion of antigen-specific B cells, and typically peaks 2 wk following initial exposure to antigen. Within the GC, activated antigen-specific B cells undergo rapid proliferation, isotype class switching, somatic hypermutation and affinity maturation.Citation3-Citation5 GC B cells express standard B cell markers such as B220, but also upregulate activation markers such as GL7 and glycoproteins that bind Peanut Agglutinin (PNA).Citation3-Citation5 GC B cells express high levels of the transcription factor BCL6, and BCL6 is required for formation of GCs.Citation3-Citation5 Additionally, GC B cells express high levels of Fas, since the majority of GC B cells die and are highly susceptible to apoptosis.Citation3-Citation5 Follicular helper T (TFH) cells are a special subset of T helper (Th) cells that are indispensable for the GC reaction.Citation6,Citation7 TFH cells are CD4+ T cells that express the chemokine receptor CXCR5, which allows for their localization within the GC.Citation6,Citation7 TFH cells also express the T cell activation markers ICOS and PD-1, and like GC B cells, also require BCL6 for their development.Citation6,Citation7 The GC leads to the development of plasma cells, which secrete high affinity antibodies, and also promotes the formation of memory B cells.Citation3-Citation5 The GC has been shown to peak earlier in a secondary response than after a primary immunization,Citation8 but nothing is known about the kinetics of the TFH response and the GC reaction with vaccines that require repeated immunizations. Since GC responses lead to high affinity antibody production and the formation of memory B cells, it is critical to study the development of TFH cells and GCs to gain insights into effective HIV vaccine strategies.
Currently, heterologous prime-boost vaccination strategies employing a DNA priming component are making headways in different disease fields, such as HIV, influenza, malaria, and tuberculosis.Citation9-Citation12 Previously, we have shown mice immunized with a DNA vector encoding the gp120 form of the HIV-1 envelope glycoprotein, followed by injection of recombinant gp120 protein, yield antibodies with higher specificity and avidity than either vaccine alone, and, more importantly, develop improved neutralizing antibodies against primary viral isolates.Citation13-Citation15 Because TFH cells are crucial for the development of plasma cells secreting mutated high affinity antibody from the GC, we hypothesized DNA priming causes more TFH cell differentiation, thereby triggering a more robust GC response. To test this, we utilized a gp120 DNA prime and gp120 protein boost vaccination scheme. Compared with protein priming, DNA priming resulted in TFH cells which were elevated earlier in the immune response and GC B cells that were significantly increased across all time points, suggesting DNA priming preferentially affected the differentiation of GC B and memory B cells.
Additionally, we utilized a novel conditional BCL6 knockout mouse system, where BCL6 can be deleted with Tamoxifen treatment after the start of the immunization regimen, to block GC formation. We found that loss of the ability to form GCs during the priming phase, paradoxically leads to a greater antibody response. Together, our data show a complex role for the GC reaction in producing high titer antibodies after immunization with an HIV vaccine preparation.
Results
Immunization with gp120-encoding DNA yields a stronger GC response than gp120 protein
To test the hypothesis that gp120-encoding DNA triggers stronger GC responses than gp120 protein alone, C57BL/6 mice were injected i.m. with either gp120-encoding DNA, gp120 protein, or empty vector DNA, 3 times, 2 wk apart (). This experimental design was based on our previous HIV-1 gp120 immunogenicity studies.Citation13-Citation15 Traditionally, GC responses tend to peak between day 7 and 10 of the immune response. In our initial experiments with this gp120 immunization scheme, we looked at these time points, only to find little evidence of an active GC (data not shown). Also, to our knowledge, no data has been published demonstrating the kinetics of TFH cell differentiation with repeated immunizations. Therefore, to determine the optimal time to analyze mice given multiple injections, we immunized C57BL/6 mice i.m. with sheep red blood cells (SRBC) 3 times, 2 wk apart and sacrificed them at different times afterward. Our data showed GC B cells and TFH cells peaking earlier, on day 3, after final injections (Fig. S1). Therefore, we chose to analyze mice for the following experiments on days 3 and 7 after final injections.
Figure 1. Experimental design for testing DNA vs. protein priming. C57BL/6 mice were primed i.m. with either gp120-encoding DNA, gp120 protein, or empty vector, 3 times, 2 wk apart. Some mice were sacrificed (S) 3 and 7 d after final priming injections. The remaining mice were rested for 4 wk, then all groups were given 2 booster injections of gp120 protein, 2 wk apart. Mice were then sacrificed 3 and 7 d after the final boosters.
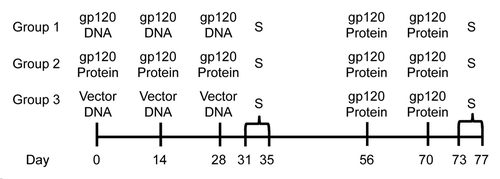
With this information in mind, we then examined TFH cells and GC B cells after just the priming phase of the immunization scheme (). Mice which received gp120-encoding DNA had increased TFH cells and significantly more GC B cells in the spleen 3 d after the last priming injections (). However, the increase in TFH cells was not significant, and by day 7 after the last priming injections, the numbers of TFH cells in mice primed with DNA equaled that of mice primed with gp120 protein. When absolute numbers of TFH cells and GC B cells were analyzed, similar trends were observed, and the increased frequency of GC B cells correlated with an increase in absolute numbers of GC B cells (Fig. S2). Antibody analysis revealed priming with gp120 DNA induced significantly higher antigen-specific IgG titers, both 3 and 7 d after final injections (day 28; ). The avidity of these antibodies was somewhat higher on day 3 with gp120 DNA priming (). This data demonstrates priming with gp120 DNA alone, without booster injections, already triggers more robust GC responses and antigen-specific IgG production than does gp120 protein.
Figure 2. Increased germinal center activity after gp120 DNA priming. Mice were immunized according to and sacrificed 3 and 7 d after final priming immunizations. (A) TFH and GC B cell populations after priming only. TFH cells gated on CD4+ CXCR5+ ICOS+ PD-1hi. GC B cells gated on B220+ Fas+ PNA+ GL7+. n = 3. Percent of total spleen; mean ± SE. (B) Representative flow plots of TFH cells and GC B cells in (A) from day 3 after final immunization. (C) Serum anti-gp120-specific IgG titers after prime only. n = 3. D) Avidity of gp120-specific IgG antibodies after priming only. n = 3; mean ± SE *P < 0.05, **P < 0.01, ***P < 0.001. Representative of 2 experiments.
Priming with gp120-encoding DNA enhances GCs and the proportion of TFH cells in spleen
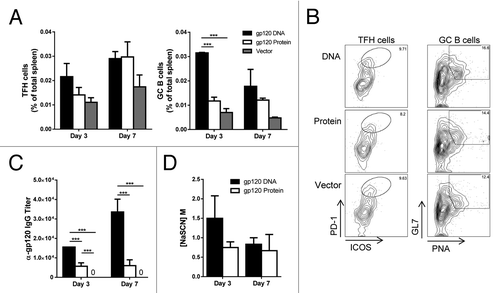
After initially priming with gp120-encoding DNA, gp120 protein, or empty vector DNA, all groups were rested for 4 wk, then received 2 gp120 protein booster immunizations, 2 wk apart (). TFH cells from spleen were elevated in mice receiving gp120 DNA 3 d after the final injections, but tapered off by day 7 (). GC B cell populations, however, remained significantly higher in mice primed with gp120 DNA on both day 3 and 7 after final immunizations (). After boosting, the increase in TFH cells in mice receiving DNA priming was not significant, and again, by day 7 after the last priming injections, the numbers of TFH cells in mice primed with DNA equaled that of mice primed with gp120 protein. The absolute numbers of TFH cells and GC B cells showed similar trends, and as with analysis after the priming phase (), the increased frequency of GC B cells () correlated with an increase in absolute numbers of GC B cells (Fig. S1). This data demonstrates the advantage of priming with gp120 DNA rather than protein, as GC cell populations were increased and arose earlier in the immune response.
Figure 3. Enhanced GC B cells and TFH cell proportions with gp120 DNA priming. Mice were immunized as in and sacrificed 3 (day 73) and 7 (day 77) d after final gp120 protein booster injections (day 70). (A) TFH and GC B cell populations after prime-boost regimen. Cells gated as in . Percent of total spleen. n = 3 – 6; mean ± SE **P < 0.01, ***P < 0.001 (B) Representative flow plots of TFH cells and GC B cells in (A) from day 3 after final protein booster. (C) Percent effector memory T cells (CD3+ CD4+ CD44hi CD62L-) in gp120 DNA and protein primed mice after protein boosters. Percent of Th cells (CD3+ CD4+). n = 5 – 6; mean ± SE. (D) Ratio of TFH to effector memory cells. n = 5 – 6; mean ± SE ***P < 0.001 by t test. Representative of 3 experiments.
When effector memory (EM) CD4+ T cells were analyzed in the spleen of these mice, no significant differences were seen on either day 3 or 7 after final immunization (). TFH cells are activated Th cells, and are found in the EM population of CD44+ CD62L− cells. Therefore, we analyzed the proportion of TFH cells within this EM population. On day 3, the proportion of TFH cells within the EM population was significantly higher with gp120 DNA priming than with protein alone (). By day 7, however, the proportion of TFH cells from gp120 protein priming caught up to the levels from gp120 DNA priming. Therefore, after boosting with gp120 protein, mice primed with DNA showed a clear advantage in TFH and GC B cell development.
Priming with gp120 DNA improves GC activity
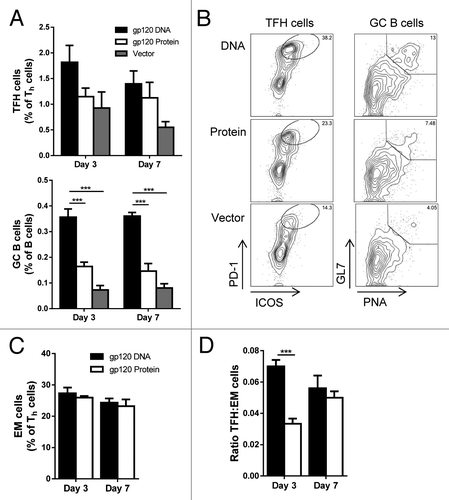
Like EM T cells, neither priming method yielded significantly higher percentages of splenic memory B cells (). Therefore, we were interested in what percentage of these memory cells were of GC origin. Recent literature has shown CD73 to be an accurate marker for determining such cells.Citation16 When the fraction of memory B cells of GC origin was examined, we saw significantly higher percentages with gp120 DNA priming both 3 and 7 d after final immunization (). Titers of anti-gp120 IgG with DNA priming were significantly higher 3 d after final immunizations, as was the avidity (). Antibody titers broadly correlate with the level of TFH and GC activity when individual mice are analyzed (Fig. S4), however this association is not as strong as the differences in TFH and GC B cells between types of immunizations. It was not until day 7 that the antibodies of mice primed with gp120 protein equaled the levels seen in gp120 DNA mice. Not only was the functionality of these B cells increased with gp120 DNA priming, but, again, the increase in antibody secretion and affinity maturation occurred earlier.
Figure 4. Priming with gp120-encoding DNA improves germinal center activity. Mice were immunized and sacrificed as in . (A) Memory B cells (gated as CD19+ IgD- GL7- CD38+) in mice primed with gp120-encoding DNA or gp120 protein. Percent of total spleen. n = 4 – 6; mean ± SE (B) Memory B cells originating from the GC. Gated as CD19+ IgD- GL7- CD38+ CD73+. The CD73+ B cells are shown as a percentage of the memory B cell population. n = 4 – 6; mean ± SE *P < 0.05, **P < 0.01 by t test. (C) Transitional B cells (gated on CD19+ IgD- GL7+ CD38+); percent of total spleen. n = 4 – 6; mean ± SE. Representative flow plots of day 3 and day 7 from mice primed with gp120 DNA. Quadrants show memory B cells (MB; top left) and transitional B cells (TB; top right). (D) Serum anti-gp120-specific IgG titers after boosting. mean ± SE. E) Avidity of serum anti-gp120-specific antibodies after boosting. mean ± SE n = 4 – 6; *P < 0.05, ***P < 0.001. Representative of 3 experiments.
While analyzing memory B cell populations, we observed a decrease in this cell population from day 3 to day 7, even though the memory cells of GC origin expanded during the same period with both immunization methods. Therefore, we utilized the surface markers CD38 and GL7 to differentiate memory B cells (MB: CD38+ GL7−) from transitional B cells (TB: CD38+ GL7+) (, flow plots). ln terms of B cell kinetics, it was interesting to observe that regardless of which priming method was used, B cells seem to be differentiating out of a memory B cell phenotype into a transitional cell state between days 3 and 7, while memory cells of GC origin retain their phenotype and proliferate. Thus, while not increasing overall memory B cell numbers, priming with gp120 DNA significantly increased the percentage of memory cells of GC origin. Furthermore, in the context of repeated immunizations, between days 3 and 7, memory B cells in all mice decreased, while those with a transitional cell phenotype increased, demonstrating a transition from a memory to a more active cell state.
Deletion of BCL6 results in a significant increase of gp120-specific IgG
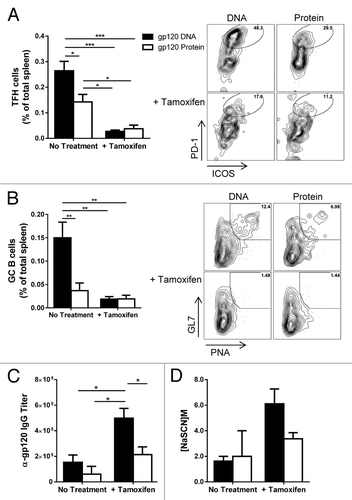
Previously, we have generated a mouse wherein exons 7 – 9 of the BCL6 gene are flanked with loxP recombination sites (BCL6fl/fl) (Fig. S2A).Citation17 In this study we created a new conditional knockout mouse model in which BCL6fl/fl mice were mated to mice expressing a Cre recombinase fused to a human estrogen receptor (Cre-ERT2). Under control of the estrogen receptor, we can functionally delete BCL6 by administering the estrogen homolog Tamoxifen to these mice. The cre-ER fusion protein resides in the cytoplasm of cells and unable to delete BCL6, but addition of Tamoxifen will cause translocation of the cre-ER protein into the nucleus where it can cause recombination of BCL6 DNA. This ER-cre system has been used in other conditional knockout systems to study the immune response.Citation18,Citation19 As in the initial experiments, mice were primed with either gp120-encoding DNA or gp120 protein, 3 times, 2 wk apart (). During the 4-wk rest period, half of the mice received i.p. injections of tamoxifen every day, for 5 d. Twelve days after the final tamoxifen injection, mice were given the first of 2 gp120 protein booster injections. Administration of tamoxifen effectively and functionally deleted BCL6 from mice (Fig. S2B). Since the most drastic changes in GC dynamics were seen 3 d after final immunizations in our earlier studies, we chose this time point to analyze these mice. Once again, gp120 DNA triggered significantly higher TFH and GC B cell populations in the spleen of mice not treated with tamoxifen (). As expected, deletion of BCL6 severely and significantly reduced the TFH and GC B cell populations (). Surprisingly, deletion of GCs led to an increase in gp120-specific IgG, (). Priming with gp120 DNA, followed by functional deletion of BCL6 significantly enhanced antigen-specific IgG titers compared with all other groups. Furthermore, the avidity of these antibodies was increased as well (). Treatment of control mice with Tamoxifen did not affect antibody titers or avidity, however, Tamoxifen-treated mice showed a non-significant trend toward higher GC and TFH responses (Fig. S6). This data verifies the necessity of BCL6 for GC development, while demonstrating the dispensability of these structures for antibody secretion and affinity maturation in the context of repeated immunizations.
Figure 5. Experimental design for deletion of BCL6. Cre-ERT2 BCL6fl/fl mice were primed with either gp120-encoding DNA or gp120 protein, followed by 2 boosters of gp120 protein, as was done for – 4. Some Cre-ERT2 BCL6fl/fl mice also received 5 i.p. injections of tamoxifen (T) between the prime and boost injections to delete BCL6. Mice were sacrificed 3 d after final injections.
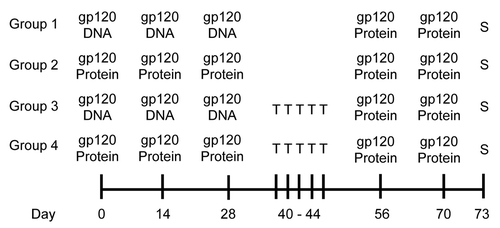
Figure 6. Germinal centers are lost with deletion of BCL6, yet antibody titers increase. Cre-ERT2 BCL6fl/fl mice were immunized as in and sacrificed 3 d after final protein booster immunizations. Some mice were given tamoxifen injections between prime and boost immunizations to delete BCL6. (A) TFH cells (gated as in ) in spleen of mice either untreated or given i.p. tamoxifen injections before boosting. Percent of total spleen. n = 3 – 4; mean ± SE. Representative flow plots of each treatment condition are also shown. (B) GC B cells (gated as in ) in spleen after prime-boost regimen with representative flow plots of each treatment condition. Percent of total spleen. n = 3 – 4; mean ± SE. (C) Serum anti-gp120-specific IgG titers and avidity of anti-gp120-specific antibodies after boosting. mean ± SE n = 3 – 4; *P < 0.05, **P < 0.01, ***P < 0.001
Discussion
While a number of research publications have shown the protective advantage of incorporating a DNA priming step into heterologous prime-boost vaccination strategies, few studies have looked into the mechanism by which DNA priming affords this advantage. Furthermore, almost nothing is known about the role of the TFH cell and the GC response in prime-boost vaccination. Here we demonstrate, using an HIV gp120 vaccine scheme, priming with gp120-encoding DNA yields increased TFH and GC B cell populations compared with priming with gp120 protein. Furthermore, this increase occurs at an earlier time-point after immunization. The early antibody response afforded by DNA priming was seen after the priming phase as well as following the boost injections. While literature exists explaining the possible different mechanisms of antigen processing and presentation with plasmid vector vaccines,Citation12,Citation20 it is not yet clear how the protein from these plasmids affords an advantage.
Human trials of HIV-1 DNA vaccines consisting only of injections of HIV-1-encoding DNA (i.e., not prime-boost approaches) have been disappointing for eliciting the production of antibodies.Citation9,Citation21 More recently, several prime-boost HIV-1 vaccines have been tested in humans that use a pure DNA component as well as protein, and these systems have succeeded in provoking a significant antibody response.Citation22-Citation25 Studies with cancer vaccines in humans have also supported the utility of DNA priming in a prime-boost system.Citation25 Thus, understanding how DNA priming can augment immunity in a prime-boost vaccination system is warranted.
Memory cell development is considered critical for an effective vaccine. We found no apparent increase in memory T cell populations after DNA priming, however, when looking at the proportion of effector Th cells which are TFH cells, we saw that gp120 DNA priming leads to more activated CD4+ T cells becoming TFH cells, rather than other Th cell subsets. For B cells, priming with DNA effectively enhanced GC memory B cell outcomes, in this case by triggering the development of more CD73+ memory B cells derived from the GC. This data, combined with the earlier peak in antigen-specific IgG, would suggest that priming with gp120 DNA not only triggers more memory B cell development from GCs, but that those memory cells can undergo affinity maturation and differentiate into plasma cells more quickly than with protein priming. The decrease in memory B cell populations between days 3 and 7, and the concurrent increase in transitional cells (), shows an interesting trend in terms of B cell kinetics. It would be worthwhile to investigate how repeated immunizations affect the timing of this cell transition; perhaps the increased number of immunizations is decreasing the time necessary to make this transition.
To investigate the necessity of GCs in the context of repeated immunizations, our inducible Cre mouse model allowed us to delete BCL6, the master transcription factor for GCs, after priming the mice. Our data suggests that once memory B cells are formed in the priming stage, further formation of GCs actually limits the antibody production of antigen-specific IgG-secreting B cells. Since, we observed more memory B cells of GC origin developing with gp120 DNA priming (), we reason that during protein booster injections, forming GCs limits the antibody-secreting potential of these memory cells, as they appear to be better able to differentiate into antibody-secreting plasma cells independent of the constrains of the GC. How these memory cells undergo additional affinity maturation, as evident by their increased avidity, outside the GC is not understood at this time. It is possible these B cells are interacting with helper T cells outside the B cell follicle.
These results have highly significant implications for the field of B cell immunology, HIV-1 vaccine development and AIDS therapy. When and where B cells can undergo affinity maturation needs to be fully understood to design an optimal immunization schedule that elicits high affinity, broadly neutralizing antibodies by a prophylactic HIV-1 vaccine. Further studies are warranted to determine how much of a role repeated immunizations play in extra-follicular antibody development. At what point GCs are no longer needed to develop antigen-specific antibodies with high avidity is of great importance to the vaccine field. Our findings also have potential impact for the design of immune therapy strategies for HIV-1 infected people. Published works have demonstrated TFH cells as being a major target and reservoir for HIV-1 (Citation26,Citation27,Citation28). Therefore, it is undesirable to stimulate these cells when treating infected patients. However, if there is a way to trigger high-affinity antibody production without involving TFH cells, patients could increase their antibody-mediated protection without activating the production of new virus from TFH cells.
One shortcoming of the study is that because mice do not produce enough serum to adequately perform a full array of HIV-1 neutralization tests, we were not able to determine if the mice given the prime-boost gp120 immunization regimen actually developed broadly neutralizing anti-HIV antibodies. Additionally, how our study of TFH cells and GC B cells in mice with prime-boost gp120 immunization relates to vaccination of humans against HIV-1 gp120 is not clear. However, currently TFH and GC B cell responses cannot be assessed in humans after vaccination. TFH-like cells in the blood have recently been used as a biomarker for the TFH response,Citation29 but the exact relationship of these blood TFH-like cells to true TFH cells remains unclear.Citation30 One clear advantage of analyzing TFH cells in mice, however, is the ability to manipulate the GC response in order to alter the antibody response, as we have done in the current study.
In conclusion, the key goal of our studies was to provide insight into how DNA priming provides an advantage in the antibody response, and here we have pinpointed, for the first time, the ability of DNA priming to augment the GC reaction, as well as to transiently increase TFH cells. Thus, the improved antibody response in a heterologous prime-boost vaccination system with DNA priming can be explained by increased TFH and GC function. At the same time, we have found that the GC reaction appears to limit antibody production in the boost phase. These results should prompt a re-evaluation of HIV vaccine design.
Materials and Methods
Mice and immunizations
Eight to 10-wk-old C57BL/6 mice were obtained commercially from The Jackson Laboratory. Experiments were done with mice of both genders, and similar results obtained with both genders. BCL6fl/fl mice were previously describedCitation17 and mated to UBC-Cre-ERT2 mice, which were also acquired from The Jackson Laboratory (stock # 007001). In UBC-cre-ERT2 mice, a fusion of the cre recombinase gene and the human Estrogen Receptor is expressed constitutively under control of the UBC gene promoter.Citation31 The floxed BCL6 allele was genotyped as previously described,Citation17 and the cre-ER allele was genotyped by PCR according to the Jackson labs protocol. Mice were bred under specific pathogen-free conditions at the laboratory animal facility at the Indiana University School of Medicine (IUSM) and were handled according to protocols approved by the IUSM Animal Use and Care Committee.
Pilot study mice were immunized i.m. with 1 × 109 sheep red blood cells (SRBC; Rockland Immunochemicals Inc.) in 100 uL of PBS. A codon optimized JR-FL gp120 DNA vaccine construct in the pJW4303 vector was used for all DNA-based immunizations, as previously reported.Citation14 Recombinant HIV-1 gp120 proteins were produced from Chinese Hamster Ovary (CHO) cells, as previously reported.Citation14 Mice were immunized i.m. with either 100 ug of DNA or 10 ug gp120 protein in ALUM (Sigma-Aldrich Corp., St. Louis). To prepare the protein/Alum, 30ug gp120 protein was absorbed onto 20ul aluminum hydroxide gel (Al(OH)3 gel, 13mg/ml) by mixing, followed by a 20 min incubation at room temperature. Protein and Al(OH)3 mixtures were then spun down at 12 000 rpm for 10 min, the supernatant was removed, and the pellet was re-suspended in the final volume of DPBS for immunization. Quantitation of protein remaining in the Alum precipitate supernatant showed that 75–80% of protein was adsorbed to the Alum. For both DNA and protein immunizations, a total of 100 uL was injected, 50 uL per hind leg.
Tamoxifen
Mice were given i.p. injections of 4 mg Tamoxifen (Sigma-Aldrich Corp., cat. # T5648) in sunflower seed oil.
Flow cytometry
All Abs were purchased from eBioscience, BD Biosciences, or Biolegend. Total spleen cells were incubated with anti-mouse CD16/CD32 (Fcγ receptor) for 20 min, followed by surface staining for the indicated markers. A viability stain (eBioscience) was used to gate out dead cells. The following antibodies were used to analyze B cells: Phycoerythrin (PE) labeled anti-mouse B220 (BD Biosciences); biotin-labeled anti-mouse Fas (BD Biosciences) + streptavidin-labeled PE-Cy7 (Biolegend); Allophycocyanin (APC)-labeled GL7 (BD Biosciences); Fluorescein isothiocyanate (FITC)-labeled Peanut Agglutinin (PNA; Vector Labs Inc.); PE-labeled anti-mouse CD38 (eBioscience); PE-Cy7-labeled anti-mouse CD73 (eBioscience). The following antibodies were used to analyze T cells: Alexa Fluor 700 labeled anti-mouse CD3 (BD Biosciences); PE-Cy7-labeled anti-mouse CD4 (BD Biosciences); PE-labeled anti-mouse CD44 (eBioscience); FITC-labeled anti-mouse CD62L (BD Biosciences); PerCP-eFluor 710-labeled anti-mouse CXCR5 (eBioscience); APC-labeled anti-mouse PD-1 (Biolegend); FITC-labeled anti-mouse ICOS (BD Biosciences).
B and T cell analysis
TFH cells were identified by the following strategy: identify % CD4+ CXCR5+ double positive T cells within the total spleen population, then identify % ICOS+ PD-1hi double positive cells within the CD4+ CXCR5+ population. The % TFH of total spleen = (% CD4+ CXCR5+ T cells) × (% ICOS+ PD-1hi double positive cells). GC B cells cells were identified by the following strategy: identify % B220+ Fas+ double positive cells B cells within the total spleen population, then identify % PNA+ GL7+ B cells within the B220+ Fas+ double positive cells B cell population. The % GC B cells of total spleen = (% B220+ Fas+ cells) x (% PNA+ GL7+ double positive cells).
Antibody quantitation analysis
Antibody titers of gp120-specific IgG were measured by ELISA as previously reported.Citation13 Antibody avidity was measured via the NaSCN displacement method, as previously described.Citation13
Statistical Analysis
All data analysis was done using SPSS Statistics 20 software. Unless otherwise stated, ANOVA with Tukey post hoc analysis was used. Only significant differences (*P < 0.05, **P < 0.01, ***P < 0.001) are indicated in Figures.
Additional material
Download Zip (533.8 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank Dr Jennifer Speth for her assistance with immunizing mice. This work was supported by NIAID grants 1R21AI090150, 1R21AI092212 and 1R21AI099825 to A.L.D., NHLBI training grant T32 HL007910-14 to K.H., NIH grants AI065250, AI082274 and AI082676, and the Bill and Melinda Gates Foundation Collaboration for AIDS Vaccine Discovery (CAVD) grant OPP1033112 to S.L., and NIH grant AI087191 to S.W.
References
- Koff WC, Burton DR, Johnson PR, Walker BD, King CR, Nabel GJ, Ahmed R, Bhan MK, Plotkin SA. Accelerating next-generation vaccine development for global disease prevention. Science 2013; 340:1232910; http://dx.doi.org/10.1126/science.1232910; PMID: 23723240
- Burton DR, Desrosiers RC, Doms RW, Koff WC, Kwong PD, Moore JP, Nabel GJ, Sodroski J, Wilson IA, Wyatt RT. HIV vaccine design and the neutralizing antibody problem. Nat Immunol 2004; 5:233 - 6; http://dx.doi.org/10.1038/ni0304-233; PMID: 14985706
- MacLennan ICM. Germinal centers. Annu Rev Immunol 1994; 12:117 - 39; http://dx.doi.org/10.1146/annurev.iy.12.040194.001001; PMID: 8011279
- Tarlinton DM. Evolution in miniature: selection, survival and distribution of antigen reactive cells in the germinal centre. Immunol Cell Biol 2008; 86:133 - 8; http://dx.doi.org/10.1038/sj.icb.7100148; PMID: 18180800
- Klein U, Dalla-Favera R. Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol 2008; 8:22 - 33; http://dx.doi.org/10.1038/nri2217; PMID: 18097447
- Fazilleau N, Mark L, McHeyzer-Williams LJ, McHeyzer-Williams MG. Follicular helper T cells: lineage and location. Immunity 2009; 30:324 - 35; http://dx.doi.org/10.1016/j.immuni.2009.03.003; PMID: 19303387
- Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol 2011; 29:621 - 63; http://dx.doi.org/10.1146/annurev-immunol-031210-101400; PMID: 21314428
- Hollowood K, Macartney J. Cell kinetics of the germinal center reaction--a stathmokinetic study. Eur J Immunol 1992; 22:261 - 6; http://dx.doi.org/10.1002/eji.1830220138; PMID: 1730253
- Vasan S, Schlesinger SJ, Huang Y, Hurley A, Lombardo A, Chen Z, Than S, Adesanya P, Bunce C, Boaz M, et al. Phase 1 safety and immunogenicity evaluation of ADVAX, a multigenic, DNA-based clade C/B’ HIV-1 candidate vaccine. PLoS One 2010; 5:e8617; http://dx.doi.org/10.1371/journal.pone.0008617; PMID: 20111582
- Dou J, Wang Y, Yu F, Yang H, Wang J, He X, Xu W, Chen J, Hu K. Protection against Mycobacterium tuberculosis challenge in mice by DNA vaccine Ag85A-ESAT-6-IL-21 priming and BCG boosting. Int J Immunogenet 2012; 39:183 - 90; http://dx.doi.org/10.1111/j.1744-313X.2011.01066.x; PMID: 22152009
- Chuang I, Sedegah M, Cicatelli S, Spring M, Polhemus M, Tamminga C, Patterson N, Guerrero M, Bennett JW, McGrath S, et al. DNA prime/Adenovirus boost malaria vaccine encoding P. falciparum CSP and AMA1 induces sterile protection associated with cell-mediated immunity. PLoS One 2013; 8:e55571; http://dx.doi.org/10.1371/journal.pone.0055571; PMID: 23457473
- Lu S. Heterologous prime-boost vaccination. Curr Opin Immunol 2009; 21:346 - 51; http://dx.doi.org/10.1016/j.coi.2009.05.016; PMID: 19500964
- Vaine M, Wang S, Hackett A, Arthos J, Lu S. Antibody responses elicited through homologous or heterologous prime-boost DNA and protein vaccinations differ in functional activity and avidity. Vaccine 2010; 28:2999 - 3007; http://dx.doi.org/10.1016/j.vaccine.2010.02.006; PMID: 20170767
- Wang S, Arthos J, Lawrence JM, Van Ryk D, Mboudjeka I, Shen S, Chou TH, Montefiori DC, Lu S. Enhanced immunogenicity of gp120 protein when combined with recombinant DNA priming to generate antibodies that neutralize the JR-FL primary isolate of human immunodeficiency virus type 1. J Virol 2005; 79:7933 - 7; http://dx.doi.org/10.1128/JVI.79.12.7933-7937.2005; PMID: 15919951
- Wang S, Pal R, Mascola JR, Chou T-HW, Mboudjeka I, Shen S, Liu Q, Whitney S, Keen T, Nair BC, et al. Polyvalent HIV-1 Env vaccine formulations delivered by the DNA priming plus protein boosting approach are effective in generating neutralizing antibodies against primary human immunodeficiency virus type 1 isolates from subtypes A, B, C, D and E. Virology 2006; 350:34 - 47; http://dx.doi.org/10.1016/j.virol.2006.02.032; PMID: 16616287
- Taylor JJ, Pape KA, Jenkins MK. A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J Exp Med 2012; 209:597 - 606; http://dx.doi.org/10.1084/jem.20111696; PMID: 22370719
- Hollister K, Kusam S, Wu H, Clegg N, Mondal A, Sawant DV, Dent AL. Insights into the role of Bcl6 in follicular Th cells using a new conditional mutant mouse model. J Immunol 2013; 191:3705 - 11; http://dx.doi.org/10.4049/jimmunol.1300378; PMID: 23980208
- Jacobs SR, Michalek RD, Rathmell JC. IL-7 is essential for homeostatic control of T cell metabolism in vivo. J Immunol 2010; 184:3461 - 9; http://dx.doi.org/10.4049/jimmunol.0902593; PMID: 20194717
- Sawamukai N, Satake A, Schmidt AM, Lamborn IT, Ojha P, Tanaka Y, Kambayashi T. Cell-autonomous role of TGFβ and IL-2 receptors in CD4+ and CD8+ inducible regulatory T-cell generation during GVHD. Blood 2012; 119:5575 - 83; http://dx.doi.org/10.1182/blood-2011-07-367987; PMID: 22496155
- Coban C, Koyama S, Takeshita F, Akira S, Ishii KJ. Molecular and cellular mechanisms of DNA vaccines. Hum Vaccin 2008; 4:453 - 6; http://dx.doi.org/10.4161/hv.4.6.6200; PMID: 18535407
- Catanzaro AT, Roederer M, Koup RA, Bailer RT, Enama ME, Nason MC, Martin JE, Rucker S, Andrews CA, Gomez PL, et al, VRC 007 Study Team. Phase I clinical evaluation of a six-plasmid multiclade HIV-1 DNA candidate vaccine. Vaccine 2007; 25:4085 - 92; http://dx.doi.org/10.1016/j.vaccine.2007.02.050; PMID: 17391815
- Koblin BA, Casapia M, Morgan C, Qin L, Wang ZM, Defawe OD, Baden L, Goepfert P, Tomaras GD, Montefiori DC, et al, NIAID HIV Vaccine Trials Network. Safety and immunogenicity of an HIV adenoviral vector boost after DNA plasmid vaccine prime by route of administration: a randomized clinical trial. PLoS One 2011; 6:e24517; http://dx.doi.org/10.1371/journal.pone.0024517; PMID: 21931737
- Bakari M, Aboud S, Nilsson C, Francis J, Buma D, Moshiro C, Aris EA, Lyamuya EF, Janabi M, Godoy-Ramirez K, et al. Broad and potent immune responses to a low dose intradermal HIV-1 DNA boosted with HIV-1 recombinant MVA among healthy adults in Tanzania. Vaccine 2011; 29:8417 - 28; http://dx.doi.org/10.1016/j.vaccine.2011.08.001; PMID: 21864626
- Koup RA, Roederer M, Lamoreaux L, Fischer J, Novik L, Nason MC, Larkin BD, Enama ME, Ledgerwood JE, Bailer RT, et al, VRC 009 Study Team, VRC 010 Study Team. Priming immunization with DNA augments immunogenicity of recombinant adenoviral vectors for both HIV-1 specific antibody and T-cell responses. PLoS One 2010; 5:e9015; http://dx.doi.org/10.1371/journal.pone.0009015; PMID: 20126394
- Lu S. Human versus HIV: round 2 defeat in AIDS vaccine development. Expert Rev Vaccines 2008; 7:151 - 3; http://dx.doi.org/10.1586/14760584.7.2.151; PMID: 18324884
- Lindqvist M, van Lunzen J, Soghoian DZ, Kuhl BD, Ranasinghe S, Kranias G, Flanders MD, Cutler S, Yudanin N, Muller MI, et al. Expansion of HIV-specific T follicular helper cells in chronic HIV infection. J Clin Invest 2012; 122:3271 - 80; http://dx.doi.org/10.1172/JCI64314; PMID: 22922259
- Perreau M, Savoye A-L, De Crignis E, Corpataux J-M, Cubas R, Haddad EK, De Leval L, Graziosi C, Pantaleo G. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J Exp Med 2013; 210:143 - 56; http://dx.doi.org/10.1084/jem.20121932; PMID: 23254284
- Xu Y, Weatherall C, Bailey M, Alcantara S, De Rose R, Estaquier J, Wilson K, Suzuki K, Corbeil J, Cooper DA, et al. Simian immunodeficiency virus infects follicular helper CD4 T cells in lymphoid tissues during pathogenic infection of pigtail macaques. J Virol 2013; 87:3760 - 73; http://dx.doi.org/10.1128/JVI.02497-12; PMID: 23325697
- Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, Su LF, Cubas R, Davis MM, Sette A, et al, International AIDS Vaccine Initiative Protocol C Principal Investigators. Human circulating PD-⁺1CXCR3⁻CXCR5⁺ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity 2013; 39:758 - 69; http://dx.doi.org/10.1016/j.immuni.2013.08.031; PMID: 24035365
- Choi YS, Yang JA, Yusuf I, Johnston RJ, Greenbaum J, Peters B, Crotty S. Bcl6 expressing follicular helper CD4 T cells are fate committed early and have the capacity to form memory. J Immunol 2013; 190:4014 - 26; http://dx.doi.org/10.4049/jimmunol.1202963; PMID: 23487426
- Ruzankina Y, Pinzon-Guzman C, Asare A, Ong T, Pontano L, Cotsarelis G, Zediak VP, Velez M, Bhandoola A, Brown EJ. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell 2007; 1:113 - 26; http://dx.doi.org/10.1016/j.stem.2007.03.002; PMID: 18371340