
Abstract
Patients (n = 34) with previously untreated, slowly progressive asymptomatic stage I/II multiple myeloma or with stage II/III multiple myeloma in stable response/plateau phase following conventional anti-tumor therapy were immunized repeatedly with the antigen-specific cancer immunotherapeutic agent tecemotide (L-BLP25). Additionally, patients were randomly allocated to either single or multiple low doses of cyclophosphamide to inhibit regulatory T cells (Treg). Immunization with tecemotide resulted in the induction/augmentation of a mucin 1-specific immune response in 47% of patients. The immune responses appeared to involve a Th1-like cellular immune response involving CD4 and CD8 T cells. The rate of immune responses was similar with single versus multiple dosing of cyclophosphamide and in patients with vs. without pre-existing mucin 1 immunity. On-treatment reductions in the slope of M-protein concentration over time (but not fulfilling clinical criteria for responses with conventional anti-tumor agents) were observed in 45% of evaluable patients, predominantly in those without versus with pre-existing mucin 1 immunity and in patients with early stage disease. No differences were seen in patients receiving single or multiple cyclophosphamide dosing. Treatment with tecemotide was generally well tolerated. Repeated vs. single dosing of cyclophosphamide had no impact on Treg numbers and was stopped after a case of fatal encephalitis that was assessed as possibly study-related. Tecemotide immunotherapy induces mucin 1-specific cellular immune responses in a substantial proportion of patients, with preliminary evidence of changes in the M-protein concentration time curve in a subset of patients.
Abbreviations
| ASCI | = | antigen-specific cancer immunotherapy |
| AUC | = | area under the curve |
| Cy | = | cyclophosphamide |
| ELISpot | = | enzyme-linked immunosorbent spot |
| GM-CSF | = | granulocyte-macrophage colony-stimulating factor |
| HR | = | hazard ratio |
| IDA | = | Immunologic Diagnostic Analysis |
| IFN-g | = | interferon-g |
| IL-17 | = | interleukin-17 |
| IQR | = | interquartile range |
| MM | = | multiple myeloma |
| MUC1 | = | mucin 1 |
| NSCLC | = | non-small cell lung cancer |
| PBMC | = | peripheral blood mononuclear cell |
| TNF-α | = | tumor necrosis factor-α |
| Treg | = | regulatory T cell |
| URR | = | upper reference range |
Introduction
Despite improvements in recent decades, management of multiple myeloma (MM) remains suboptimal. There is no established therapy for asymptomatic MM and the standard of care for most patients following an initial response to primary therapy is ‘watchful waiting’.Citation1 Studies of conventional chemotherapy as maintenance therapy proved disappointing.Citation2,3 Newer agents including thalidomide,Citation4 lenalidomideCitation5,6 or bortezomibCitation7 can improve progression-free – and sometimes overall – survival but can also be associated with toxicity, and are not approved in most countries for maintenance therapy.Citation1
A potential new strategy in the early disease phase is the use of immunotherapy to direct an immune response against malignant cells. Tecemotide is a liposomal antigen-specific cancer immunotherapeutic agent targeting mucin 1 (MUC1). It incorporates a synthetic, 25 amino acid, non-glycosylated MUC1 lipopeptide (BLP25) and monophosphoryl lipid A immunoadjuvant in a liposomal (L) delivery system. Results from a global, randomized, placebo-controlled trial of tecemotide in stage III non-small cell lung cancer (NSCLC) have been reported recently.Citation8 While prolongation of overall survival in the primary analysis population failed to achieve statistical significance, there was a trend toward prolonged survival and time-to-tumor progression with tecemotide. Also, tecemotide maintenance therapy resulted in a notable improvement in survival in the predefined subgroup of patients who had previously received concurrent chemoradiotherapy, but not when administered after sequential chemoradiotherapy.
The potential utility of tecemotide in treating MM is supported by studies reporting MUC1 expression on myeloma cells,Citation9,10 recognition of MUC1-positive MM cell lines by cytotoxic T lymphocytes from MM patientsCitation11 and elevated levels of MUC1-specific CD8+ T-cells in peripheral blood and bone marrow from patients with MM.Citation12 MUC1 normally carries extensive O-linked glycans, but tumor-associated MUC1 is frequently hypoglycosylated. Furthermore, antigen processing by dendritic cells is more efficient when MUC1 is less extensively glycosylated, leading to stronger T cell responses.Citation13-15 Therefore, it seems reasonable to anticipate that immunization with non-glycosylated MUC1 peptides may induce a specific immune response to tumor-associated MUC1.
The primary objective of this exploratory study was to investigate the MUC1-specific immune response to tecemotide in patients with previously untreated, asymptomatic stage I/II MM or with stage II/III disease in stable response/plateau phase after primary anti-tumor therapy. Secondary objectives were to clarify the nature of the immune response and gain a preliminary assessment of the safety and clinical efficacy of tecemotide combined with single or repeated administration of low-dose cyclophosphamide (Cy).
Multiple myeloma is associated with alterations in immune status, including increased regulatory T (Treg) cells that could suppress anti-tumor immune responses.Citation16 Low-dose Cy might reduce Treg numbersCitation17 and was administered as a single dose before the first immunization in all clinical trials with tecemotide.Citation8,18,19 Identical single dosing of Cy in a recent phase II study in renal cell carcinoma of immunization with multiple tumor-associated peptides led to a 20% reduction in Tregs 3 d after Cy administration that persisted for at least 3 weeks. Treg levels were not reduced in the absence of Cy.Citation20 Animal studies suggest that a single administration of Cy induces a durable Treg depletion that may persist for only a few weeksCitation21 and repeated administration of low-doses of Cy improved survival versus single dosing in a mouse model of MM.Citation22 Therefore, in this study we explored the effects of single or repeated low doses of Cy to gain preliminary insights on whether repeated Cy dosing could affect Treg levels and possibly enhance the response to tecemotide.
Results
Patient characteristics
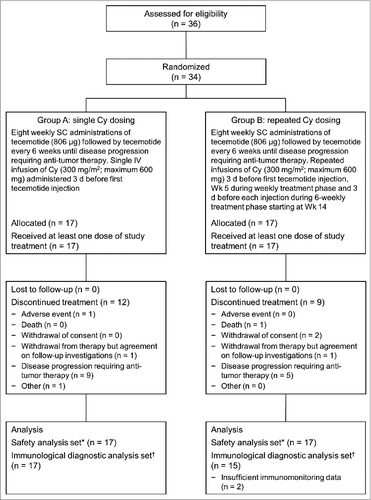
Thirty-four patients were enrolled and randomly allocated to receive tecemotide with either single or repeated Cy dosing (17 per group). Two patients in the repeated Cy group were missing immunologic samples up to week 9 and were excluded from the Immunologic Diagnostic Analysis (IDA) Set (). Patient demographics and baseline characteristics are presented in . The 2 groups were comparable in terms of gender, age, performance status and MM duration. However, the proportion of previously treated stage II/III patients was higher, and of chemotherapy-naïve stage I/II patients was lower, in the group with single compared with repeated Cy dosing.
Table 1. Patient demographics and baseline disease characteristics (Safety Analysis Set)
Figure 1. CONSORT flow diagram for enrolment and treatment of patients. Cy: cyclophosphamide; d: day; IV: intravenous; SC: subcutaneous; Wk: week. *Patients who received at least one dose of study treatment; †Patients with at least one complete set of baseline, week 5 and week 9 data of either ELISpot, proliferation or cytokine assay.
Treatment duration and dosing
Median treatment duration was 54 weeks (interquartile range [IQR] = 50–93) and 87 weeks (39-116) and median number of tecemotide administrations was 15 (IQR = 12–19) and 21 (13-23) for Groups A and B respectively. All patients in Group A received a single Cy infusion per protocol and the median number of infusions in Group B was 11 (IQR = 3–13). The study protocol allowed the investigator to reduce the Cy dose at their discretion. While this occurred frequently in Group B, most deviations were minor and at each administration only 0–2 patients received <50% of the target dose. The median number of Cy infusions in Group B was less than expected from the treatment duration, partly reflecting suspension of Cy dosing after the clinical hold. All patients had completed the weekly tecemotide treatment before the hold. The 6-weekly treatment phase was interrupted in 25 of 34 patients (74%), 16 of whom restarted treatment after the lift of the clinical hold after a median of 175 d (range 154–215).
Frequency of MUC1-specific immune responses
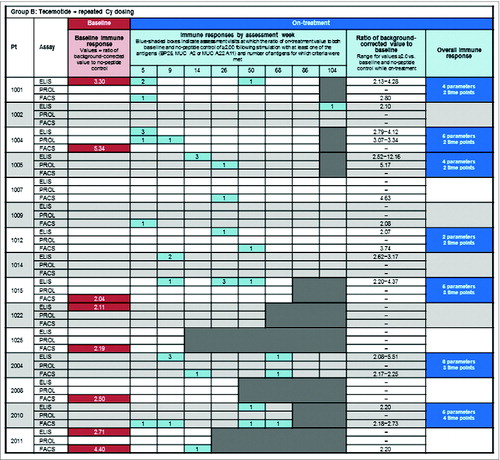
Seventeen of 32 (53%) patients showed a spontaneous MUC1-specific immune response at study entry prior to starting tecemotide treatment. Rates of pre-existing MUC1 immune responses were somewhat higher in Group A (10 of 17; 59%) than Group B (7 of 15; 47%) and in patients with previously treated stage II/III disease (11 of 19, 58%) compared with chemotherapy-naïve stage I/II MM (6 of 13, 46%).
Specific on-treatment induction or augmentation of MUC1 overall induced immune responses occurred in 15 of 32 (47%) patients. On-treatment induction of an overall immune response was seen in 8 of 17 (47%) patients with, and 7 of 15 patients (47%) without, a baseline MUC1-specific immune response. The rate of on-treatment induction/augmentation was somewhat higher among patients with chemotherapy naïve stage I/II MM than those with previously treated stage II/III disease ().
Table 2. Summary of on-treatment induction of overall MUC1-specific immune responses
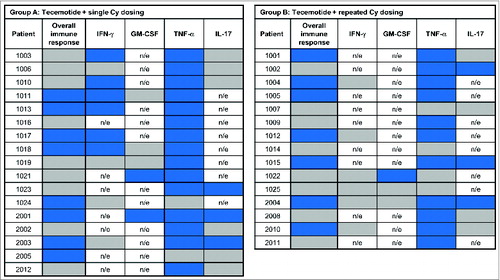
shows the frequency of MUC1-specific immune responses in each assay at baseline (red shading) and on-treatment (blue shading) for each patient in group A () and group B (). For the display of responses by assessment week, the number of antigens (parameters) for which the criteria for an immune response were met is indicated by the numbers in the blue boxes. Induction or augmentation of MUC1-specific immune responses occurred early in the course of treatment, typically within the first 9 weeks. The proportion of patients with an early induced MUC1-specific immune response (i.e. in any assay at ≥1 time-point within the first 9 weeks) was 47% in both groups, similar to the proportion of patients with an overall tecemotide-induced immune response (in any of the individual assays at 2 time-points up to week 50). Appendix S5 shows the time course of changes in the ELISpot assay for a representative patient with an overall immune response.
Figure 2. Baseline MUC1 immune responsiveness and on-treatment induction of MUC1-specific immune responses. (A) Group A: tecemotide with single Cy dosing and (B) Group B: tecemotide with repeated Cy dosing. Baseline MUC1-specific immune response (![]()
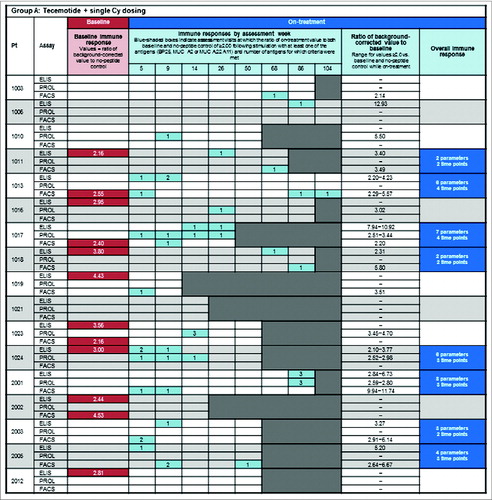
Figure 2. Continued.
Characterization of the nature of the immune response
There were no apparent differences in the rates of overall induced MUC1-specific immune responses according to HLA subtype (Appendix S6). The absolute count of CD8+ effector T cells in peripheral blood tended to increase over time in Group B but decrease in Group A (median normalized AUC = 0.246 and −0.243, respectively; p = 0.08). There were no other significant differences in the normalized AUC values of effector and memory T cells between treatment groups (data not shown).
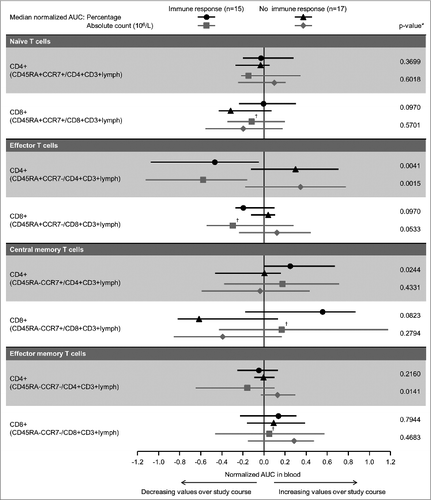
There were no clear differences between the treatment groups in Treg frequencies. Although the normalized AUC of several Treg subpopulations tended to be higher in Group B, none of the p-values from the Mann-Whitney U-test of between-group differences was <0.05 (). Treg levels decreased modestly in the days immediately following repeat Cy administrations in some patients, but tended to increase in the long term (data not shown). Some differences in effector and memory T cells in blood were seen between those patients with, compared to those patients without, an overall induced MUC1 immune response ().
Figure 3. Median normalized AUC values for T cell subsets in blood. Comparison of median normalized AUC values of percentages and absolute counts for (A) Treg cells in Groups A and B; and, (B) naïve, effector and memory CD4+ and CD8+ T cells for patients with and without an immune response. Bars = interquartile range. AUC: area under the curve. *Mann-Whitney U-test testing similarity of distribution of AUC values regarding treatment arms. †n = 14.
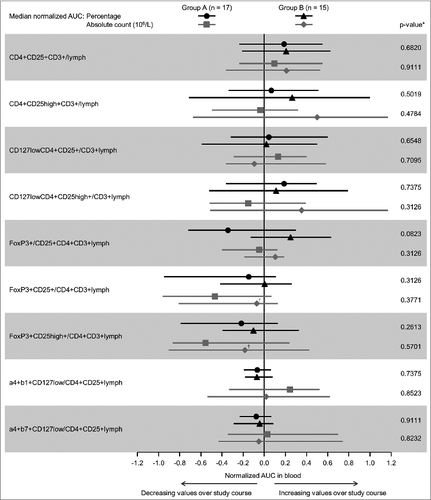
Figure 3. Continued.
Cytokine production following in vitro stimulation of PBMC with MUC1-derived peptides appeared to involve a Th1/T-cytotoxic 1-like response (). The majority of patients (26 of 32; 81%) across both groups showed an on-treatment induction (≥50% increase, blue shading) of TNF-α production and in Group A IFN-γ production was induced for 6 of 10 (60%) patients with evaluable samples.
Figure 4. Immunotherapy-induced production of non-disease-related cytokines by PBMC. Cytokine production determined by Luminex® assay following in vitro stimulation of PBMC with MUC1-derived peptides (BP25, MUC-A2 and MUC-A22-A11). Induction (![]()
Safety and tolerability
All patients reported at least one treatment-emergent AE; summarizes the most frequently reported. Adverse events related to Cy (nausea, constipation, fatigue) occurred more frequently with repeated than single Cy dosing. The most common AEs reported as related to tecemotide were injection-site reactions (ISRs) (Group A: 8/17 [47%]; Group B: 10/17 [59%]) and flu-like symptoms (both Groups: 2/17 [12%]). The majority of ISRs involved nodulation or erythema. All ISRs were mild-to-moderate in severity. One patient in each group had an ISR that lasted >72 d and one patient (Group B) had an ISR lasting 56–72 d. All other ISRs lasted <56 d. At least one injection site nodule was reported in 23.5% of patients in both groups. When present, the median nodule size was 20 mm in Group A and 16 mm in Group B.
Table 3. Treatment-emergent adverse events (Safety Analysis Set). (A) Treatment-emergent AEs of any grade reported in ≥15% of patients in either treatment group. (B) All grade 3/4 treatment-emergent AEs
Two of the 34 patients permanently discontinued study treatment owing to treatment-emergent AEs: one patient in Group A due to an injection site ulcer (grade 2) and one patient in Group B with mood alteration (grade 1) who discontinued Cy but not tecemotide. Three (18%) patients in Group A and 5 (29%) in Group B reported ≥1 grade 3/4 treatment-emergent AE. In one instance in Group B – a case of encephalitis with secondary symptoms of aphasia, cerebral hemorrhage, hypoxia, loss of consciousness and status epilepticus – this was assessed as possibly related to study medication. The patient was initially diagnosed with stage II MM and previous treatment with several regimens had included high-dose melphalan followed by autologous stem cell transplantation and maintenance chemotherapy with bortezomib (discontinued prior to study entry). The event occurred during the 6-weekly treatment phase after the patient had received 13 tecemotide injections and 7 Cy infusions, and started 27 and 24 d after the last administrations of Cy and tecemotide, respectively. Study treatment was discontinued but the patient died approximately 2 months after the onset of neurologic symptoms. Post-mortem assessments were inconclusive as to the cause. The event led to the clinical hold after which repeated dosing of Cy was halted.
Clinical effects
There were no objective clinical responses according to Bladé criteria.Citation23 Disease progression occurred in 9 of 17 (53%) patients in Group A and 5 of 15 (33%) patients in Group B (IDA Set). Median time to tumor progression (TTP) was 15.2 months in Group A and was not reached in Group B (hazard ratio [HR] = 0.378; 95% CI 0.121, 1.180; p = 0.08), although the small number of events should be noted.
The frequency of progressive disease was 33% (5 of 15 patients) among patients with an overall induced immune response vs. 53% (9 of 17 patients) of those without. Median TTP was not reached for immune responders and was 15.9 months for non-responders (HR = 0.486; 95% CI 0.162, 1.458, p = 0.19).
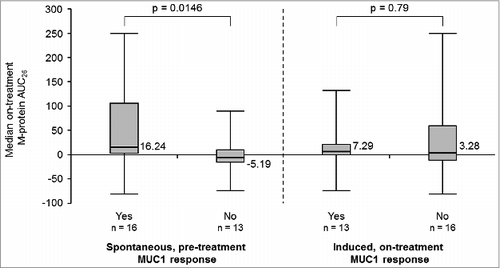
The slope of M-protein concentration over time was analyzed comparing per patient the slope before enrolment to the slope after study entry. A down-shift in slope was assessed as reflecting reduced tumor activity. While receiving tecemotide the M-protein slope was down-shifted compared to that prior to study enrolment in 13 of 29 (45%) evaluable patients (). On-treatment reduction in the M-protein slope occurred in 9 of 12 (75%) patients with chemotherapy-naïve MM stage I/II and 4 of 17 (24%) patients with previously treated MM stage II/III. On-treatment changes in M-protein concentration did not differ significantly between Groups A and B. The presence of a treatment-induced MUC1 response was not associated with M-protein changes during treatment. However, the median area under the curve (AUC) of M-protein concentration changes up to week 26 of treatment (M-protein AUC26) was positive (increasing) in patients with, but negative (decreasing) in patients without, a pre-existing baseline MUC1 response (p = 0.01; ). Appendix S7 shows the time course of M-protein changes for a representative patient with an on-treatment slope reduction.
Figure 5. M-protein concentration changes. (A) Comparison of the slope of M-protein changes over time before and during study treatment. Negative M-protein slope/decrease (![]()
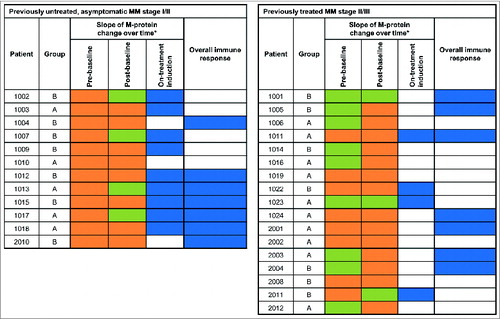
Figure 5. Continued.
Discussion
This is the first exploratory trial of the antigen-specific cancer immunotherapy (ASCI) tecemotide in patients with MM. The primary objective was to determine whether tecemotide induced a MUC1-specific T cell response. We also sought to characterize the nature of any immune response, and to obtain preliminary insights into the effects of single versus repeated low-doses of Cy on Treg levels and the response to immunotherapy, as well as to gain a preliminary assessment of the possible clinical, disease-stabilizing effects of tecemotide.
MUC1 expression is present in ∼40–60% of patients with MM,Citation25 including those with newly diagnosed disease.Citation26 This study included only patients with MUC1-expressing MM cells. The frequency of spontaneous MUC1-specific T cell responses prior to starting study treatment suggested that the immune system was often already primed to tumor-derived MUC1 as a result of host immunosurveillance.
Our results support the ability of tecemotide immunotherapy to elicit MUC1-specific responses in a substantial proportion of people with MM, with ∼50% of patients in the study showing an induced or augmented response during the study. The rate of treatment-induced MUC1-specific immune responses was similar in both treatment groups and in patients with or without a pre-existing MUC1 immune response. However, immune responses were generally weak with poor durability and did not correlate with a reduction in M-protein, possibly due to the poor magnitude of the immune response. Given the lack of previous studies investigating immune responses to peptide-based immunotherapies at multiple timepoints, it is difficult to speculate on the reasons for poor durability. However, poor immunogenicity of active immunotherapies has often been recognized as a limitation of this therapeutic strategy,Citation27 and is likely to be explained, at least in part, by the up-regulation of immunosuppressive entities within the tumor microenvironment.Citation28 As such, combination therapies that enhance immune effector mechanisms may be of value to achieve maximum clinical benefit. Cytokines such as interleukins, interferons and granulocyte-macrophage colony-stimulating factor (GM-CSF) are potential immune stimulants. Of note, GM-CSF has been used effectively in combination with several active immunotherapies.Citation29-31 Toll-like receptors have been shown to potentiate the immune response to vaccination in preclinical studies, and clinical trials are now warranted to investigate this strategy further.Citation27 Immune checkpoint inhibitors represent another promising area of combination immunotherapy research.Citation32
The assessment of leukocyte populations over time is difficult to interpret and may have been impacted by the clinical hold. However, on-treatment induction of a MUC1-specific immune response appeared to be associated with reduced CD4+ effector/effector memory T cells and to involve a Th1/T-cytotoxic 1-like response.
Repeated administration of low-dose Cy was halted after the clinical hold, confounding the secondary objective of comparing the 2 Cy dosing schedules. Consequently, any impact of repeated vs. single low doses of Cy remains unclear. Results from before the hold show that repeated low doses of Cy reduced Treg levels in the days immediately following each infusion in some patients. However, in the long-term, Tregs tended to increase. While studies in humans have shown that low intravenous doses of Cy can decrease Treg number and function,Citation33 this observation is sometimes limited to a subset (∼50%) of patientsCitation34 and others report no such effect.Citation35 These differences may be explained by the considerable variation in Cy pharmacokinetics between individuals. Genetic factors,Citation36 weight,Citation37,38 ageCitation39 and organ dysfunctionCitation40,41 have all been shown to influence Cy pharmacokinetics.
The study was exploratory and was not designed or powered to provide a definitive assessment of clinical safety and efficacy. However, the preliminary findings are compatible with a potential treatment effect of tecemotide in multiple myeloma. Study treatment was generally well-tolerated, consistent with experience in other malignancies.Citation19,Citation42-45 The adverse events (AEs) most frequently reported as possibly related to tecemotide administration were injection-site reactions and flu-like symptoms. Few serious AEs were reported and, with one exception, were not considered as related to study treatment. The underlying cause of the case of fatal encephalitis could not be clearly established, even after autopsy.
No objective clinical responses were seen according to Bladé criteria. This was not unexpected as, based on the mechanism of action of tecemotide, disease stabilization might be anticipated but not objective tumor regressions. It is now widely recognized that the standard response criteria for assessing the effects of cytotoxic chemotherapy have important limitations when applied to immunotherapeutic agents and do not provide a complete description of their effects.Citation46 Larger controlled trials are needed to explore the possibility of prolonged TTP in patients with an induced/augmented MUC1-specific immune response after treatment with tecemotide. The results from the START trial of tecemotide in unresectable stage III NSCLC, in which prolonged survival with tecemotide was observed in a subgroup of patients following concurrent but not sequential chemoradiotherapy,Citation45 support the potential for clinical benefit with tecemotide in selected patients and are consistent with the findings from this study.
M-protein concentrations were determined as a biochemical indicator of disease stabilization, with reduction over time thought to reflect reduced tumor activity.Citation23 The on-treatment reductions in M-protein changes over time in nearly 50% of patients provide early support for a possible clinical benefit of tecemotide particularly in early stage MM patients. We found no clear evidence that repeated Cy administration explained the downshift in M-protein concentration changes over time.
The reductions in M-protein concentration (as shown by negative AUC) in the majority of patients without a pre-existing MUC1-specific immune response are consistent with a disease-stabilizing effect. There was no evidence of disease stabilization among the patients with spontaneous MUC1-specific immune responses prior to treatment. The significance of a pre-treatment immune response for patient outcomes is likely to be complex. Our results and especially the subgroup results should be interpreted with caution given the small sample size but could reflect previous escape of the tumor from immune surveillance, rendering it less susceptible to a subsequent treatment-induced immune response. Other studies reporting baseline immune responses have shown variable effects on patient outcome. In a phase I study of patients with stage III/IV melanoma treated with PD1 blockade and a multipeptide vaccine, high numbers of pre-treatment MART-1 tetramer positive CD8+ T cells were associated with disease progression, while lower numbers were associated with treatment response or stable disease. However, there was considerable variability in T cell numbers among the patients in each group.Citation47 Moreover, a phase I study of peptide vaccination in hepatocellular carcinoma indicated that those with a baseline immune response were more likely to experience stable disease than disease progression.Citation48 Thus it appears unlikely that pre-existing immunity can reliably predict patient outcomes.
Tecemotide has been evaluated in epithelial cancers with preliminary evidence of clinical benefit.Citation45 By demonstrating for the first time induction/augmentation of MUC1-specific immune responses in a substantial proportion of patients with MM, a malignancy frequently associated with cellular and humoral immunodeficiency, our findings suggest that, in principle, the clinical utility of tecemotide may extend to haematological malignancies. However, the immune responses we observed were weak and of poor durability. Our results provide preliminary evidence that the most robust responses and greatest clinical benefit may be seen in patients with early stage disease, and suggest that future investigations of ASCI should focus on this subgroup of patients. Furthermore, it appears that combining ASCI with appropriate immunomodulatory therapy, for example to overcome immunosuppressive mechanisms deployed in the tumor microenvironment, is critical to optimizing immune responses directed against tumor antigens and that exploration of the most effective combinations should be a priority for future studies.
The study had a number of limitations. It did not include a non-treated control or tecemotide-only arm. The inclusion criteria for the study appear wide, with both pre-treated and treatment-naïve patients being recruited. However, the patients had a similar clinical presentation with none showing an emerging need for therapy as evidenced by the large proportion of patients that continued into an extended immunization protocol. There was an imbalance between study groups in the MM disease stage at baseline, which limits conclusions that can be drawn regarding the differences in TTP between the 2 groups. The comparison of single versus repeated Cy dosing on immune effects including Treg frequency was also compromised due to the clinical hold. A further confounding effect is the interruption to all study treatments for the duration of the clinical hold, which occurred at different points in the 6-weekly tecemotide treatment phase for individual patients. Due to the exploratory nature of the study, all findings need to be confirmed in further studies.
In summary, MUC1-specific immune responses were induced or augmented in a substantial proportion of patients with MUC1-expressing MM cells during this study of tecemotide and Cy, and preliminary evidence is consistent with clinical stabilization in a subset of these patients.
Patients and Methods
Patients
Eligible patients had MUC1-expressing MM cells (MUC1 expression by ≥10% of plasma MM cells, estimated by immunostaining of clot sections of formalin-fixed, paraffin-embedded bone marrow biopsies using monoclonal antibodies against CD138 (clone MI15; Dako A/S, Copenhagen, Denmark) and against CD227 (MUC1; clone HMPV; BD Biosciences - PharMingen, San Diego CA, USA)) and either:
Previously untreated, slowly progressive, asymptomatic stage I/II MM with rising monoclonal protein (M-protein) concentrations (≥10%) on 2 occasions separated by ≥4 weeks within the last 18 months.
Or:
Stage II/III MM with a stable response/plateau phase (Bladé criteriaCitation23) and a treatment-free interval of ≥3 months after prior anti-tumor therapy.
These patient groups were selected because of the relatively stable/slowly progressive state of MM, which may allow for better response to immunotherapy, and because of feasibility considerations for this single-center trial. Patients were ≥18 years, with Eastern Cooperative Oncology Group performance status ≤1 and life expectancy ≥6 months. Other inclusion criteria included platelet count ≥100 × 109/L, white blood cell count ≥2.5 × 109/L, hemoglobin ≥90 g/L, total bilirubin ≤1.5 × the upper reference range (URR), aspartate aminotransferase ≤2.5 × URR and serum creatinine ≤2 × URR.
Key exclusion criteria included previous exposure to MUC1-targeting therapy, radiotherapy, immunotherapy or any investigational drug in the preceding 30 d and presence of any pre-existing medical condition requiring chronic steroid or immunosuppressive therapy other than maintenance therapy with prednisone ≤10 mg/day or equivalent. Medical conditions excluding participation included hereditary/congenital immunodeficiencies, autoimmune disease that could compromise patient safety, clinically significant cardiac disease, other previous malignancy within 5 y (excluding basal cell carcinoma of the skin, in situ carcinoma of the uterine cervix or gastrointestinal intramucosal carcinoma), known hepatitis B and/or C, and splenectomy.
Study design and conduct
All patients in this single-center study received tecemotide and were randomly allocated (1:1 ratio) to single (Group A) or repeated (Group B) dosing of Cy using a randomization list with permutated blocks of randomly varying block sizes. Tecemotide was given as 8 consecutive weekly subcutaneous administrations followed by 6-weekly administration until disease progression requiring anti-tumor therapy (Appendix S1). Each tecemotide dose was administered as 4 injections to different anatomical sites, each containing one quarter of the total dose (806 μg lipopeptide, Appendix S2). In Group A, a single intravenous infusion of Cy (300 mg/m2; maximum total dose of 600 mg) was administered 3 d before the first tecemotide injection. In Group B, Cy was additionally administered 3 d prior to the tecemotide injection at week 5, and 3 d before each injection during the 6-weekly treatment phase starting at week 14.
Patients were enrolled between January 2008 and February 2010, and the primary analysis was performed in January 2011 after the last patient on treatment had reached Week 50. The study was impacted by a clinical hold due to a serious AE of fatal encephalitis in a patient in Group B that was possibly treatment-related. At the time of the clinical hold, patient recruitment had been completed and all patients had completed at least the weekly treatment phase. Treatment was interrupted for 5 months at various stages for individual patients. Repeated dosing of Cy in Group B was stopped when the study resumed.
Recruitment of 15 patients per treatment arm was considered sufficient for the exploratory analysis of immunologic effects and power calculations were not performed.
Ethics statement
All patients gave written, informed consent. The study was conducted in compliance with the principles of the International Conference on Harmonisation guidelines on Good Clinical Practice, the Declaration of Helsinki and local regulatory requirements, and was approved by the Karolinska Ethical Committee Review Board (EPN 2009/1765–32) and the Swedish Medical Products Agency (151:2007/52348). The trial was registered at EudraCT as #2006–001810–33 and at www.clinicaltrials.gov as #NCT01094548.
Immunomonitoring
The primary objective was to evaluate the MUC1-specific immune response to tecemotide in patients with MM. Peripheral blood mononuclear cells (PBMC) were collected at baseline, before the first Cy infusion, prior to tecemotide administration at week 5, one week after the last weekly treatment (week 9), at weeks 14, 26 and 50 during the 6-weekly treatment phase, and every 18 weeks thereafter.
The primary target variable was the specific anti-MUC1 T cell response. The following parameters were considered after short-term (5 d for ELISpot/lymphoproliferation assays, 6 hours for intracellular interferon-γ [IFN-γ] staining) in vitro stimulation of PBMC with MUC1-derived peptides:
ELISpot: in vitro stimulation with either BP25, MUC-A2 or MUC-A11
Proliferation assay: stimulation with either BP25, MUC-A2 or MUC-A11
Intracellular cytokine staining by FACS:
IFN-γ+CD69+/CD8+CD3+lymphocytes following stimulation with BP25
IFN-γ+CD69+/CD4+CD3+lymphocytes following stimulation with BP25.
BP25 is a synthetic MUC1 peptide consisting of 25 amino acids (STAPPAHGVTSAPDTRPAPGSTAPP). It differs from lipopeptide BLP25 by the absence of a palmitoyl side chain that facilitates binding of BP25 to a liposome. MUC-A2Citation11 and MUC-A22-A11Citation24 are synthetic MUC1 peptides consisting of 9 amino acids (MUC-A2: TSAPDTRPA; MUC-A22-A11: STAPPAHGV) and known to bind to HLA-A2 and HLA-A11, respectively. These were used to assess immune responses to peptides with different HLA specificities. The main criteria for assessing MUC1-specific immune responses were the following:
Overall induced immune response: for at least 2 timepoints: at least one parameter in at least one assay with ratio to background ≥2, and ratio of background-corrected value to baseline ≥2
Early increase of MUC1-specific immune response: for at least one parameter and timepoint up to Week 9: ratio to background ≥2, and ratio of background-corrected value to baseline ≥2
MUC1-specific immune response at baseline: for at least one parameter and at least one baseline sample: ratio to background ≥2
Further details on the criteria for immune responses can be found in Appendix S3.
Determination of Treg frequencies and further T-cell phenotyping in blood was performed by FACS at baseline, prior to the first Cy infusion, prior to tecemotide administration at weeks 5 and 9, and throughout the 6-weekly treatment phase. Naive and memory, central and effector T cells were characterized using flow cytometry by their surface expression of CD45RA and CCR7, as initially proposed by Sallusto.Citation49 AUC of the different cell frequencies over time was calculated to assess increases (positive AUC) or decreases (negative AUC) of cell populations. Induction of T helper (Th) 1 cytokine secretion by PBMC was assessed by Luminex® assay (Luminex, Austin TX, USA) following short-term in vitro stimulation of PBMC with the MUC1-derived peptides. Induction of cytokine production following stimulation by MUC1-derived peptides was retrospectively defined as a 50% increase over baseline for the specified cytokine on ≥1 occasion throughout immunomonitoring.
Immunomonitoring and tumor cell analyses were performed with freshly isolated cells at the Cancer Center Karolinska, Stockholm, Sweden. Data management was performed by Quintiles, Illkirch, France and statistical analysis by PRA International, Mannheim, Germany, both under the supervision of Merck KGaA, Darmstadt, Germany.
Safety and tolerability
Safety and tolerability were assessed in terms of incidence, severity and relatedness of AEs, including injection site reactions, physical examination findings, vital signs, and laboratory and other assessments. Vital signs were monitored before each tecemotide administration and for 1 hour and 0.5 hours after each administration in the weekly and 6-weekly treatment phases, respectively. Injection sites were inspected at baseline and at each tecemotide administration visit.
Clinical efficacy
Clinical efficacy assessments included response and TTP determined using Bladé criteria,Citation23 as well as time to anti-tumor therapy and M-protein concentrations.
Statistical analysis
The main analysis of immunologic response, safety and efficacy was performed after all patients had reached week 50 of treatment or had discontinued study medication. Immunologic parameters were analyzed in the IDA Set comprising patients with complete data during the primary treatment phase (i.e., baseline, week 5 and week 9) for ≥1 of the MUC1-specific immunomonitoring assays. The Safety Analysis Set comprised all subjects that received ≥1 dose of study treatment.
M-protein linear regression coefficients prior to enrolment vs. on study were calculated for each patient to explore changes in M-protein over time. M-protein concentration was measured pre-screening, at week 9, prior to every 6-weekly tecemotide administration and at the end of treatment visit. M-protein concentrations prior to enrolment were used to assess historical MM status. Slope analysis of M-protein data excluded patients with <5 pre- or <5 post-baseline values and used 2 separate linear regression models on pre- and post-baseline values for each patient versus treatment day, including intercept. ‘Slope’ refers to the regression coefficient for the slope parameter with negative slope indicating decreasing M protein concentrations over time and positive slope indicating rising M protein concentrations.
On-treatment changes over time were assessed as area under the curve (AUC), normalized to account for differences in treatment duration (Appendix S4). Between-group comparisons of AUC were based on the Mann-Whitney U-test. Time to progression was analyzed with the Kaplan-Meier method. Univariate Cox proportional hazards regression for the estimation of hazard ratios and their 95% confidence intervals, as well as the log-rank test were used for between-group comparisons.
All statistical analyses were performed separately for the 2 treatment arms, unless otherwise specified. The study was not powered to detect differences in responses between the arms. Statistics for continuous variables include means, medians, ranges and appropriate measures of variability. Qualitative variables were summarized by means of counts and percentages. Unless otherwise stated the calculation of proportions included the missing category. Statistical analyses are considered as descriptive even when statistical tests were conducted. No adjustment for multiplicity was performed.
Trial team:
Coordinating Investigator: Håkan Mellstedt
Study coordination: Eva Rossmann
Clinical investigators: Anders Österborg, Eva Löfvenberg
Laboratory personnel: Ingrid Eriksson, Barbro Näsman-Glaser, Ann Svensson, Fariba Mozaffari, Kia Heimersson, Barbro Larsson, Belinda Reinhold Pannagel, Aniruddha Choudhury
Clinical personnel:
Lotta Hansson, Jeanette Lundin, Sonja Sönnert-Husa, Marzia Palma
Sponsor team:
Study physicians: Veit Erpenbeck, Martin Falk, Roberto Carlesi, Andreas Schröder
Biostatistics: Stefanie Senger, Anja Loos, Anja von Heydebreck
Biomarkers and immunology: Michael Wolf, Ulf Forssmann, Mauro D’Antonio, Marco Minerdi, Sonia Quaratino
Drug safety: Ingrid Jörg, Regina Schick, Joachim Gerloff, Nicola Wallis
Data management: Marcel Mackiewicz
Study operations: Marie-Louice Willberg, Mandana Ashkzari, Alan Zanich
Clinical Research Organization: Quintiles
Disclosure of Potential Conflicts of Interest
AÖ received research funding from Merck KGaA. AS and AvH held employment positions at, and stock in, Merck KGaA, and AS holds stock in Oncothyreon. HM acted as a consultant for, and received research funding and honoraria from, Merck KGaA. UF held an employment position at Merck Serono S.A. Geneva. AC, EL and ER do not have any conflicts of interest to disclose.
Supplementary_Appendices.docx
Download MS Word (361.9 KB)Acknowledgments
The authors would like to thank patients, investigators and the study teams. Medical writing assistance was provided by Ian Faulkner, International Medical Press, UK.
References
- Ludwig H, Durie BG, McCarthy P, Palumbo A, San Miguel J, Barlogie B, Morgan G, Sonneveld P, Spencer A, Andersen KC, et al. IMWG consensus on maintenance therapy in multiple myeloma. Blood 2012; 119: 3003-15; PMID:22271445; http://dx.doi.org/10.1182/blood-2011-11-374249
- Belch A, Shelley W, Bergsagel D, Wilson K, Klimo P, White D, Willan A. A randomized trial of maintenance versus no maintenance melphalan and prednisone in responding multiple myeloma patients. Br J Cancer 1988; 57:94-9; PMID:3279997; http://dx.doi.org/10.1038/bjc.1988.17
- Shustik C, Belch A, Robinson S, Rubin SH, Dolan SP, Kovacs MJ, Grewal KS, Walde D, Barr R, Wilson J, et al. A randomised comparison of melphalan with prednisone or dexamethasone as induction therapy and dexamethasone or observation as maintenance therapy in multiple myeloma: NCIC CTG MY.7. Br J Haematol 2007; 136:203-11; PMID:17233817; http://dx.doi.org/10.1111/j.1365-2141.2006.06405.x
- Kagoya Y, Nannya Y, Kurokawa M. Thalidomide maintenance therapy for patients with multiple myeloma: meta-analysis. Leuk Res 2012; 36:1016-21; PMID:22579366; http://dx.doi.org/10.1016/j.leukres.2012.04.001
- Attal M, Lauwers-Cances V, Marit G, Caillot D, Moreau P, Facon T, Stoppa AM, Hulin C, Benboubker L, Garderet L, et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. N Engl J Med 2012; 366:1782-91; PMID:22571202; http://dx.doi.org/10.1056/NEJMoa1114138
- McCarthy PL, Owzar K, Hofmeister CC, Hurd DD, Hassoun H, Richardson PG, Giralt S, Stadtmauer EA, Weisdorf DJ, Vij R, et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N Engl J Med 2012; 366: 1770-81; PMID:22571201; http://dx.doi.org/10.1056/NEJMoa1114083
- Sonneveld P, Schmidt-Wolf IG, van der HB, El Jarari L, Bertsch U, Salwender H, Zweegman S, Vellenga E, Broyl A, Blau IW, et al. Bortezomib induction and maintenance treatment in patients with newly diagnosed multiple myeloma: results of the randomized phase III HOVON-65/ GMMG-HD4 trial. J Clin Oncol 2012; 30:2946-55; PMID:22802322; http://dx.doi.org/10.1200/JCO.2011.39.6820
- Butts C, Socinski MA, Mitchell PL, Thatcher N, Havel L, Krzakowski M, Nawrocki S, Ciuleanu TE, Bosquée L, Trigo JM, et al. Tecemotide (L-BLP25) versus placebo after chemoradiotherapy for stage III non-small-cell lung cancer (START): a randomised, double-blind, phase 3 trial. Lancet Oncol 2014; 15:59-68; PMID:24331154; http://dx.doi.org/10.1016/S1470-2045(13)70510-2
- Takahashi T, Makiguchi Y, Hinoda Y, Kakiuchi H, Nakagawa N, Imai K, Yachi A. Expression of MUC1 on myeloma cells and induction of HLA-unrestricted CTL against MUC1 from a multiple myeloma patient. J Immunol 1994; 153:2102-9; PMID:8051415
- Treon SP, Mollick JA, Urashima M, Teoh G, Chauhan D, Ogata A, Raje N, Hilgers JH, Nadler L, Belch AR, et al. Muc-1 core protein is expressed on multiple myeloma cells and is induced by dexamethasone. Blood 1999; 93:1287-98; PMID:9949172
- Brossart P, Heinrich KS, Stuhler G, Behnke L, Reichardt VL, Stevanovic S, Muhm A, Rammensee HG, Kanz L, Brugger W. Identification of HLA-A2-restricted T-cell epitopes derived from the MUC1 tumor antigen for broadly applicable vaccine therapies. Blood 1999; 93:4309-17; PMID:10361129
- Choi C, Witzens M, Bucur M, Feuerer M, Sommerfeldt N, Trojan A, Ho A, Schirrmacher V, Goldschmidt H, Beckhove P. Enrichment of functional CD8 memory T cells specific for MUC1 in bone marrow of patients with multiple myeloma. Blood 2005; 105:2132-4; PMID:15561890; http://dx.doi.org/10.1182/blood-2004-01-0366
- Hiltbold EM, Alter MD, Ciborowski P, Finn OJ. Presentation of MUC1 tumor antigen by class I MHC and CTL function correlate with the glycosylation state of the protein taken Up by dendritic cells. Cell Immunol 1999; 194:143-9; PMID:10383817; http://dx.doi.org/10.1006/cimm.1999.1512
- Hanisch FG, Schwientek T, Bergwelt-Baildon MS, Schultze JL, Finn O. O-Linked glycans control glycoprotein processing by antigen-presenting cells: a biochemical approach to the molecular aspects of MUC1 processing by dendritic cells. Eur J Immunol 2003; 33:3242-54; PMID:14635032; http://dx.doi.org/10.1002/eji.200324189
- Purcell AW, van Driel IR, Gleeson PA. Impact of glycans on T-cell tolerance to glycosylated self-antigens. Immunol Cell Biol 2008; 86:574-9; PMID:18626489; http://dx.doi.org/10.1038/icb.2008.48
- Beyer M, Kochanek M, Giese T, Endl E, Weihrauch MR, Knolle PA, Classen S, Schultze JL. In vivo peripheral expansion of naive CD4+CD25high FoxP3+ regulatory T cells in patients with multiple myeloma. Blood 2006; 107:3940-9; PMID:16410445; http://dx.doi.org/10.1182/blood-2005-09-3671
- Traverso I, Fenoglio D, Negrini S, Parodi A, Battaglia F, Kalli F, Conteduca G, Tardito S, Traverso P, Indiveri F, et al. Cyclophosphamide inhibits the generation and function of CD8(+) regulatory T cells. Hum Immunol 2012; 73:207-13; PMID:22285846
- Chu CS, Boyer J, Schullery DS, Gimotty PA, Gamerman V, Bender J, Levine BL, Coukos G, Rubin SC, Morgan MA, et al. Phase I/II randomized trial of dendritic cell vaccination with or without cyclophosphamide for consolidation therapy of advanced ovarian cancer in first or second remission. Cancer Immunol Immunother 2012; 61:629-41; PMID:22021066; http://dx.doi.org/10.1007/s00262-011-1081-8
- Butts C, Murray N, Maksymiuk A, Goss G, Marshall E, Soulières D, Cormier Y, Ellis P, Price A, Sawhney R, et al. Randomized phase IIB trial of BLP25 liposome vaccine in stage IIIB and IV non-small-cell lung cancer. J Clin Oncol 2005; 23:6674-81; PMID:16170175; http://dx.doi.org/10.1200/JCO.2005.13.011
- Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, Staehler M, Brugger W, Dietrich PY, Mendrzyk R, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med 2012; 18:1254-61; PMID:22842478
- Lutsiak ME, Semnani RT, De Pascalis R, Kashmiri SV, Schlom J, Sabzevari H. Inhibition of CD4(+)25 +T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 2005; 105:2862-8; PMID:15591121; http://dx.doi.org/10.1182/blood-2004-06-2410
- Sharabi A, Laronne-Bar-On A, Meshorer A, Haran-Ghera N. Chemoimmunotherapy reduces the progression of multiple myeloma in a mouse model. Cancer Prev Res (Phila) 2010; 3:1265-76; PMID:20719903; http://dx.doi.org/10.1158/1940-6207.CAPR-10-0138
- Bladé J, Samson D, Reece D, Apperley J, Bjorkstrand B, Gahrton G, Gertz M, Giralt S, Jagannath S, Vesole D. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high-dose therapy and haemopoietic stem cell transplantation. Myeloma subcommittee of the EBMT. European group for blood and marrow transplant. Br J Haematol 1998; 102:1115-23; PMID:Can't; http://dx.doi.org/10.1046/j.1365-2141.1998.00930.x
- Doménech N, Henderson RA, Finn OJ. Identification of an HLA-A11-restricted epitope from the tandem repeat domain of the epithelial tumor antigen mucin. J Immunol 1995; 155:4766-74.
- Baldus SE, Palmen C, Thiele J MUC1 (EMA) expressing plasma cells in bone marrow infiltrated by plasma cell myeloma. Histol Histopathol 2007; 22:889-93; PMID:17503346
- Lemancewicz D, Bolkun L, Porowska H, Galar M, Semeniuk J, Kloczko J, Dziecioł J. The levels of sMUC-1 in patients with multiple myeloma. Folia Histochem Cytobiol 2011; 49:654-8; PMID:22252760; http://dx.doi.org/10.5603/FHC.2011.0089
- Schlom J. Therapeutic cancer vaccines: current status and moving forward. J Natl Cancer Inst 2012; 104:599-613; PMID:22395641; http://dx.doi.org/10.1093/jnci/djs033
- McGray AJ, Hallett R, Bernard D, Swift SL, Zhu Z, Teoderascu F, Vanseggelen H, Hassell JA, Hurwitz AA, Wan Y, et al. Immunotherapy-induced CD8+ T cells instigate immune suppression in the tumor. Mol Ther 2014; 22:206-18; PMID:24196579; http://dx.doi.org/10.1038/mt.2013.255
- Borrello IM, Levitsky HI, Stock W, Sher D, Qin L, DeAngelo DJ, Alyea EP, Stone RM, Damon LE, Linker CA, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF)-secreting cellular immunotherapy in combination with autologous stem cell transplantation (ASCT) as postremission therapy for acute myeloid leukemia (AML). Blood 2009; 114:1736-45; PMID:19556425; http://dx.doi.org/10.1182/blood-2009-02-205278
- Emens LA, Asquith JM, Leatherman JM, Kobrin BJ, Petrik S, Laiko M, Levi J, Daphtary MM, Biedrzycki B, Wolff AC, et al. Timed sequential treatment with cyclophosphamide, doxorubicin, and an allogeneic granulocyte-macrophage colony-stimulating factor-secreting breast tumor vaccine: a chemotherapy dose-ranging factorial study of safety and immune activation. J Clin Oncol 2009; 27:5911-8; PMID:19805669; http://dx.doi.org/10.1200/JCO.2009.23.3494
- Lutz E, Yeo CJ, Lillemoe KD, Biedrzycki B, Kobrin B, Herman J, Sugar E, Piantadosi S, Cameron JL, Solt S, et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma. A Phase II trial of safety, efficacy, and immune activation. Ann Surg 2011; 253:328-35; PMID:21217520; http://dx.doi.org/10.1097/SLA.0b013e3181fd271c
- Madan RA, Mohebtash M, Arlen EM, Vergati M, Steinberg SM, Tsang KY, Dahut WL, Schlom J, Gulley JL Overall survival (OS) analysis of a phase I trial of a vector-based vaccine (PSA-TRICOM) and ipilimumab (Ipi) in the treatment of metastatic castration-resistant prostate cancer (mCRPC). [abstract]. J Clin Oncol 2010; 28: Abstract number 2250.
- Greten TF, Ormandy LA, Fikuart A, Hochst B, Henschen S, Hörning M, Manns MP, Korangy F. Low-dose cyclophosphamide treatment impairs regulatory T cells and unmasks AFP-specific CD4+ T-cell responses in patients with advanced HCC. J Immunother 2010; 33:211-8; PMID:20139774; http://dx.doi.org/10.1097/CJI.0b013e3181bb499f
- Greten TF, Forner A, Korangy F, N’Kontchou G, Barget N, Ayuso C, Ormandy LA, Manns MP, Beaugrand M, Bruix J. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer 2010; 10:209; PMID:20478057; http://dx.doi.org/10.1186/1471-2407-10-209
- Cerullo V, Diaconu I, Kangasniemi L, Rajecki M, Escutenaire S, Koski A, Romano V, Rouvinen N, Tuuminen T, Laasonen L, et al. Immunological effects of low-dose cyclophosphamide in cancer patients treated with oncolytic adenovirus 2. Mol Ther 2011; 19:1737-46; PMID:21673660; http://dx.doi.org/10.1038/mt.2011.113
- Kim IW, Yun HY, Choi B, Han N, Kim MG, Park S, Oh JM. Population pharmacokinetics analysis of cyclophosphamide with genetic effects in patients undergoing hematopoietic stem cell transplantation. Eur J Clin Pharmacol 2013; 69:1543-51; PMID:23588565
- Batey MA, Wright JG, Azzabi A, Newell DR, Lind MJ, Calvert AH, Boddy AV. Population pharmacokinetics of adjuvant cyclophosphamide, methotrexate and 5-fluorouracil (CMF). Eur J Cancer 2002; 38:1081-9; PMID:12008196; http://dx.doi.org/10.1016/S0959-8049(02)00024-2
- Powis G, Reece P, Ahmann DL, Ingle JN. Effect of body weight on the pharmacokinetics of cyclophosphamide in breast cancer patients. Cancer Chemother Pharmacol 1987; 20:219-22; PMID:3315280; http://dx.doi.org/10.1007/BF00570489
- de Jonge ME, Huitema AD, Rodenhuis S, Beijnen JH. Clinical pharmacokinetics of cyclophosphamide. Clin Pharmacokinet 2005; 44:1135-64; PMID:16231966; http://dx.doi.org/10.2165/00003088-200544110-00003
- Juma FD, Rogers HJ, Trounce JR. Effect of renal insufficiency on the pharmacokinetics of cyclophosphamide and some of its metabolites. Eur J Clin Pharmacol 1981; 19:443-51; PMID:7250178; http://dx.doi.org/10.1007/BF00548589
- Juma FD. Effect of liver failure on the pharmacokinetics of cyclophosphamide. Eur J Clin Pharmacol 1984; 26:591-3; PMID:6468474; http://dx.doi.org/10.1007/BF00543491
- Ohyanagi F, Horai T, Sekine I, Yamamoto N, Nakagawa K, Nishio M, Senger S, Morsli N, Tamura T. Safety of BLP25 liposome vaccine (L-BLP25) in Japanese patients with unresectable stage III NSCLC after primary chemoradiotherapy: preliminary results from a Phase I/II study. Jpn J Clin Oncol 2011; 41:718-22; PMID:21393255; http://dx.doi.org/10.1093/jjco/hyr021
- Butts C, Murray RN, Smith CJ, Ellis PM, Jasas K, Maksymiuk A, Goss G, Ely G, Beier F, Soulières D. A multicenter open-label study to assess the safety of a new formulation of BLP25 liposome vaccine in patients with unresectable stage III non-small-cell lung cancer. Clin Lung Cancer 2010; 11:391-5; PMID:21071331; http://dx.doi.org/10.3816/CLC.2010.n.101
- North SA, Graham K, Bodnar D, Venner P. A pilot study of the liposomal MUC1 vaccine BLP25 in prostate specific antigen failures after radical prostatectomy. J Urol 2006; 176:91-5; PMID:16753376; http://dx.doi.org/10.1016/S0022-5347(06)00494-0
- Butts C, Socinski MA, Mitchell PL, Thatcher N, Havel L, Krzakowski M, Nawrocki S, Ciuleanu TE, Bosquée L, Trigo JM, et al. Tecemotide (L-BLP25) versus placebo after chemoradiotherapy for stage III non-small-cell lung cancer (START): a randomised, double-blind, phase 3 trial. Lancet Oncol 2014; 15:59-68; PMID:24331154; http://dx.doi.org/10.1016/S1470-2045(13)70510-2
- Hoos A, Eggermont AM, Janetzki S, Hodi FS, Ibrahim R, Anderson A, Humphrey R, Blumenstein B, Old L, Wolchok J. Improved endpoints for cancer immunotherapy trials. J Natl Cancer Inst 2010; 102:1388-97; PMID:20826737; http://dx.doi.org/10.1093/jnci/djq310
- Weber JS, Kudchadkar RR, Yu B, Gallenstein D, Horak CE, Inzunza HD, Zhao X, Martinez AJ, Wang W, Gibney G, et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J Clin Oncol 2013; 31:4311-8; PMID:24145345; http://dx.doi.org/10.1200/JCO.2013.51.4802
- Sawada Y, Yoshikawa T, Nobuoka D, Shirakawa H, Kuronuma T, Motomura Y, Mizuno S, Ishii H, Nakachi K, Konishi M, et al. Phase I trial of a glypican-3-derived peptide vaccine for advanced hepatocellular carcinoma: immunologic evidence and potential for improving overall survival. Clin Cancer Res 2012; 18:3686-96; PMID:22577059; http://dx.doi.org/10.1158/1078-0432.CCR-11-3044
- Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999; 401:708-12; PMID:10537110; http://dx.doi.org/10.1038/44385