
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
This lead candidate selection study compared two anti-(+)-methamphetamine (METH) monoclonal antibodies (mAb) to determine their ability to reduce METH-induced locomotor effects and redistribute METH and (+)-amphetamine (AMP) in a preclinical overdose model. Both mAbs have high affinity for METH, but mAb4G9 has moderate and mAb7F9 has low affinity for AMP. In the placebo-controlled behavioral experiment, the effects of each mAb on the locomotor response to a single 1 mg/kg intravenous (IV) METH dose were determined in rats. The doses of mAb binding sites were administered such that they equaled 1, 0.56, 0.32, and 0.1 times the molar equivalent (mol-eq) of METH in the body 30 min after the METH dose. METH disposition was determined in separate animals that similarly received either a 1 or 0.32 mol-eq dose of mAb binding sites 30 min after a 1 mg/kg METH dose. Total METH-induced distance traveled was significantly reduced in rats that received the highest three doses of each mAb compared with saline. The duration of METH effects was also significantly reduced by mAb7F9 at the highest dose. The disposition of METH was altered dose-dependently by both mAbs as shown in reductions of volume of distribution and total clearance, and increases in elimination half-life. These data indicate that both mAbs are effective at reducing METH-induced behavior and favorably altering METH disposition. Both were therefore suitable for further preclinical testing as potential human medications for treating METH use; however, due to results reported here and in later studies, mAb7F9 was selected for clinical development.
Abbreviations:
- AMP, (+)-amphetamine
- AUC, area under the concentration vs. time curve
- ClT, total body clearance
- Cmax, maximum concentration
- id, inner diameter
- IV, intravenous
- JVC, jugular venous catheter
- KD, dissociation constant
- KI, concentration of inhibitor which prevents 50% of the target ligand from binding
- LMA, locomotor activity
- mAb, monoclonal antibody
- MDMA, (+)-3, 4-methylenedioxy-N-methylamphetamine
- METH, (+)-methamphetamine
- mol-eq, molar equivalent
- SD, standard deviation
- t1/2λz, terminal elimination half-life
- Tmax, time of maximum concentration
- Vd, volume of distribution
Introduction
Anti-drug monoclonal antibodies (mAbs) effectively reverse and prevent many pharmacologic effects of the abused drug methamphetamine (METH) in preclinical studies. These antibody medications reduce stimulant-induced locomotor activity (LMA)Citation1,2 and hemodynamic effectsCitation3 in rats, and antagonize METH effects in pigeon drug discriminationCitation4 and rat self-administration models.Citation5 Furthermore, anti-METH mAbs are effective when given before or after the METH is administered.Citation2
Anti-METH mAbs function via pharmacokinetic antagonism of METH effects. By dramatically altering the disposition of METH via high-affinity binding and redistribution,Citation1,6 they reduce its stimulant effects.Citation1,2 The binding interactions between METH and mAb are, however, complex. Characterizing the factors that determine the efficacy of anti-drug antibodies continues to be a major goal of the development of these molecules as medications. Several of these factors are known, including mAb affinity for its target ligand and the ability to rapidly alter drug pharmacokinetics.Citation2,7,8
An additional important characteristic is the duration of in vivo activity. We have shown that despite having a similar biological half-life to other mAbs (i.e., ∼6–7 d in the ratCitation6), one of our highest affinity anti-METH mAbs only effectively reduces METH effects for a short time period.Citation2 The reasons for this in vivo inactivation have not been completely determined,Citation9 but the ability to reduce the effects of repeated METH doses over an extended time period is important for long-term success of new mAbs.
Through targeted hapten design, we developed two murine anti-METH mAbs that are not inactivated in vivo and have approximately equivalent, long functional half-lives.Citation6,9,10 Furthermore, both mAbs have high affinity for METH and differing affinities for the related compounds (+)-amphetamine (AMP) and (+)-3,4-methylenedioxymethamphetamine (MDMA). MAb4G9 has a KD for METH of 16 nM and a KI for MDMA of 68 nM.Citation11 MAb7F9 has a KD for METH of 8 nM and a KI for MDMA of 8 nM.Citation12 They are both also known to bind AMP and this characteristic was directly compared for this report.
Since both mAb4G9 and mAb7F9 are long acting, have high affinity for METH and also bind a class of related substances, they were chosen for further study as possible candidates for clinical development. The purpose of these studies was to test these candidates for their ability to reduce METH stimulant effects and favorably alter METH kinetics. A secondary goal was to understand the differences that mAb affinity for AMP (a pharmacologically active METH metabolite) may have on both measures. To do this, we measured the changes in METH-induced LMA resulting from administration of these high affinity anti-METH mAbs after a METH dose. In placebo controlled studies of LMA, rats were given METH (1 mg/kg) followed by one of four mAb doses that ranged from 0.1 mol-eq binding sites to the amount of METH in the body at the time of mAb administration (36.2 mg/kg) up to a 1 mol-eq mAb dose (362 mg/kg). In addition, the effects on METH and AMP (as a metabolite of METH) pharmacokinetics of the lowest (0.32 mol-eq) and highest (1 mol-eq) active or effective mAb doses in the LMA studies were quantified in separate groups of rats.
These are the first reported experiments comparing the efficacy of two anti-METH mAbs that also have affinity for AMP in a model of METH overdose. This comparison of murine mAb4G9 and mAb7F9 was part of a series of tests that resulted in the selection of mAb7F9 for conversion to a chimeric form as described in Stevens et al.Citation12 and subsequent clinical development. Importantly, these results were part of the data set reviewed by the US Food and Drug Administration prior to their allowing the initiation of a recently completed Phase 1a human safety study of the human-mouse chimeric form of mAb7F9 called ch-mAb7F9.
Results
Relative affinities for AMP
Previous studies have reported values representing the affinities of mAb4G9 and mAb7F9 for METH, AMP, and MDMA. Their affinities for METH (described by the dissociation constant, KD) are similar, 16 and 8 nM, respectively. The KD for mAb4G9 binding to AMP (50 nM) has been reported,Citation11 however, only a KI for mAb7F9 binding to AMP (370 nM) has been published to date.Citation12 Because these studies aimed to consider the effects of AMP affinity on reduction of stimulant effects and alteration of METH pharmacokinetics, we determined the KD for AMP of both mAb7F9 and mAb4G9 using the same modified radioimmunoassay method as done previously.Citation11 Our results show that mAb4G9 has an approximately 2.5-fold greater affinity for AMP than mAb7F9. The KD values obtained were 110 and 270 nM, respectively. Please note, this value for mAb4G9 differs from the previously published report due to a 2-fold error in the earlier AMP stock preparation. This error resulted from the mistaken assumption that the amphetamine sulfate salt used contained only one AMP molecule when it actually contained two.
In vivo health effects
All rats tolerated the dosing of METH and mAb well. There were no changes in body weights over the course of the study.
Effect of each mAb on METH-induced LMA
When rats received METH followed 30 min later by saline as a control for antibody dosing (study day 7), average horizontal locomotion and rearing behavior increased over time to peak at approximately 20–30 min after mAb dosing (, open circles; graphical representation of the rearing behavior is not shown, but the results were similar to the horizontal locomotion data). Conversely, when the rats received either antibody at the highest three doses, horizontal locomotion and rearing behavior decreased soon after mAb administration (, closed circles). This is in contradistinction to the 0.1 mol-eq mAb dose groups, where the effects were similar to the saline controls.
Figure 1. The total distance traveled after METH administration (open circles) and after METH plus mAb (filled circles) for mAb4G9 (left panels) and mAb7F9 (right panels). There were 6 rats per group. The values represent the central moving averages (± SD) for each time point. Thus, for the y value at time t = 42, for example, the average of the y values at 40, 42 and 44 were used. Percent reductions in total distance traveled between saline and mAb treatment days are indicated. Gray shading indicates time between METH administration (time = −30) and saline or mAb treatment (time = 0). The results for total rearing events were similar.
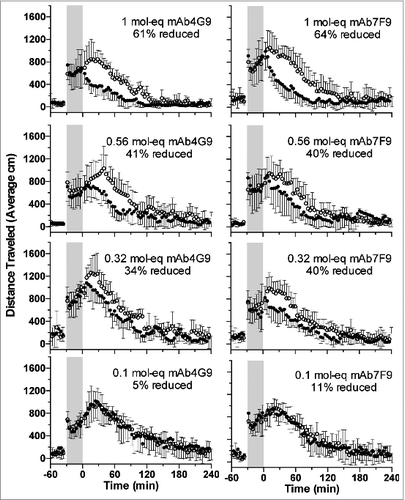
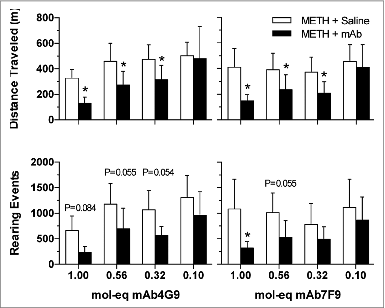
The rapid reduction in METH-stimulant effects was reflected in the time at which the maximum behavioral effects were measured after mAb administration. For sessions during which both mAb were administered at the 1 mol-eq dose, the peak horizontal locomotor value occurred significantly earlier than during the saline control session (). In addition, the highest measured value was significantly reduced by the highest two mAb doses for distance traveled for mAb4G9, and for rearing events at the 1 mol-eq dose of both mAbs ().
Table 1. Time of peak activity
Table 2. Highest measured value
The duration of METH effect on horizontal locomotion was significantly reduced by mAb7F9 at the highest dose and was also near significance at the 0.56 mol-eq dose (). None of the mAb4G9 results reached significance for duration of activity. At each dose, there were no differences between the antibodies for duration of METH effects, or total activity. The total distance traveled was significantly reduced compared with placebo control values for both mAb at the three highest doses (). Total rearing events were statistically less than placebo for the 1 mol-eq mAb7F9 dose and very near significance at the 0.56 mol-eq dose.
Figure 2. The average duration (± SD) of stimulant effects as reflected by the duration of increased LMA above baseline for METH followed by mAb4G9 (left panels) or mAb7F9 (right panels). The duration of METH action was calculated for each parameter starting at the time of mAb or saline (mAb placebo) administration until the LMA had returned to baseline levels. *P < 0.05, compared with saline for that mAb dose.
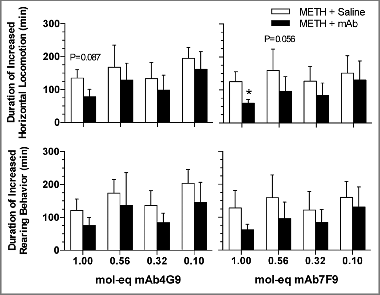
Figure 3. The average total distance traveled and number of rearing events (± SD) from the time of mAb or saline (mAb placebo) administration until activity returned to baseline for METH followed by mAb4G9 (left panels) or mAb7F9 (right panels). *P < 0.05, compared with saline for that mAb dose.
Effect of each mAb on METH pharmacokinetics
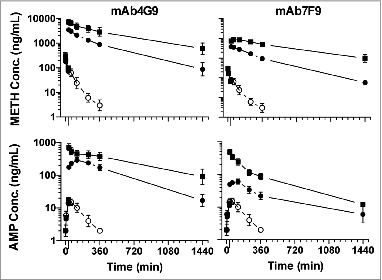
The two antibodies had different effects on METH pharmacokinetics that revealed dose- and antibody-dependent effects. Compared with vehicle treatment, both mAbs increased serum METH concentrations (, top panels) with higher concentrations resulting from mAb7F9 treatment than mAb4G9. MAb7F9 increased METH area under the concentration vs. time curve from 34–360 min () significantly more than mAb4G9 at the 1 mol-eq dose, but not at the 0.32 mol-eq dose, at which the
were similar (). Both average
were greater than saline by more than 100-fold at the 0.32 mol-eq dose. At the 1 mol-eq dose, the average
for mAb4G9 and mAb7F9 was over 300- and about 500-fold higher than placebo, respectively.
Table 3. Pharmacokinetic parameters of METH after vehicle, mAb4G9 or mAb7F9 administration
Figure 4. Average (± SD) METH and AMP serum concentrations over time after METH administration followed by vehicle (n = 3, open circles), mAb4G9 (n = 4–5, left panels) or mAb7F9 (n = 4, right panels) at 1 (squares) or 0.32 (closed circles) mol-eq doses. METH concentrations are in the upper panels, and AMP concentrations are in the lower panels. METH was given at time t = 0 and mAb or vehicle was given at time t = 30 (long tick mark on the x-axis).
The sequestration of METH in the serum was reflected by reductions in Vd and even greater reductions in ClT that were significant compared with placebo treatment and antibody dose-dependent (). This resulted in an overall increase in METH elimination half-life. While not statistically different, the half-life of METH in the presence of mAb7F9 was shorter than with mAb4G9 at both doses tested. Volume of distribution was reduced by 30- to 40-fold for both antibodies at the 0.32 mol-eq dose and by 60- or 100-fold for mAb4G9 and mAb7F9 at the 1 mol-eq dose, respectively. At the 0.32 mol-eq dose, clearance was similarly reduced for both mAbs, at approximately 100-fold, but mAb4G9 reduced clearance only 300-fold while mAb7F9 reduced total clearance by 560-fold at the 1 mol-eq dose. The percent of the METH dose eliminated in the urine during the first 24 h after dosing was decreased significantly by both mAb at the 1 mol-eq dose compared with the vehicle treated rats (). There was an unexpected increase in the METH urine clearance following the 0.32 mAb7F9 dose.
Effect of each mAb on AMP pharmacokinetics
AMP concentrations were slightly different as a function of mAb administration, but dramatically different compared with placebo administration. Opposite to the METH results, mAb4G9 raised the serum AMP concentration more than mAb7F9 at both dose levels (). MAb4G9 increased AMP significantly more than mAb7F9 at both doses, however even mAb7F9 at the 1 mol-eq dose increased the
significantly compared with vehicle control ().
Table 4. Pharmacokinetic parameters of AMP (as a metabolite) after METH administration followed by vehicle, mAb4G9 or mAb7F9 administration
These increases in were reflected by increases in the elimination half-life of AMP. All mAb treatments raised the half-life by at least 3-fold and it went up by 5-fold after the 1 mol-eq dose of mAb4G9. It is surprising that the AMP half-life was longer following the 0.32 mol-eq dose of mAbF79 compared with the 1 mol-eq dose, but the highly variable value of the last point (24 h) likely had too much influence on this parameter.
As expected based on the relative affinities of the mAbs for AMP, the Cmax was highest following a 1 mol-eq dose of mAb4G9 and was significantly different from vehicle after all mAb doses except the low mAb7F9 dose (). Following a 1 mol-eq dose, this peak (Tmax) typically occurred at the first measured time point after antibody administration and was not different from vehicle, however, after a 0.32 mol-eq dose of either mAb, the time to peak was extended 2- to 3-fold.
Effect of each mAb on METH and AMP brain concentrations
Brain METH and AMP concentrations at 1440 min (24 h) after vehicle dosing were not measureable, as expected given the short METH and AMP half-life in rats. With mAb dosing, METH brain concentrations were low but detectable (). The amount in the brain preparation was about 45-fold less than that in the serum at the same time point for both mAbs at the 1 mol-eq dose. At the 0.32 mol-eq dose for both mAbs, brain concentrations were about 20-fold less than serum concentrations. While these ratios are not absolute comparisons, they do indicate that both antibodies function similarly and dose-dependently to keep METH out of the brain. AMP brain concentrations were not as similar between the mAbs (). After mAb4G9 dosing, AMP concentrations in the brain preparations were 13- and 7-fold less than serum concentrations at the 1 and 0.32 mol-eq doses, respectively. Following mAb7F9 dosing, AMP brain concentrations were 2- and 3-fold less than serum concentrations at the 1 and 0.32 mol-eq doses, respectively.
Discussion
The purpose of this study was to characterize two anti-METH mAbs to aid in selection of a mAb for clinical development as a treatment for METH use. While the two potential therapeutic indications, treatment of METH overdose and reduction of relapse to METH use, may require different mAbs with contrasting characteristics, therapeutic considerations for both indications are similar. The results from each mAb were therefore compared with both indications as possibilities.
Reduction of stimulant effects
In this model of METH effects, the two mAbs had similar effects on METH-induced hyperactivity. While horizontal locomotion and rearing behavior are only two of the many behavioral effects that can be studied, the effects on the reduction of METH-induced hyperactivity suggest that both mAb have potential for the treatment of METH effects in humans.
Both mAbs rapidly reduced the acute effects of METH. One of the reasons for giving the mAb at 30 min after the METH dose is because this is the time of peak locomotor effects after a 1 mg/kg IV METH dose.Citation13 Both mAb reduced the stimulatory effects of METH within 4 min after their administration. This is of critical importance because mAbs must work rapidly to reverse an acute overdose in humans. Rapid reversal of hypertension, tachycardia or intense CNS stimulation would be of advantage in the treatment of medically unstable patients. In addition, the rapid action of these mAbs on METH effects suggests that they may be useful in reducing or preventing the positive reinforcement that accompanies METH administration.
Despite the differences in affinity for AMP, both mAbs reduced stimulant-induced hyperactivity similarly and the effects of AMP cannot be discerned from those of METH in this model. No additive behavior changes from mAb4G9 were observed that would suggest it reduced AMP effects in addition to METH effects, but this was not directly tested. The effects of the mAbs may be similar because mAb4G9 reduced AMP effects better than mAb7F9, and mAb7F9 reduced METH effects better than mAb4G9 due to the minor METH KD difference of 8 and 16 nM, respectively. That the results are similar suggests that there was no substantial benefit to mAb4G9 having higher affinity for AMP.
Favorable alterations in METH PK
Even though both mAbs were similarly effective in reducing locomotor effects of METH in rats, there were subtle differences in the effects on METH serum concentrations. The maximal METH serum concentrations and METH in the mAb7F9 1 mol-eq group were significantly higher than in the 1 mol-eq mAb4G9 treated group perhaps because mAb7F9 reduced both estimated Vd and CLT more than mAb4G9. This may have occurred because mAb7F9 has higher affinity for METH or because it binds METH more effectively; alternatively, AMP may have competed for and occupied a significant portion of the mAb4G9 METH binding capacity. Regardless, greater binding by mAb7F9 could redistribute METH more rapidly from the periphery back into the bloodstream, as demonstrated by the reduced Vd, which resulted in higher early METH concentrations. The lower METH clearance from mAb7F9 was apparently compensated for by the more effective restricted METH distribution, which together resulted in a slightly faster elimination rate. The significance of this is not clear, since the measured effects were not different between the two mAbs.
Alterations in AMP PK
There were differences in AMP serum concentrations with the two mAbs that should also be considered. AMP concentrations were much higher in the presence of mAb4G9 (; lower panels), though serum AMP concentrations were still much higher in the mAb7F9 treated animals than the vehicle treated animals.
The difference between the AMP PK results could be that the higher affinity binding of AMP by mAb4G9 raised the AMP concentrations more than mAb7F9 by redistributing and retaining AMP in the blood. A different possible explanation is that mAb7F9 binding to METH prevents formation of AMP. In both instances, the choice of animal model is important. In male rats, 35–40% of a dose of METH is metabolized to AMP.Citation14 If METH binding by either mAb prevents some of the metabolism, large differences in AMP PK could be observed. Regardless of the reasons for the differences in AMP concentrations between the two mAbs, humans do not metabolize METH to AMP as extensively as rats.Citation13,15 Further, since there was no additional reduction in stimulant effects by mAb4G9, the differences in AMP binding are likely of limited clinical importance.
Reduction of METH in the brain
Brain METH and AMP concentrations were measureable at the 24 h time point after treatment with both mAbs, whereas they were not measurable in the vehicle treated animals. Previous studies and general pharmacokinetic principles suggest that brain concentrations are always greater than serum concentrations in the absence of mAb (∼8:1) while the reverse is true after mAb treatment (e.g., see ref. 16). The ability of the mAb to redistribute and retain METH in the serum provides enough of a pool of free METH that during the normal on and off process of mAb binding, a small amount of the METH is allowed to distribute into the brain. However, the concentrations of METH that renter the brain after mAb dosing are negligible and the rate at which the METH enters the brain at this point after METH administration is so slow that effects are not measurable.
Feasibility of doses
With both mAbs, doses that resulted in fewer binding sites than the number of METH molecules in the body still reduced METH effects. The mAbs reduced METH effects even at doses that were 1/3rd mol-eq to the amount of METH in the rat at the time of mAb dosing. Similarly, we have previously shown that an anti-PCP mAb effectively reduces brain PCP concentrations and locomotor effects even when the body burden of PCP is many times greater than the number of mAb binding sites,Citation17,18 and analogous data have been shown for anti-nicotine mAbs, e.g., ref. 19. Though the highest mAb dose tested in these current studies (362 mg/kg) was the most effective, the intermediate doses (202 and 115 mg/kg) significantly altered METH effects compared with saline treatment. These doses are substantially larger on a per kg basis than those of other antibodies approved for human use. For instance, mAb doses in the range of 10–40 mg/kg are used for the treatment of cancer and infectious diseases in humans.Citation20,21
The projected effective human antibody doses for our mAbs will be smaller and in line with those for approved mAbs. This is in part because of the differences in cerebral blood flow in man compared with rats leading to a larger amount of mAb reaching the human brain, resulting in a more effective reduction in METH CNS effects. Additionally, we have calculated that the amount of mAb in serum required to bind all of a 30 mg IV dose of METH in humans, with a resulting METH serum concentration of ∼110 ng/mL,Citation22 is only 50 μg/mL. Assuming normal human IgG PK parameters, this would suggest that doses of 10 to 20 mg/kg in humans could result in effective serum concentrations with dosing once per half-life.
Potential for scale-up
One of the final considerations for choosing a mAb for clinical development is its laboratory performance. Drug product intended for human use is subjected to many tests to indicate identity, purity, potency and other defining characteristics. All of these are repeated to show stability of the medication over time. As these studies were conducted, it was noted that mAb7F9 performed better than mAb4G9. For instance, solutions of mAb4G9 form visible precipitates upon buffer exchange and at times during concentrating steps. Since the intention was to choose a murine mAb, then replace its constant domains with those from a human IgG to form a chimeric mAb, the original isotype of the murine antibody was not a selection consideration, but may have contributed to laboratory performance problems for mAb4G9. Although changing the isotype might solve this issue, it is not a guarantee of success. Scale-up potential was a serious consideration when selecting a clinical candidate.
Conclusions
In summary, both new anti-METH mAbs were effective in reducing the effects of an acute IV dose of METH in a rat model and causing favorable changes in METH pharmacokinetics. These and many other factors should be considered when making a decision about what medications to take forward to human studies. Because of their similarities in efficacy, neither mAb should be ruled out based only on these studies though mAb7F9 has a slight edge based on better METH PK and laboratory handling ease. Further studies of the effects of these mAbs in other efficacy models such as self-administration and multiple METH challenges have also been conducted (unpublished studies) to give a greater understanding of their effects in chronic users, and mAb7F9 was eventually chosen as the candidate for clinical development.
Materials and Methods
Drugs and reagents
METH hydrochloride as powder, AMP sulfate as powder, and liquid tritiated AMP were obtained from the Research Triangle Institute through the National Institute on Drug Abuse. All other reagents were purchased from Sigma Aldrich unless specified otherwise.
Anti-METH mAb production and purification
Synthesis of METH-like haptens, hapten conjugation to carrier protein and immunization of mice, and production of hybridoma cell lines are described in previous studies.Citation1,10 Large scale production of anti-METH mAb, purification and formulation are also described elsewhere.Citation10 The mAb4G9 is an IgG2b, κ light chain mAb and the mAb7F9 is an IgG1, κ light chain.Citation11
AMP binding study
To determine the dissociation constants (KD) for AMP, both antibodies were tested as in Carroll et al.Citation11 using a modified radioimmunoassay in which unlabeled AMP competes with [3H]-AMP for binding to the mAb.
Animals
Adult male Sprague Dawley rats with dual indwelling jugular vein catheters (medical-grade tubing polyurethane tubing; 0.025 in id × 0.040 in od) were obtained from Charles River Laboratories. On the day before the studies began, the catheters were flushed with 0.2 mL sterile saline followed by 0.05 mL of sterile glycerol containing 25 units of heparin. Catheters were maintained by flushing with the glycerol/heparin solution every 3–4 d. The rats were housed separately and fed 3 food pellets (∼20 g) each day, which resulted in weights between 235–270 g throughout the experiment. Body weight was monitored throughout the course of the studies. All animal experiments were conducted with the approval of the Institutional Animal Care and Use Committee of the University of Arkansas for Medical Sciences and were in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health.
Locomotor Activity Studies
After arrival from the vendor, the rats were allowed to rest for 3 d in their home cages. Three days before each experiment began, the rats were acclimated for 6 h to the chambers in which the behavioral experiments were conducted. On experimental days, rats were placed in the open-top polyethylene experimental chambers (60 × 45 × 40 cm) and behavior was recorded by a video camera located above the chambers. Analysis of the behavior videotapes was performed with Ethovision software (Noldus Information Technology, Inc.) as previously described.Citation3,23 The parameters that were measured included horizontal locomotion in the form of distance traveled, and the number of rearing events.
In these experiments, rats underwent 4 study sessions, and the sessions were separated by 3 d. On study day 1, all groups received saline to obtain baseline (non-drug-induced) behavior. On study day 4, all rats received a 1 mg/kg METH dose (calculated as the free base form). On study day 7, all rats received a 1 mg/kg METH dose followed 30 min later by saline, equal to 1 mL/kg as a control for antibody administration. Because of the behavioral sensitization known to occur after repeated amphetamine treatments,Citation24 the second METH dose (study day 7) was used to determine the METH-induced LMA baseline. On Day 10, METH (1 mg/kg) was administered and followed 30 min later by an IV injection of anti-METH mAb4G9 or mAb7F9.
Four doses of mAb were tested. These were chosen so that the ratio of mAb binding sites (2 per mAb molecule) to METH molecules in the body at 30 min after the METH dose was 1, 0.56, 0.32, and 0.1. These doses are stated with units of binding site:METH molecule molar equivalents (mol-eq). The amount of METH remaining was determined using pharmacokinetic data from Rivière et al.Citation13 and by the equation: body burden = dose * e−kt.Citation25 The 30 min time point was chosen for treatment because previous behavioral and pharmacokinetic data from our laboratory have shown that METH-induced LMA and active metabolite concentrations (AMP) in tissues are near maximum at 30 min in male Sprague Dawley rats.Citation13,16
All control buffer and mAb solutions were administered via a jugular venous catheter in a 3 mL volume at a rate of 2 mL/min. The rats were randomly assigned to their antibody and dose groups (n = 6/group). The antibody was always given at the end of the study (day 10) because the long elimination half-life of murine mAb in rats (at least 6 dCitation6) makes it possible that a single antibody dose could alter METH effects for as much as a one month (4 mAb elimination half-lives).
Behavioral data (i.e., distance traveled and number of rearing events) were recorded in consecutive 2 min bins. Because of the variability of the rat responses across 2 min bins, central moving averages for each time point were computed using the bin value before and after each time point as a generic smoothing operation. For example, the reported y value at time t = 42 min was the average of the y values from the bins at 40, 42, and 44 min.
The total METH-induced activity (total distance traveled and number of rearing events) and the duration of METH action were calculated starting at the time of mAb or mAb placebo (saline) administration until the LMA had returned to baseline levels. LMA was considered to have returned to baseline when the values from 4 consecutive 2 min intervals were less than or equal to the mean + 1 standard deviation (SD) of the 30 min behavioral baseline data observed just prior to drug administration on each study day. The value of the peak effect and the time at which this occurred were recorded from the individual effect vs. time curves.
Pharmacokinetic studies
The highest (1 mol-eq) and lowest (0.32 mol-eq) effective doses of both mAb were tested. Adult male Sprague Dawley rats (Charles River Laboratories) with dual jugular venous catheters (JVC) were allowed to rest for 3 d after arrival. The rats were then acclimated to metabolic cages where the pharmacokinetic studies were conducted. Following acclimation, rats received METH (1 mg/kg) and were then placed in the metabolic cages for 6 h. Three days later, rats received 1 mg/kg METH via the left JVC. Blood samples (100 μL) were collected via the right JVC at 1, 5, and 30 min after METH. Immediately after the 30 min time point, either mAb4G9 (n = 4–5/group), mAb7F9 (n = 4/group), or saline (n = 3; 3 mL) was administered via the left JVC. Blood samples were then collected at 34 min, 1, 2, 4, and 6 h after METH. At the 24 h post-METH time point, rats were anesthetized with halothane, sacrificed by decapitation, and blood and brains were collected. The brains were immediately frozen in liquid nitrogen. Blood samples were allowed to clot and serum was collected after centrifugation. Urine samples were also collected over the 24 h period post-METH. All samples were stored at −80°C until analysis.
Determination of METH and AMP concentrations in serum, brain, and urine
A solid phase extraction method combined with ultra performance liquid chromatography (UPLC) separation and tandem mass spectrometer detection was used for the determination of METH and AMP concentrations. The method utilizes solid phase extraction via Oasis® MCX sorbent technology (Waters Corp) which has a cation exchange capacity of 1 meq/gram and is designed to extract basic compounds from various matrices. Validation consisted of extracting triplicates of calibration and quality control standards (METH, AMP, 4-OH METH, and 4-OH AMP concentrations ranging from 0.3–1000 ng/mL), along with serum and brain samples from previous experiments with known METH and AMP concentrations, with 400 μL 12% phosphoric acid (85%; HPLC grade). Each sample and standard contained internal standard (10 ng/mL METH-d5 and AMP-d11). Samples and standards were subsequently vortex-mixed and spun by centrifugation at 14 000 rpm for 5 min. The supernatant was loaded onto each well of Oasis® MCX μElution 96-well plates (30 μm; Waters Corp) on a vacuum manifold with a flow rate of 1–3 mL/min. The plates were pre-conditioned with 200 μL methanol and pre-equilibrated with 200 μL Milli-Q® water. The analytes were then washed with 100 μL 5% methanol in 0.1 M HCl followed by 100 μL methanol, and eluted with two 60 μL aliquots of 5% NH4OH/ACN. Once validated, serum standards and serum, brain and urine samples were extracted and injected (5 μL) onto an ACQUITY UPLC® system (Waters Corp) interfaced with a Waters Quattro Premier XE mass spectrometer (the HPLC-MS system). They were then chromatographically separated with an ACQUITY UPLC® BEH C18 column (1.7 μm particle size, 2.1 × 100 mm id; Waters Corp) maintained at 45°C. The analytes were eluted off the column with a binary gradient of 0.5 mL/min and a total run time of 4.5 min. The gradient elution profile was as follows: 0–2 min, 99% Mobile Phase A and 1% Mobile Phase B; 2–4 min, 100% Mobile Phase A, and 4–4.5 min, 99% Mobile Phase A and 1% Mobile Phase B. Mobile Phase A was made up of 10 mM NH4OHAc (pH 3.7), 2% methanol, and 5% ACN, whereas Mobile Phase B was made up of 10 mM NH4OHAc (pH 3.7), 4% methanol, and 95% ACN.
For mass spectrometric analysis, the conditions were optimized for precursor ion signal using the following parameters: capillary voltage, 2.5 kV; cone voltage, 18 V; source block temp, 150°C; desolvation gas (nitrogen) delivered at 600 L/hr gas flow rate at 500°C. Argon gas pressure was used to dissociate the precursor ions (METH, AMP) and the collision energy was optimized to enhance the signal of the product ions used in quantification. The ion transitions were m/z 150.1 → 90.8 for METH, 136 → 90.8 for AMP, 155 → 92 for METH-d5, and 147 → 98 for AMP-d11. A standard curve was formulated using the calibration standards and the lower and upper limits of quantification were found to be 0.3 and 1000 ng/mL respectively. The predicted concentrations from the standards and samples from previous studies were reproducible (both within-day and between-day precision and accuracy) and statistically similar to their expected concentrations (within ± 20%).
Pharmacokinetic analysis
Pharmacokinetic parameters were derived from the log concentration vs. time graph of METH and AMP (a metabolite) in the terminal-elimination phase by a model-independent method with a first-order input function and no weighting using WinNonlin (Pharsight Corporation). The best-fit line was determined visually. Due to insufficient sampling of the later portions of the terminal elimination phase in the mAb treated rats, all pharmacokinetic parameters other than should be considered estimates and used with caution.
Estimated pharmacokinetic parameters were determined using the following equations: t1/2λz = 0.693 / λz; Vd = ClT / λz; and ClT = Dose / where t1/2λz is the terminal elimination half-life, λz is terminal elimination rate-constant, ClT is total clearance, Vd is the volume of distribution, and
is area under the serum concentration vs. time curve from time zero to infinity. Percent METH in urine for each rat was the total cumulative amount of METH in the urine divided by the total administered dose. Maximum serum concentrations (Cmax) and time of maximum serum concentrations (Tmax) were determined by observed values. Harmonic mean and “pseudo” standard deviation of estimated t1/2λz values were calculated as described by Lam et al.Citation26
The was calculated by the trapezoidal rule. One rat in the 1 mol-eq mAb4G9 treated group was missing a serum concentration measurement at 34 min and another was missing a measurement at 360 min. These values were estimated using the equation for each rat's estimated t1/2λz.
Statistical analysis
Pharmacokinetic data were analyzed using a Two-Way ANOVA. For the behavior data analysis, a repeated measures factorial arrangement of treatments analysis of variance was fit to the data. Significance was declared at 5%. For both analyses, pre-planned contrasts of interest among treatment combinations were adjusted for multiplicity using the Bonferroni multiple comparison post hoc test.
Disclosure of Potential Conflicts of Interest
S.M.O. and W.B.G. are full time faculty members at the University of Arkansas for Medical Sciences. They have financial interests in and serve as Chief Scientific Officer (S.M.O.) and Chief Medical Officer (W.B.G.) for InterveXion Therapeutics, LLC (Little Rock, AR), a pharmaceutical biotechnology company focused on treating human drug addiction with antibody-based therapy.
Acknowledgments
The authors would like to thank Rachel Tawney for technical assistance performing the in vitro affinity comparisons and Sherri Wood for her expert assistance with the animal studies. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Funding
This work was supported by grants from the National Institute on Drug Abuse (R01DA011560, P01DA014361, and T32DA022981) and the Translational Research Institute (grant 660 UL1TR000039) through the National Center for Research Resources and National Center for Advancing Translational Sciences of the National Institutes of Health.
References
- Byrnes-Blake KA, Laurenzana EM, Carroll FI, Abraham P, Gentry WB, Landes RD, Owens SM. Pharmacodynamic mechanisms of monoclonal antibody-based antagonism of (+)-methamphetamine in rats. Eur J Pharmacol 2003; 461:119-28; PMID:12586207; http://dx.doi.org/10.1016/S0014-2999(03)01313-X
- Byrnes-Blake KA, Laurenzana EM, Landes RD, Gentry WB, Owens SM. Monoclonal IgG affinity and treatment time alters antagonism of (+)-methamphetamine effects in rats. Eur J Pharmacol 2005; 521:86-94; PMID:16182279; http://dx.doi.org/10.1016/j.ejphar.2005.08.016
- Gentry WB, Laurenzana EM, Williams DK, West JR, Berg RJ, Terlea T, Owens SM. Safety and efficiency of an anti-(+)-methamphetamine monoclonal antibody in the protection against cardiovascular and central nervous system effects of (+)-methamphetamine in rats. Int Immunopharmacol 2006; 6:968-77; PMID:16644483; http://dx.doi.org/10.1016/j.intimp.2006.01.008
- McMillan DE, Hardwick WC, Li M, Owens SM. Pharmacokinetic antagonism of (+)-methamphetamine discrimination by a low-affinity monoclonal anti-methamphetamine antibody. Behav Pharmacol 2002; 13:465-73; PMID:12394422; http://dx.doi.org/10.1097/00008877-200209000-00019
- McMillan DE, Hardwick WC, Li M, Gunnell MG, Carroll FI, Abraham P, Owens SM. Effects of murine-derived anti-methamphetamine monoclonal antibodies on (+)-methamphetamine self-administration in the rat. J Pharmacol Exp Ther 2004; 309:1248-55; PMID:14993256; http://dx.doi.org/10.1124/jpet.103.061762
- Laurenzana EM, Hendrickson HP, Carpenter D, Peterson EC, Gentry WB, West M, Che Y, Carroll FI, Owens SM. Functional and biological determinants affecting the duration of action and efficacy of anti-(+)-methamphetamine monoclonal antibodies in rats. Vaccine 2009; 27:7011-20; PMID:19800446; http://dx.doi.org/10.1016/j.vaccine.2009.09.072
- Pentel PR, Dufek MB, Roiko SA, Lesage MG, Keyler DE. Differential effects of passive immunization with nicotine-specific antibodies on the acute and chronic distribution of nicotine to brain in rats. J Pharmacol Exp Ther 2006; 317:660-6; PMID:16407464; http://dx.doi.org/10.1124/jpet.105.097873
- Gentry WB, Rüedi-Bettschen D, Owens SM. Anti-(+)-methamphetamine monoclonal antibody antagonists designed to prevent the progression of human diseases of addiction. Clin Pharmacol Ther 2010; 88:390-3; PMID:20668443; http://dx.doi.org/10.1038/clpt.2010.155
- Owens SM, Atchley WT, Hambuchen MD, Peterson EC, Gentry WB. Monoclonal antibodies as pharmacokinetic antagonists for the treatment of (+)-methamphetamine addiction. CNS Neurol Disord Drug Targets 2011; 10:892-8; PMID:22229314; http://dx.doi.org/10.2174/187152711799219370
- Peterson EC, Gunnell M, Che Y, Goforth RL, Carroll FI, Henry R, Liu H, Owens SM. Using hapten design to discover therapeutic monoclonal antibodies for treating methamphetamine abuse. J Pharmacol Exp Ther 2007; 322:30-9; PMID:17452421; http://dx.doi.org/10.1124/jpet.106.117150
- Carroll FI, Abraham P, Gong PK, Pidaparthi RR, Blough BE, Che Y, Hampton A, Gunnell M, Lay JO Jr., Peterson EC, et al. The synthesis of haptens and their use for the development of monoclonal antibodies for treating methamphetamine abuse. J Med Chem 2009; 52:7301-9; PMID:19877685; http://dx.doi.org/10.1021/jm901134w
- Stevens MW, Tawney RL, West CM, Kight AD, Henry RL, Owens SM, Gentry WB. Preclinical characterization of an anti-methamphetamine monoclonal antibody for human use. MAbs 2014; 6:547-55; PMID:24492290; http://dx.doi.org/10.4161/mabs.27620
- Rivière GJ, Byrnes KA, Gentry WB, Owens SM. Spontaneous locomotor activity and pharmacokinetics of intravenous methamphetamine and its metabolite amphetamine in the rat. J Pharmacol Exp Ther 1999; 291:1220-6; PMID:10565845
- Milesi-Hallé A, Hendrickson HP, Laurenzana EM, Gentry WB, Owens SM. Sex- and dose-dependency in the pharmacokinetics and pharmacodynamics of (+)-methamphetamine and its metabolite (+)-amphetamine in rats. Toxicol Appl Pharmacol 2005; 209:203-13; PMID:15916788; http://dx.doi.org/10.1016/j.taap.2005.04.007
- Cook CE, Jeffcoat AR, Hill JM, Pugh DE, Patetta PK, Sadler BM, White WR, Perez-Reyes M. Pharmacokinetics of methamphetamine self-administered to human subjects by smoking S-(+)-methamphetamine hydrochloride. Drug Metab Dispos 1993; 21:717-23; PMID:8104133
- Rivière GJ, Gentry WB, Owens SM. Disposition of methamphetamine and its metabolite amphetamine in brain and other tissues in rats after intravenous administration. J Pharmacol Exp Ther 2000; 292:1042-7; PMID:10688621
- Laurenzana EM, Gunnell MG, Gentry WB, Owens SM. Treatment of adverse effects of excessive phencyclidine exposure in rats with a minimal dose of monoclonal antibody. J Pharmacol Exp Ther 2003; 306:1092-8; PMID:12829731; http://dx.doi.org/10.1124/jpet.103.053140
- Pitas G, Laurenzana EM, Williams DK, Owens SM, Gentry WB. Anti-phencyclidine monoclonal antibody binding capacity is not the only determinant of effectiveness, disproving the concept that antibody capacity is easily surmounted. Drug Metab Dispos 2006; 34:906-12; PMID:16507651
- Carrera MRA, Ashley JA, Hoffman TZ, Isomura S, Wirsching P, Koob GF, Janda KD. Investigations using immunization to attenuate the psychoactive effects of nicotine. Bioorg Med Chem 2004; 12:563-70; PMID:14738965; http://dx.doi.org/10.1016/j.bmc.2003.11.029
- Grillo-López AJ, White CA, Dallaire BK, Varns CL, Shen CD, Wei A, Leonard JE, McClure A, Weaver R, Cairelli S, et al. Rituximab: the first monoclonal antibody approved for the treatment of lymphoma. Curr Pharm Biotechnol 2000; 1:1-9; PMID:11467356; http://dx.doi.org/10.2174/1389201003379059
- Migone T-S, Subramanian GM, Zhong J, Healey LM, Corey A, Devalaraja M, Lo L, Ullrich S, Zimmerman J, Chen A, et al. Raxibacumab for the treatment of inhalational anthrax. N Engl J Med 2009; 361:135-44; PMID:19587338; http://dx.doi.org/10.1056/NEJMoa0810603
- Cruickshank CC, Dyer KR. A review of the clinical pharmacology of methamphetamine. Addiction 2009; 104:1085-99; PMID:19426289; http://dx.doi.org/10.1111/j.1360-0443.2009.02564.x
- Hardin JSJ, Wessinger WDW, Wenger GRG, Proksch JWJ, Laurenzana EME, Owens SMS. A single dose of monoclonal anti-phencyclidine IgG offers long-term reductions in phencyclidine behavioral effects in rats. J Pharmacol Exp Ther 2002; 302:119-26; PMID:12065708; http://dx.doi.org/10.1124/jpet.302.1.119
- Robinson TE, Becker JB. Enduring changes in brain and behavior produced by chronic amphetamine administration: a review and evaluation of animal models of amphetamine psychosis. Brain Res 1986; 396:157-98; PMID:3527341; http://dx.doi.org/10.1016/0165-0173(86)90002-0
- Rowland M, Tozer TN. Clinical pharmacokinetics: concepts and applications. Baltimore: Williams & Wilkins; 1995.
- Lam FC, Hung CT, Perrier DG. Estimation of variance for harmonic mean half-lives. J Pharm Sci 1985; 74:229-31; PMID:3989700; http://dx.doi.org/10.1002/jps.2600740229