
Abstract
To date there is no explanation why the development of almost all types of solid tumors occurs sharing a similar scenario: (1) creation of a cancer stem cell (CSC), (2) CSC multiplication and formation of a multicellular tumor spheroid (TS), (3) vascularization of the TS and its transformation into a vascularized primary tumor, (4) metastatic spreading of CSCs, (5) formation of a metastatic TSs and its transformation into metastatic tumors, and (6) potentially endless repetition of this cycle of events. The above gaps in our knowledge are related to the biology of cancer and specifically to tumorigenesis, which covers the process from the creation of a CSC to the formation of a malignant tumor and the development of metastases. My Oncogerminative Theory of Tumorigenesis considers tumor formation as a dynamic self-organizing process that mimics a self-organizing process of early embryo development. In the initial step in that process, gene mutations combined with epigenetic dysregulation cause somatic cells to be reprogrammed into CSCs, which are immortal pseudo-germline cells. Mimicking the behavior of fertilized germline cells, the CSC achieves immortality by passing through the stages of its life-cycle and developing into a pseudo-blastula-stage embryo, which manifests in the body as a malignant tumor. In this view, the development of a malignant tumor from a CSC is a phenomenon of developmental biology, which we named a desperate asexual self-cloning event. The theory explains seven core characteristics of malignant tumors: (1) CSC immortality, (2) multistep development of a malignant tumor from a single CSC, (3) heterogeneity of malignant tumor cell populations, (4) metastatic spread of CSCs, (5) invasive growth, (6) malignant progression, and (7) selective immune tolerance toward cancer cells. The Oncogerminative Theory of Tumorigenesis suggests new avenues for discovery of revolutionary therapies to treat, prevent, and eradicate cancer.
Introduction
The growing evidences about strikingly similarities between CSCs and embryonic stem cells.Citation1,Citation2 have generated new interest in the old “embryonal” theories of cancer proposed more than 150 y ago. In 1858 Rudolf Virchow proposed the “embryonal-rest hypothesis” of tumor development, based on histological similarities between tumors and embryonic tissues. He suggested that tumors develop from the activation of “dormant” cells present in mature tissue that are remainders of embryonic cells.Citation3 In 1877 Julius Cohnheim postulated that the remaining embryonic cells, from which tumors form, were “lost” during organogenesis.Citation4 In 1890 David Paul von Hansemann proposed the concept of anaplasia that implies dedifferentiation, or loss of structural and functional differentiation of normal cells. He suggested that, due to anaplasia, somatic cells may transform into cancer cells which acquire “egg-like” features (for a review see ref. Citation5). In 1902 John Beard published “Embryological Aspects and Etiology of Carcinoma” in The Lancet, in which he proposed his Trophoblastic Theory of Cancer.Citation6 Based on behavioral similarities of cancer and trophoblastic cells he postulated that cancer was identical to the trophoblast. According to Beard remaining trophoblastic cells are lying dormant throughout the body. In mature organism the cells may receive a signal that causes them to begin growing as a malignant tumor. Various embryonal theories of cancer have been proposed in subsequent years.Citation7-Citation10
Although the above embryonal theories were the first attempts to explain the origin and biological behavior of malignant cells but they do not explain the mechanisms that drive the normal cell to be transformed into cancer cell. This led to scientific interest into elucidation of the essence of malignant transformation of cells.
During the last few decades, two cancer-development paradigms that are focused on cancer cell properties have driven cancer research. According to the first paradigm, the “somatic mutation theory,” the transformation of a normal somatic cell into a malignant cell is the result of sequential genomic mutations and epigenetic alterations of a stochastic nature.Citation11-Citation14 Douglas Hanahan and Robert A Weinberg suggested 10 hallmarks that were essential to the transformation of normal cells into malignant cells: (1) sustained proliferative signaling, (2) evasion of the effects of growth suppressors, (3) resistance to programmed cell death, (4) replicative immortality, (5) induction of angiogenesis, (6) invasiveness, (7) metastasis, (8) reprogramming of energy metabolism, (9) evading destruction by host defense cells, and (10) recruiting normal cells that create the “tumor microenvironment.” Each sequential genomic mutation or epigenetic alteration increases the affected cell’s repertoire of aberrant behaviors (for a review see refs. Citation14 and Citation15).
The second paradigm, which emerged more recently, is the discovery of cancer stem cells (CSCs) and the development of “the CSC theory of oncogenesis.” CSCs are stem cells (SCs) that have, through a series of genomic mutations or epigenetic alterations, been transformed. These cells have been isolated from both hematopoietic tumors (e.g., leukemias) and solid tumors. CSCs are tumorigenic in contrast to other non-stem-cell cancer cells, which are non-tumorigenic. CSCs are thought to persist in tumors as a distinct sub-population.Citation16-Citation18 The above-noted genomic mutations or epigenetic alterations are thought to cause reprogramming of a normal stem cell into a CSC. CSCs are the only cells that have the ability to generate primary and metastatic tumors. The above view represents the frontier of our current knowelege of the origin and the properties of CSCs.
However, the appearance of a CSC in the body is still not a disease. Cancer as a disease begins when a CSC implements the development of a malignant tumor. Neither old emryonal theories or current cancer theories can explain why the development of almost all types of solid tumors occurs sharing a similar scenario: (1) creation of a CSC (from an SC) due to one or more genetic alterations, (2) CSC multiplication and formation of a multicellular tumor spheroid (MCTS) with a heterogeneous population of cells, (3) vascularization of the MCTS and its transformation into a vascularized primary tumor, (4) disaggregation of CSCs from the bulk tumor and their metastatic spreading to local lymph nodes and other body tissues, (5) formation of a metastatic MCTS, and its vascularization and transformation into metastatic tumors, and (6) potentially endless repetition of this cycle of events.
The above gaps in our knowledge are related to the biology of cancer and specifically to tumorigenesis, which covers the process from the creation of a CSC to the formation of a malignant tumor and the development of metastases.
In the period 1989–1993 we published the Oncogerminative Hypothesis of Tumor FormationCitation19-Citation22 in which we proposed an explanation why the above mentioned tumorigenic scenario is common for most or all solid tumors. The main idea of our original hypothesis was that the key to discerning the nature of cancer development is to understand the mechanism underlying the expression of the potential immortality of the cancer cell. We hypothesized that the only way for a malignant cell to transform its potential immortality into actual immortality in vivo is to mimic the mechanism of development of natural immortality of germline cells.Citation19-Citation22 Over the past 20 years, numerous studies (discussed below) have confirmed the correctness of the main idea of our oncogerminative hypothesis of tumor formation and provided a basis for our development of a substantially expanded version: the Oncogerminative Theory of Tumorigenesis. In this article we present that theory, review recent data that validate and develop the core ideas that we presented 20 years ago, and introduce several fundamentally new concepts to explain tumorigenesis and natural selective immune tolerance to cancer.
An Oncogerminative Cell is a CSC that Mimics the Genetic Program of a Germline Cell
In 1989 we introduced the concept of an “oncogerminative cell” to describe specific types of cancer cells that exclusively possess the ability to develop into primary and metastatic tumors.Citation20 This concept anticipated the emergence of evidence of the existence of CSCs. The first conclusive evidence for the existence of CSCs was published in 1997 in Nature Medicine. Bonnet and Dick isolated a subpopulation of leukemic cells that express a specific surface marker CD34, but that lack the CD38 marker.Citation23 The first solid tumor CSC was isolated from breast cancer by Michael Clarke's group in 2003.Citation24 CSCs are considered to be the only cells within a tumor that possesses the capacity to self-renew and to cause the heterogeneous lineages of cancer cells that comprise the tumor.Citation25 Thus, we can now state that the oncogerminative cell is identical to the CSC. Hereafter we will use the term “oncogerminative cell” as a synonym for a “cancer stem cell.”
Our hypothesis considered malignant transformation as a process of somatic cell reprogramming into a potentially immortal oncogerminative cell. This oncogerminative cell (i.e., this CSC) achieves immortality by passing through its life cycle, which mimics the natural life cycle of the fertilized germ cell. The result of this CSC cycling is the development of a malignant tumor. We predicted that during tumor development the oncogerminative cell divides asymmetrically [1 CSC → 1 CSC + 1 non-CSC ] to produce not only oncogerminative cells, but also other types of daughter cells, including differentiated cells. This prediction was confirmed by recent data that CSCs share many characteristics with normal stem cells, including the abilities to self-renew and differentiate.Citation6 Moreover, there is growing evidence that some solid tumors also follow a differentiation program similar to that of the normal tissue of origin.Citation26 The daughter cells produced by parent CSCs follow many of the rules observed by daughter cells in normal tissues. The CSCs are like a caricature of normal cells: they display many of the same features as normal tissues, but in a distorted way.Citation27 In 1987 we hypothesized that the only way for a somatic cell to acquire the potential for immortality is to be reprogrammed so that it mimics a germline cell, which implies expression of the dormant genes that determine the behavioral properties of a normal germline cell when that cell is achieving immortality.Citation20-Citation22 The Oncogerminative Theory states that the essence of malignant transformation is the germinalization of a normal somatic cell, i.e., its reprogramming into a CSC, which is pseudo-germline cell. .
This claim is now supported by numerous studies. Janic and colleaguesCitation28 recently showed that some brain tumor cells mimic the genetic program of germline cells. These authors demonstrated that Drosophila lethalCitation3 malignant brain tumors (L(3)mbt) exhibit a soma-to-germline transformation through the ectopic expression of genes normally required for germline stemness, fitness, or longevity. Inactivation of any of the germline genes (nanos, vasa, piwi, or aubergine) suppressed the malignant growth of L(3)mbt. Marilyn Monk and Cathy HoldingCitation29 hypothesized that human pre-implantation embryonic cells are similar in phenotype to cancer cells. Both types of cell undergo reprogramming to a proliferative stem cell state and become potentially immortal and invasive. To test the hypothesis that embryonic genes are re-expressed in cancer cells, the authors prepare amplified cDNA from human individual preimplantation embryos and isolate embryo-specific sequences. Then these isolated embryo-specific genes were tested for their expression in a panel of human cancers. It was found that three of the five embryo-expressed cDNA sequences tested were re-expressed in cells of different tumors. The authors also tested a range of cancer cell lines for expression of embryo and/or cancer genes C and E and of OCT4. All three gene sequences were expressed in various cancer cell lines but not in immortalized fibroblasts.Citation29 Therefore, it might be expected that cancer cells will express genes that are expressed in very early embryonic cells, especially genes specifically associated with reprogramming, and will return to the undifferentiated and proliferative stem cell state that is associated with immortality and invasiveness. Genes that are specific to this unique early phase of the human life cycle and that are not expressed in committed somatic cells and immortalized normal cells (fibroblasts) may have greater potential for being targeted in cancer treatment.
A similar genetic event occurs in the early embryo during establishment of its germ cell lineage. As is well known, the pluripotent epiblast cells in the early embryo are destined to form both somatic cells and primordial germ cells. In the few cells that undergo specification to establish the germ cell lineage, there is a repression of the somatic program. So, the general characteristic of germ cell specification is that the expression of somatic genes must be repressed for the germ cell program to eventually be initiated.Citation30 Akira Nakamura and colleagues described the cell biology of germ cell formation, along with how the germplasm leads to the repression of somatic gene expression (for a review see ref. Citation31). Recent evidence shows that Blimp1, a known transcriptional repressor with a SET/PR domain, is crucial for the specification of primordial germ cells (PGCs). Blimp1 (Prdm1), the key determinant of PGCs, plays a combinatorial role with Prdm14 during PGC specification from postimplantation epiblast cells. They together initiate epigenetic reprogramming in early germ cells toward an underlying pluripotent state, which is equivalent to the state of embryonic stem cells (ESCs).Citation32,Citation33 Blimp1 has the role, at least in part, of suppressing the cell’s somatic program.Citation34
Jekaterina Erenpreisa and Mark S Cragg observed the activation of meiotic genes in cancer cells during reversible polyploidy induced by DNA damage.Citation25,Citation26 The upregulation of key meiotic genes (MOS, REC8, SGO1, SGO2, DMC1, SPO11, SCYP1,2,3, STAG3) was found, and was associated with reversible polyploidy in TP53-deficient lymphoma, breast, colon, ovarian, and cervical cancer cell lines after irradiation or spindle damage (for a review see refs. Citation35–Citation38). The ectopic expression of key meiotic genes such as MOS was also found in primary non-small cell lung carcinoma, such as DMC1Citation39—in cervical cancer, such as SPO11, REC8, SGO1, and HORMAD1,Citation37 and in melanoma.Citation40
Landmark studies conducted by Yamanaka and others recently demonstrated that they could create pluripotent stem cells from adult human cellsCitation41 These artificially created pluripotent stem cells are termed induced pluripotent stem cells (iPSCs) and were first described by Kazutoshi Takahashi and Shinya Yamanaka in mice in 2006.Citation42 These researchers found that just four factors normally produced in the embryo are sufficient to reprogram differentiated, non-embryonic cells into pluripotent stem cells. The four Yamanaka reprogramming factors, Oct4, Sox2, Klf4, and c-Myc, when introduced into differentiated cells, reverses cell fate and induces these cells to revert back to a progenitor-like or ESC-like state. It is believed that this is a stochastic process whose mechanisms lie in the ability of these four transcription factors to induce epigenetic restructuring of the genome, thereby enabling expression of previously silenced genes (for a review see refs. Citation42–Citation44). Shi-Lung Lin and colleagues showed that a cancer cell could also be reprogrammed into an ESC-like pluripotent cell.Citation45 There is evidence that the closest in vivo equivalent of an ESC is an early germ cell (refs. Citation46 and Citation47). The mir-302s microRNA (miRNA) family was investigated as one of the key factors essential for the reprogramming of cancer cells into ESC-like pluripotent cells. Members of the mir-302 family (mir-302s) are expressed in slow-growing human ESCs, and their numbers quickly decrease after cell differentiation and proliferation. Therefore, mir-302s were investigated as one of the key factors essential for maintenance of ESC renewal and pluripotency. The Pol-II-based intronic miRNA expression system was used to transgenically transfect mir-302s into several human cancer cell lines. mir-302s-transfected cells, especially miRNA-induced pluripotent stem (mirPS) cells, not only expressed all sorts of hESC markers such as Oct3/4, SSEA-3, SSEA-4, Sox2, and Nanog, but also, they had a highly demethylated genome, similar to the reprogrammed genome of a fertilized egg.Citation48 Onder and Daley showed that the expression of the miR-302/367 cluster of miRNAs can directly reprogram somatic cells into induced pluripotent stem cells without the use of any transcription factors.Citation48,Citation49 In contrast, it was recently revealed that stable miR-302–367 cluster expression is sufficient to suppress the stemness signature, self-renewal, and infiltration of glioma stem cells within a host brain tissue, through inhibition of the CXCR4 pathway. The inhibition of CXCR4 leads to the disruption of the sonic hedgehog (SHH)-GLI-NANOG network, which is involved in self-renewal and expression of the embryonic stem cell-like signature. Others have demonstrated that the miR-302–367 cluster is able to efficiently trigger a cascade of inhibitory events leading to the disruption of the tumorigenic properties of glioma stem-like cells.Citation50 miRNAs are emerging as important regulators of cellular differentiation, their importance underscored by the fact that they are often dysregulated during carcinogenesis. At present, a strong link between miRNA and Blimp1dysregulation and human cancer has been established.Citation51-Citation53 We anticipate that Oct4, Sox2, Klf4, c-Myc, Blimp1, and miRNA types of regulators, along with as yet unknown ones, will be found to play a crucial role in malignant transformation by the reprogramming of somatic cells into cancer stem cells.
The oncogerminative theory states that a crucial genetic event leading to malignant transformation of a somatic cell is its “germinalization,” i.e., its reprogramming into a pseudo-germline cell, which is a CSC (i.e., an oncogerminative cell). This means that, like the germline cell, or its closest in vivo equivalent, the embryonic stem cell,Citation44,Citation53,Citation54 the CSC achieves immortality by passage through its life cycle. Thus, the oncogerminative cell (i.e., the CSC) is a reprogrammed somatic cell that has acquired basic phenotypic properties of a germline cell—the ability to realize its potential immortality by passing through its life cycle. The oncogerminative cell (i.e., the CSC) is the only cell that possesses the ability to generate a malignant tumor. However, the oncogerminative cell, which arises from a committed somatic cell, does not possess the pluripotency of the initial embryonic stem cells due to crucial differences between the morphogenetic potencies of the egg cell’s cytoplasm and of the oncogerminative cell’s cytoplasm. During embryogenesis, the egg cell’s cytoplasm is distributed among committed cells. This distribution is accompanied by a gradual restriction of morphogenetic abilities in the cytoplasm of such cells.Citation55,Citation56 Any proliferating cell of the body, including a wide variety of somatic stem cells and different progenitor cells, may undergo malignant transformation into a CSC. CSCs, consequently, may lead to a great variety of malignant tumors of different histological types.
Thus the oncogerminative cells (i.e., the CSCs) have different normal precursor cells and belong to different histological types of cells. Despite this fact, all oncogerminative cells have a fundamentally common basis that is grounded in the acquired ability of each of these cells to generate a malignant tumor by passing through its own life cycle, which mimics the life cycle of normal germline cells.
The creation of a CSC in the body is still not a disease. A CSC is still only a potential threat for the body, since it could be eliminated by an innate immune response such as increased production of natural killer cells. To initiate the disease of cancer, the oncogerminative cell must generate a malignant tumor.
Cancer Development is a Desperate Asexual Self-Cloning Event
The oncogerminative theory of tumorigenesis considers tumor formation as a dynamic self-organizing process that mimics a self-organizing process of early embryo development. This approach allows us to explain the common biological and behavioral properties of almost all solid cancers. These include multistep development of a malignant tumor involving: (1) avascular and vascular steps of tumor growth, (2) tumor cell heterogeneity, (3) disaggregation of the cells of the tumor compartment including CSCs, (4) transformation of the CSCs, into metastatic CSCs, and their spreading within the body’s tissues, (5) a common pattern of development of metastatic tumors of different origin, (6) common patterns of malignant progression, (7) constant expression of stage-specific embryonic antigens by different malignant tumors, and (8) development of selective immune tolerance to cancer cells.
The oncogerminative theory holds that the CSC does not invent an alternative mechanism to ensure its immortality. Instead, oncogerminative cells co-opt a pre-existing mechanism of potential immortality of a germline cell along with its early life cycle pattern ().
Figure 1. Stages of the life cycles of germline cells (A) and oncogerminative cells (B). (A) Z, zygote; CSE, cleavage stage embryo; MSE, morula stage embryo; ABSE, avascular blastocyst-stage embryo; IBSE, implanted blastocyst-stage embryo; F, fetus. SMB, sexually mature body. (B) CSC, cancer stem cell (i.e., oncogerminative cell); PCSC, parthenogenetic cancer stem cell (a pseudo-cleavage-stage embryo); TG, tumor germ (a morula-stage embryo-like structure); TS, tumor spheroid (an imitation avascular blastocyst-stage embryo); VTS/VT, vascularized tumor spheroid and/or vascularized tumor (an implanted blastocyst-stage embryo-like entity).
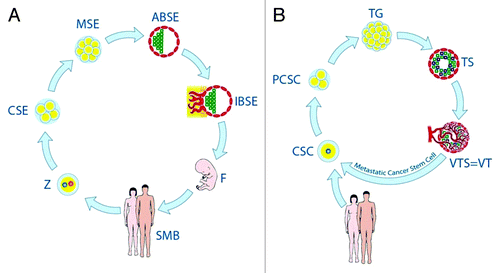
The theory postulates five stages of development of a malignant tumor from an oncogerminative cell:
Stage 1: reproduction of oncogerminative cells. (This stage mimics a cleavage-stage embryo.)
Stage 2: aggregation of oncogerminative cells and formation of the tumor germ, which consist of oncogerminative cells only. (This stage mimics a morula-stage embryo.)
Stage 3: the transformation of the tumor germ to a tumor spheroid with a heterogeneous cell population. (This stage mimics the avascular blastocyst-stage embryo.)
Stage 4: vascularization of the tumor spheroid and its further growth as a vascularized tumor. (This stage mimics the post-implantation blastocyst-stage embryo.)
Stage 5: disaggregation of oncogerminative cells and their metastatic spreading into body tissues. (This stage mimics primordial germ cell migration.). Metastatic oncogerminative cells can go through their life cycle again and generate additional metastatic tumors.
The theory offers a new explanation—that cancer development is associated with a unique sort of biological immortality. It is based on the core principle upon which nature creates biological life: the potential immortality of the populations of living organisms. The single-celled organisms are potentially immortal due to their ability to undergo an infinite number of cell divisions without a distinction between parent cells and offspring cells.Citation57,Citation58 Some simple multicellular animals such as hydra, jellyfish (Turritopsis dohrnii), and planarian flatworms do not undergo senescence and, as such, are potentially immortal.Citation59,Citation60 Populations of mammals survive indefinitely even though individuals in those populations grow old and die. That requires that young mammalian offspring be produced at the expense of old parents, and this has classically been explained in terms of an immortal germ line passing through a transient and disposable soma, or body.Citation61 Thus, mammals possess two general types of cells: mortal cells made of soma and a lineage of potentially immortal germline cells that is passed from one generation to the next, indefinitely. The germline cells can achieve immortality only through embryogenesis, by creation of an endless line of intermediate generations of individuals.
The oncogerminative theory of tumorigenesis postulates that cancer is a desperate attempt of the mortal somatic cell to achieve immortality through germinalization: reprogramming of that cell into a pseudo-germline cell.
Here we would like to present the evidence, which has appeared during the last decade, for an alternative way by which normal somatic cells may achieve immortality by being transformed into immortal tumor cells. Sundaram M, at al. (2004) studied early cellular events during carcinogen-induced transformation of C3H10T1/2 cells. Multinucleate and/or polyploid giant cells (MN/PGs) formed due to DNA damage were expected to die via mitotic catastrophe. However before they die, some MN/PGs undergo a novel type of cell division, which the authors termed “neosis”Citation62. They viewed neosis as a parasexual somatic reduction division that is characterized by DNA damage-induced senescence and/or mitotic crisis and polyploidization. That is followed by production of aneuploid daughter cells via nuclear budding; with asymmetric cytokinesis and cellularization conferring extended, but limited, mitotic life span to the offspring. The immediate neotic progeny were termed, by the authors, Raju cells, which transiently display stem cell properties. The Raju cells immediately undergo symmetric mitotic division and become mature tumor cells. The authors believed that the neosis paradigm of self-renewal of cancer cells is consistent with telomere attrition, aging, and the origin of cancer cells after reactivation of telomerase, and constitutes an alternative to the cancer stem cell hypothesis.Citation63 Erenpreisa J, Cragg MS in their review “Cancer: a matter of life cycle?” (2007), attempted to reconcile both the cancer stem cell hypothesis and the neosis hypothesis with the idea that cycling polyploidy, intermitting senescence, and rejuvenation may be features of a life cycle analogous to the life cycles of certain unicellular organisms. The authors suggested that mitotic catastrophe may represent a mechanism through which the cell can switch from the usual mitotic cell-cycle to this evolutionarily conserved life cycle.Citation64
We believe that both cancer stem cells and Raju cells, which were mentioned above, represent tumor-initiating cells, the only cells that can give rise to malignant tumors. The theory states that tumor-initiating cells, in order to proceed with tumor development, should acquire the characteristics of germline cells. We named such tumor-initiating cells oncogerminative or pseudo-germline cells. The pseudo-germline cell (i.e., a CSC) achieves immortality through pseudo-embryogenesis, by creation of an endless line of intermediate generations of pseudo-clones that are malignant tumors. Therefore, a malignant tumor is a product of the life cycle of an oncogerminative cell just as an embryo is a product of the life cycle of a germline cell.
Thus, we consider cancer development in an innovative way: as a phenomenon of developmental biology, which we named a “desperate asexual self-cloning process.” In mammals this process manifests as cancer development.
Parthenogenetic-Like Reproduction of an Oncogerminative Cell
Based on the view, in the Oncogerminative Theory of Tumorigenesis, that an oncogerminative cell is a pseudo-germline cell, it is reasonable to assume that an oncogerminative cell may undergo parthenogenetic-like activation when it develops into the tumor germ (TG).
Parthenogenesis is a form of asexual clonal reproduction. In animals, parthenogenesis means development of an embryo from an unfertilized egg cell. According to Suomalainen et al., three systems of sex determination may be considered in animal offspring derived from parthenogenesis: (1) female parthenogenesis, or thelytoky, in which unfertilized egg cells develop into females; (2) arrhenotoky, in which unfertilized egg cells develop parthenogenetically into males; and (3) deuterotoky (amphitoky), in which unfertilized egg cells stimulated parthenogenetically give rise to both females and males.Citation65,Citation66 Parthenogenesis is also classified as either (1) generative, or haploid (n), parthenogenesis or (2) somatic parthenogenesis, which may be diploid (2n) or polyploidy (>2n). In generative or haploid (meiotic) parthenogenesis, individuals are developed from chromosomally reduced egg cells, which thus have the haploid chromosome number. In somatic (ameiotic) parthenogenesis, offspring have the unreduced (zygotic; zygoid, if diploid or polyploidy) chromosome number place.Citation67
Whereas parthenogenesis is a common mode of reproduction in lower organisms, the mammalian parthenote (activated egg) fails to produce a successful pregnancy. This does not mean, however, that embryonic tissues cannot be generated from parthenogenetically activated oocytes in humans. Indeed, it recently was shown that pluripotent stem cells can be created from parthenogenetically activated eggs, either nonhuman primate or human. Vrana KE at al. described in vitro parthenogenetic development of monkey (Macaca fascicularis) eggs to the blastocyst stage, and their use to create a pluripotent line of stem cells. These monkey stem cells (Cyno-1 cells) are positive for telomerase activity and are immunoreactive for alkaline phosphatase, octamer-binding transcription factor 4 (Oct-4), stage-specific embryonic antigen 4 (SSEA-4), tumor rejection antigen 1–60 (TRA 1–60), and tumor rejection antigen 1–81 (TRA 1–81) (traditional markers of human embryonic stem cells). Cyno-1 cells can be differentiated in vitro into dopaminergic and serotonergic neurons, contractile cardiomyocyte-like cells, smooth muscle, ciliated epithelia, and adipocytes. When Cyno-1 cells were injected into severe combined immunodeficient mice, teratomas with derivatives from all three embryonic germ layers were obtained.Citation68
In 2007 Elena Revazova, and her research team were the first to intentionally create human stem cells from unfertilized human eggs using parthenogenesis. These stem cells are homozygous in the HLA region of DNA. Accordingly, these stem cells have been named human parthenogenic stem cells (hpSCs).Citation69,Citation70
The idea that parthenogenesis may be a key feature of carcinogenesis was proposed more than 90 years ago.Citation71,Citation72 The original and subsequent parthenogenetic versions of the embryological theory of cancer were considered in detail by Janis Erenpreiss in his review “Current concepts of malignant growth”Citation73 Common to these versions were the suggestion that because only germ cells are subject to parthenogenetic activation, the process of carcinogenesis resembles gametogenesis. This suggestion has been supported by numerous lines of evidence that are consistent with a parthenogenetic origin of germ cell tumors.
Thus, human ovarian germ cell tumors (including teratomas) develop directly from oocytes. The fact that the 46,XX karyotype is found in almost all mature teratomas strengthens the theory of its parthenogenetic origin.Citation74 A high frequency of ovarian teratomas has been described in LT, an inbred strain of miceCitation75 and in some strains of mice (LT/Sv and Mos-null) with inherited mutations . In these strains, ovarian teratomas are derived from oocytes that undergo maturation and spontaneous parthenogenetic activation followed by embryonic development within the ovarian follicles.Citation76
A large majority of primary testicular tumors originate from germ cells. The germ cell tumors of an adult testis are classified into two types of entities on the basis of histological, serological, and clinical data. These two types are seminoma, and non-seminoma germ cell tumors (NSGCT), which include embryonal carcinoma, yolk sac tumors, polyembryoma, choriocarcinoma, and teratomasCitation77.. The major differences between the two groups suggest that seminoma tumors recapitulate some aspects of spermatogenesis, whereas NSGCT represent descendants of pluripotent embryonic cells that were formed from the germ cell through parthenogenetic activation.Citation78,Citation79
We assume that the development of a variety of non-germ cell tumors is also due to parthenogenetic-like activation of CSCs, which we believe are pseudo-germline cells. The basis of this assumption is numerous experimental findings that are consistent with the possibility of reprogramming differentiated somatic cells into pluripotent cells. These reprogrammed cells had features similar to features of embryonic stem (ES) cells and their closest equivalent, embryonic germ (EG) cells, in terms of their gene expression profiles and epigenetic profiles. Reprogramming of mouse and human somatic cells into pluripotent ES and EG cells, which was done to generate induced pluripotent stem (iPS) cells, has proved possible with the expression of only four transcription factors: Oct4, Sox2, c-Myc, and Klf4 (for a review see refs. Citation44, Citation80).
Zaehres H, Wu GM, Gentile L, Ko K, et al. have shown that neural stem cells in adult mice express higher endogenous levels of Sox2 and c-Myc than do embryonic stem cells, and that exogenous Oct4 together with either Klf4 or c-Myc is sufficient to generate iPS cells from neural stem cells. These two-factor iPS cells are similar to embryonic stem cells at the molecular level, contribute to development of the germ line, and form chimeras.Citation81
Our assumption is also supported by data recently published by Chunfang Liu, Zhan Ma, Jun Hou et al.Citation82 The authors showed that genes that are specific to germline cells and embryonic development are activated in many tumors. Specifically, they showed that germline traits that are present in human hepatoblastoma cells are associated with the malignant behaviors of these tumor cells. In culture, single human hepatoblastoma cells differentiated into germ cell-like cells, which further developed into oocyte-like cells and formed parthenogenetic blastocyst-like structures. The germ cell-like cells and their embryonic derivatives from hepatoblastoma cells can give rise to xenograft tumors with embryonal and/or germline traits and intrahepatic metastasis. The authors suggested that germline potential is spontaneously activated in human hepatoblastoma cells, which might be important for tumor formation and metastasis.
Another set of supporting data came from the observation that in a great many cases parthenogenesis appears in connection with polyploidy. Polyploid forms occur especially in animals with apomictic (ameiotic) parthenogenesis.Citation83 Although the formation of polyploid cells is part of the developmental program of several tissues, polyploidization also accompanies some pathological processes and malignant transformation of cells. It is believed that tetraploid cells precede aneuploid ones in the early phases of tumor development.Citation84
In their review “Three steps to the immortality of cancer cells: senescence, polyploidy and self-renewal,” Erenpreisa J and Cragg MS (2013) summarized data on the key role of the c-Myc oncogen in polyploidization of normal cell and cancer cell immortality.Citation35 c-Myc, which is one of the four Yamanaka transcription factors originally described to be essential for the generation of iPSC, directly activates DNA replication; its overexpression uncouples DNA replication from mitosis, thereby favoring polyploidy.Citation85 Erenpreisa and Cragg also showed that prolonged growth arrest of damaged tumor cells in G2 and their transition, through aborted mitoses, into polyploidy (with its enhanced capacity for DNA repair) as well as arrest reversal, may be associated with the induced meiosis-like program.Citation35
Thus, we consider the pathenogenetic-like activation of oncogerminative (i.e., CS) cells as an option during somatic (ameiotic) parthenogenesis.
We also assume the existence of an alternative variant of tumor formation by the oncogerminative cells. We believe that a tumor may also be formed as a result of the aggregation of oncogerminative cells, which, in this case, mimics the behavior of cells of the morula. The ability to aggregate is a property of the embryonic cells of the morula-stage embryo. This property was demonstrated by experiments in which a re-aggregated chimeric morula was obtained from the cells of two disaggregated mouse embryos at the morula stage. Genetically mosaic mice can be produced by aggregating, during cleavage stages, the blastomeres of two embryos of different genotypes into a single cluster, and by transferring the developing aggregates to the uterus of a surrogate mother.Citation86,Citation87
Similarities Between the Development of a Tumor Spheroid and the Development of an Early-Stage Embryo
There are a great number of similarities between (1) the earliest evolutionarily conserved stages of the development of the blastocyst-stage embryo from a fertilized oocyte and (2) the development of a tumor spheroid from a CSC. After the vascularization of both the blastocyst and the tumor spheroid, the fundamental differences between them rapidly manifest. In the case of embryo development, precise morphogenetic processes take place during tissue differentiation that results in the development of a normal fetus. In the case of tumor development, one only observes an increase in tumor cell mass.
Early stages of embryo development of different triploblastic forms of metazoans remain rather similar and follow similar time patterns in species at different stages of evolution. Initially a totipotent embryonic stem cell (zygote) gives rise to three-dimensional multicellular structures of an early embryo: morula and blastocyst. The early stages of embryogenesis are the point at which embryonic stem cell lines are derived. The fertilized egg (day 1) undergoes cell division to form a 2-cell embryo, followed by a 4-cell one, and so on until a ball of cells called a morula are formed by the fourth day. Day 4 morula cells are still totipotent. The morula becomes hollow, forming the blastocyst. This is the stage at which pluripotent embryonic stem cell lines, cells of the trophoblast, and predecessors of germline cells, are generated. Following the blastocyst stage, the tissues of the embryo start to form and the cells become multipotent.
According to my theory, the oncogerminative cell, being a pseudo-germline cell, gives rise to the three-dimensional multicellular structures of early malignant tumors: to the tumor germ (TG), which mimics the morula-stage embryo, and to the multicellular tumor spheroid, which mimics the blastocyst-stage embryo) (). A TG is a precursor of a tumor spheroid (like a morula is a precursor of a blastocyst). A TG is a multicellular structure that is composed of CSCs. Each of the cells of an artificially disaggregated TG may give rise to a new TG just as each of the cells of an artificially disaggregated morula-stage embryo may develop into a new embryo.Citation86 Unlike the tumor spheroid, the TG does not have a structural organization similar to the tumor and is unable to initiate neovascularization.
The tumor germ is transformed into the tumor spheroid. Tumor spheroids are heterogeneous cellular aggregates that, when greater than 500 μm in diameter, are frequently characterized by hypoxic regions and necrotic centers.Citation88,Citation89 Tumor spheroids display three distinct cell layers defined by their local microenvironment: an outer layer comprised of mitotic cells, a middle layer of quiescent non-dividing cells, and a dense necrotic core. These concentric spheroid regions are generated by the gradients of nutrients, oxygen, and growth factors that produce different microenvironments within the tumor mass that are associated with the build-up of acidic metabolic waste and hypoxic conditions in the tumor core.Citation90
The tumor spheroid is considered an in vitro micro-model for the avascular stage of the in vivo growing tumor.Citation88,Citation91 This idea comes from findings on the identity of the basic features of the in vitro tumor spheroid and in vivo tumor nodule: the heterogeneity of cell populations, the capability for three-dimensional growth, the presence of a proliferation gradient, similar growth-kinetics patterns, similar extracellular matrix formation, similar glucose content, similar oxygenation conditions of the tissues, the presence of central necrosis, the identity of antigenic composition, and the ability to secrete angiogenesis factors and other growth factors.Citation92 Tumor spheroid cells are able to initiate neovascularization.Citation93
Tumor Cell Heterogeneity Mimics Cell Heterogeneity of the Blastocyst
All malignant tumors are monoclonal in origin. However, a fundamental property of both tumor spheroids and tumors is the heterogeneity of their cell populations. Different subpopulations of cells may show different capacities for growth, differentiation, metastasis, and different sensitivities to radiation and chemotherapy. There are only tiny populations of cells, specifically CSCs, in each malignancy that are capable of tumor initiation. The proportion of CSCs in solid tumors is less than 1%.Citation94,Citation95 CSCs drive tumorigenesis, and give rise to a large population of differentiated progeny that make up the bulk of the tumor, but that lack tumorigenic potential.Citation64 Multiple studies have reported the presence, in malignant tumors, of cells with different degrees of differentiation,Citation96-Citation99 including benign and normal cells.Citation100 The mechanisms that underlie the heterogeneity of tumor cells, which derive from a single CSC, are not fully understood until present.
In 1989 we hypothesized that, when developing into a tumor, the oncogerminative cell obeys the same biological laws as an embryonic stem cell does when developing into a blastocyst.Citation20 This allowed us to explain the heterogeneity of tumor cell populations. A single fertilized cell gives rise to the pre-implantation blastocyst, which is a multicellular spherical structure with a heterogeneous cell population. It consists of three major types of cells: trophoblastic cells, cells of the epiblast (precursors of somatic cells and germline cells), and cells of the hypoblast (precursors of extra-embryonic cells) ().
Figure 2. Types of cells of the pre-implanted blastocyst (A) and tumor spheroid (B). (A) Types of cells of the pre-implanted blastocyst. Red, trophoblast; green, precursor somatic cells; blue, precursor primordial germ cells. (B) Types of cells of the tumor spheroid. Red, oncotrophoblastic cells or pseudo-trophoblasts; green, oncosomatic cells or pseudo-somatic cells; blue, oncogerminative cells or pseudo-germline cells.
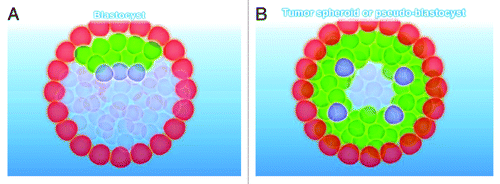
In mammalian embryos, the direction of blastomere differentiation is determined solely by the localization of the blastomere in the pre-implantation blastocyst. A cell of the outer cell layer of the blastocyst becomes part of the trophoblast, and a cell localized inside the blastocyst gives rise to the inner cell mass. When labeled blastomeres are transferred into the inner or outer sites of the blastocyst, they give rise to trophoblasts or inner cell mass, respectively.Citation101,Citation102
We hypothesized that the development of a tumor spheroid, which mimics the development of a blastocyst, results in the development of three major types of cells (): oncotrophoblastic cells, pseudo-trophoblast cells; oncogerminative cells (i.e., cancer stem cells), pseudo-germline cells; and oncosomatic cells, pseudo-somatic cells
Below we present the data that support our hypothesis.
Oncotrophoblastic Cells in Malignant Tumors
In several types of tumors of embryonic and germ-cell origin, the presence of trophoblastic cells has been established.Citation103,Citation104 There is indirect evidence of the presence of trophoblastic cells in tumors of non-embryonic and non-germinal cell origin. Such evidence is based on the persistent detection, in different malignant tumors, of placental markers such as pregnancy-associated plasma protein A (PAPP-A), pregnancy-specific β1-glycoprotein (SP1), human placental lactogen (HPL), human chorionic gonadotropin (hCG), oncomodulin or calcium binding protein-15 (CBP-15), carcinoembryonic antigen (CEA), placental alkaline phosphatase, and growth factors. Growth factors include fibroblast growth factors (FGFs) and vascular endothelial growth factor (VEGF). These markers are common in the fetal placenta.Citation105,Citation106
Trophoblastic cells of the blastocyst and cells localized to the superficial layer of the tumor spheroid exhibit invasive properties, an ability to lyse surrounding host tissues, and the ability to phagocytize the lysed cells. When a blastocyst is cultured in vitro, its gigantic trophoblastic cells are able to invade and to lyse not only the simultaneously cultured other tissues, but also the cells of the embryo itself.Citation107 This is reminiscent of the ability of tumor spheroids to lyse other cells in the invasion of other tissues during co-culturing.Citation108
We assume that the oncotrophoblastic cells of the tumor spheroid in vivo are mainly responsible for its implantation, invasiveness, and ability to induce angiogenesis.
Oncogerminative Cells (i.e., CSCs) in Malignant Tumors
Oncogerminative cells are solely responsible for the development of malignant tumors with heterogeneous cell populations, for the spreading of tumors within the body’s tissues, and for their development into metastatic tumors.
Germline cells may give rise to gonadal and extragonadal germ cell tumors.Citation109-Citation111 Extragonadal germ cell tumors are found in areas of the body other than the ovary or testicle such as the brain, chest, abdomen, or tailbone. The stem cells of teratocarcinomas are embryocarcinoma cells (EC-cells). EC cells are nearly identical to pluripotent embryonic stem cells (ES cells) and similar in size to inner cell mass (ICM) cells. ES cells are cells of the epiblast or inner cell mass of the blastocyst.Citation112 Under special conditions, germ cells can acquire properties similar to those of embryonic stem cells (ESCs). The underlying mechanism of that change is still unknown. These changed cells are then called embryonic germ (EG) cells. Both EG and ES cells are pluripotent. Recent studies have demonstrated that it is possible to generate primordial germ cells from ES cells.Citation33,Citation113 Thus, EC cells, which are identical to ES cells, are also closely related to embryonic germline cells. Thus, EC cells, which are the stem cells of teratocarcinomas, represent the oncogerminative cell fraction of those carcinomas. The ES-cells, when implanted into ectopic sites, give rise to teratocarcinomas.Citation114 During normal embryogenesis, ES-cells exist during a short period of early embryo development—from 5.5 to 6.5 days after fertilization. According to A.P. Dyban,Citation115 the short existence of ES-cells in the epiblast is a manifestation of mechanisms that prevent the transformation of an embryo into a teratocarcinoma. In teratocarcinomas, the EC-cells give rise to two types of cells: daughter pluripotent EC cells (we consider these cells as oncogerminative ones), and another type of EC cell that may differentiate into the cells of normal tissues.Citation116
There is direct evidence of the presence of germline cells, namely primordial germ cells, in non-germ cell malignancies. For example, in hematological malignancies primordial germ cells were identified by their morphology, an intense PAS, PAS-D reaction, and the presence of calcium-activated neutral proteinase.Citation117 Germline-like malignant cells were found in malignant brain tumors in Drosophila.Citation28 Orthologs of some of these genes were also expressed in human somatic tumors. There is indirect evidence of the presence of germline cells in malignant tumors of different origins. Only in mammals do germline cells continue to express high levels of the enzyme telomerase, while most somatic cells, regardless of their rate of proliferation, do not possess telomerase activity.Citation118 In contrast to somatic cells, telomerase is found in the overwhelming majority of human cancers and is usually responsible for allowing cancer cells to grow indefinitely.Citation119 So, telomerase is a common marker of both types of immortal cells: normal germline cells and oncogerminative cancer stem cells. Another example of indirect evidence is the Cancer/Testis Associated (CTA) family of genes that is expressed in germ cells of the testis and generally not in other normal tissues. In contrast, CTA gene expression is found in a number of malignant tumors of various histological types.Citation120
Oncosomatic Cells in Malignant Tumors
Cancer stem cells as well as normal embryonic and adult stem cells are defined by both their ability to make more stem cells (stem cells self-renewal) and their ability to produce cells that differentiate. One strategy by which cancer stem cells can accomplish these two tasks is asymmetric cell divisionCitation121..Asymmetric divisions of the CSCs gives rise to one cell of the same potency (CSC self-renewal), and another that maybe of the same potency or stimulated to further differentiation. Thus cancer stem cells are known to be capable of generating multiple lineages of normal and neoplastic cells.Citation122 Recently Zhang S, Mercado-Uribe I, and Liu J.Citation123 have reported that the polyploidy giant cancer cells (PGCCs) induced by paclitaxel from an established invasive breast ductal carcinoma cell line MCF-7Citation124 generated multiple lineages of tumor stromal and differentiated cancer cells and formed cancer organotypic structure (COS) in vitro. The PGCCs not only grew into well-differentiated cancer cells that formed COS in vitro but also trans-differentiated into multiple tumor stromal cells including myoepithelial, endothelial and erythroid cells. These results demonstrated that paclitaxel-induced PGCCs have properties of cancer stem cells that can generate both epithelial cancer cells and multilineage of normal stromal cells.Citation123 Differentiated “normal” cells, i.e., oncosomatic cells, are ubiquitously observed in tumors of different origins.Citation125 Thus we believe that tumor-initiating (oncogerminative) cells due to it asymmetric division are capable to give rise to the new generation of oncogerminative cells as well as to the cells with different grade of differentiation including oncotrophoblastic and oncosomatic cells.
A Cancer Stem Cell (CSC), When Developing into a Metastatic Tumor, Mimics the Behavior of a Fertilized Germline Cell
The oncogerminative theory of tumorigenesis states that oncogerminative cells (i.e., CSCs), which mimic primordial germ cells, possess the same phenotypic property as primordial germ cells—the ability to aggregate and then disaggregate. CSCs distribute in a host body by metastatic spread. The oncogerminative CSC is the only cell of the heterogeneous tumor-cell population that is able to metastasize and develop into a metastatic tumor. However, the oncogerminative cell does not have a monopoly on this behavior. The ability to metastasize is a natural behavior of germline cells, more specifically primordial germ cells. A phenotypic property of mammalian primordial germ cells (PGCs) is their ability to aggregate and form germinative tissue in early embryogenesis. Subsequently, this tissue disaggregates into primordial germ cells, which then migrate to the embryo's genital ridges within a definite time period of embryo development.
More than 20 years ago we hypothesized that oncogerminative cells, which mimic germline cells, possess the same basic property: the ability to aggregate and form an oncogerminative-cell-compartment within the malignant tumor.Citation20,Citation22 Subsequently, this cell-compartment may disaggregate into individual oncogerminative cells, which migrate into the host’s target tissues. We assumed that the ability of oncogerminative cells to aggregate reversibly is centrally important to metastatic spreading and formation of metastatic tumors. Strong evidences for our assumption was the discovery of the epithelial-to-mesenchymal transition (EMT) mechanism.
The EMT is a process in which cells lose epithelial characteristics and acquire a migratory, mesenchymal phenotype.Citation126 EMT induces profound morphological and phenotypic changes in a cell: the epithelial cells lose their cell polarity and cell-cell adhesion, and gain migratory and invasive properties to become mesenchymal stem cells.Citation127 It has been shown that the EMT, and the reverse process, the mesenchymal-to-epithelial transition (MET), play crucial roles in embryogenesis, wound healing, cancer metastasis, and induced pluripotent stem cell reprogramming.Citation44,Citation128,Citation129 Molecular markers of EMT include E-cadherin, vimentin, epidermal growth factor receptor (EGFR), platelet-derived growth factor (PDGF) D, and nuclear factor κB (NF-κB). Mesenchymal cells do not make mature cell-cell contacts, can invade through the extracellular matrix, and express markers such as vimentin, fibronectin, N-cadherin, Twist, and Snail.Citation126,Citation128 The gene expression changes associated with MET including the suppression of key mesenchymal genes, such as Snail, Slug, Zeb1, Zeb2, Twist1, Twist2, N-cad, and Fn and the upregulation of key epithelial genes, such as E-cadherin, Ocln, Cldn3, Dsp, Pkp1, Pkp3, Ep-CAM, Krt8, and Krt19.Citation44,Citation130
It is widely believed that the ability to reversibly shift from an epithelial cell, via an EMT, to a mesenchymal cell, and back to an epithelial cell, via an MET, is an evolutionarily conserved molecular mechanism that underlies cell migration in normal and pathogenic processes including metastasis.Citation128 During embryogenesis, this mechanism coordinates cell migration involving movement of cells of different lineages over short and long distances throughout the body. In particular the migration of the mammalian primordial germ cells (PGCs) begins when 50 gonocytes and about 5,000 PGCs arrive at the genital ridge. PGCs come from the epiblast and subsequently migrate into the mesoderm, the endoderm and the posterior of the yolk sac, where they aggregate. In 4.5 weeks postconception (wpc) embryos disaggregation and migration of PGCs then takes place from the epigaster along the gut and across the dorsal mesentery to reach the genital ridges.Citation131 PGCs in 4–5 wpc embryos are thought to leave the gut epithelium by an EMT-like transition.Citation132
Recent evidence has demonstrated that the EMT, induced by different factors, is associated with tumor aggressiveness and metastasis and that these cells share molecular characteristics with CSCs, and thus are often called cancer stem-like cells or tumor-initiating cells.Citation126 Thus, upon transfection by activated Ras, the subpopulation of CD44high/CD24low immortalized human mammary epithelial cells, as well as cancer cells that possess stem-like properties, increases in number with the concomitant induction of the EMT.Citation133 The acquisition of an EMT phenotype is a critical process for switching early stage carcinomas into invasive malignancies, which is often associated with the loss of epithelial differentiation and gain of mesenchymal phenotype. Later, when these circulating tumor cells (CTCs) exit the bloodstream to form micrometastases, they undergo a MET for clonal outgrowth at these metastatic sites. Thus, EMT and MET form the initiation and completion of the invasion-metastasis cascade.Citation134
Therefore, we believe that the oncogerminative cell (i.e., the CSC) and the PGC use the same EMT/MET mechanism when implementing their migratory-tissue-formation cascades. The shifting between EMT and MET phenotypes underlie the migration of PGCs from the epigaster to the genital ridges and subsequent formation of the medullary tissue of the future gonad. Similar EMT/MET mechanisms initiate the migration of the oncogerminative cells (i.e., CSCs) from the primary tumor to the target tissue and subsequent formation of the metastatic tumor.
The evidence indicating similarities between oncogerminative cells and primordial germ cells comes from research into common markers that are only found in germline cells and metastasizing cancer stem cells. Numerous markers—germ cell alkaline phosphatase (GCAP), the matrix metalloproteinase (MMP) family of proteins, Oct4, several microRNA (miRNA) families, and the recently found Sall4 protein—that are developmentally expressed in cells of pre-implantation blastocysts and in primordial germ cells also serve as markers of metastasizing cells from different types of tumors.Citation135-Citation137 These markers were found in both germ cell tumors and in non-germ-cell tumors (breast carcinoma, renal adenocarcinoma, melanoma, non-small-cell lung carcinoma, colon adenocarcinoma, leukemic cells, and others).Citation138-Citation140
Although we consider the oncogerminative cell (i.e., the CSC) as the only cell that is able to initiate the development of a metastatic tumor we would like to emphasize that a circulating oncogerminative cell alone is unable to implant, invade, and to develop into a metastatic tumor. Just as a fertilized germline cell must create a blastocyst before the implantation, a circulating oncogerminative cell must first create a multicellular structure (a tumor spheroid) in order to generate the new metastatic tumor ().
Figure 3. (A) Stages of development of the preimplantation blastocyst. (B) Stages of hematogenous metastasis.
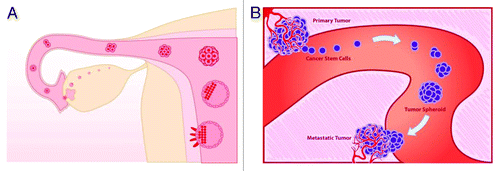
After settling in a host tissue, a metastatic oncogerminative cell may go through its life cycle again and develop a metastatic tumor that has a heterogeneous cell population. Disaggregation of the oncogerminative cells of the metastatic tumor may occur again, initiating, in the same manner, development of the next generation of metastatic tumors with a different ratio of oncogerminative, oncotrophoblastic, and oncosomatic cells. As a result of such a clonal selection “vehicle,” the proportions of the oncogerminative, oncotrophoblastic, and oncosomatic cells may change in favor of the oncogerminative and oncotrophoblastic ones.
We believe that a progressive decrease in the size of the fraction of the most differentiated cells, the oncosomatic cells, in subsequent generations of metastatic tumors underlies malignant progression.
Phylogenetic Immune Tolerance is the Essence of Selective Immune Tolerance to Cancer
The problem of overcoming immune tolerance to tumor self-antigens remains a most important unresolved issue. The theory suggests a new approach to understanding the biological nature of immune tolerance to cancer cells. This approach involves the following points. We consider a malignant tumor to be a pseudo-blastocyst-stage-embryo developed by an oncogerminative cell (i.e., a CSC). Therefore, the nature of the interrelation between host and tumor has to be considered in the context of the interrelation between the host and pseudo-embryo. So, in order to understand how and why cancer cells escape immune surveillance, we have to elucidate how and why the fetus and placenta escape immune rejection during pregnancy. More than 50 years ago, Medawar described pregnancy as an immunological paradox because the fetus is normally accepted by the maternal immune system despite expression of immunogenic embryonic antigens, including paternal alloantigens.Citation141 A fetus is, in genetic terms, a semi-allograft that escapes rejection. At present, it is well known that the embryo and cancers express a wide spectrum of common embryonic antigens.Citation19,Citation21
Below we discuss the similarities and differences between the immune events associated with embryo development and those associated with cancer development ().
Table 1. Comparison of host immune alterations during embryonic development and cancer development
Comparison of host immune alterations during embryogenesis and tumorigenesis
Stage I: The development of a “critical mass” of embryonic cells and cancer cells. The embryonic stem cells and oncogerminative cells (i.e., CSCs) are only the cells that are capable of going through its life cycle, which results in the development of a multicellular structures: the blastocyst and tumor spheroid (a pseudo-blastocyst), respectively.
In both cases, the development of a multicellular structure occurs under avascular conditions. The essence of the first stage of both embryo development and cancer development is the developing of multicellular structures with a “critical mass” of cells that are able to protect themselves against host immunity and are sufficiently secure to be in intimate association with maternal blood without the risk of being rejected due to a host-vs.-graft reaction.
Stage II: First shift of host immunity: development of selective immune tolerance to the blastocyst-stage embryo and to the tumor spheroid. During embryonic development, two groups of potentially immunogenic embryonic antigens appear: (1) antigens of the early embryo and (2) antigens of the placenta. Mammals possess a mechanism that underlies maternal immune tolerance to the antigens of the early embryo and thus guarantees survival and normal development of a semi-alien embryo in a maternal body. The essence of maternal selective immune tolerance to an early embryo is the absence of the immune response to early embryonic antigens due to the absence, in the host, of the appropriate clones of the immunocompetent lymphocytes. The recognition of what is “foreign” and what belongs to ones “own” body (i.e., what is “self”) is a fundamental property of the immune system. The achieving of this self-tolerance mainly occurs during T cell maturation in the thymus. About 95% of all the cells formed there die before they are ready to mature and emigrate into the peripheral lymphatic organs. This wastefulness is due to the extremely stringent selection criteria that are applied to the T cells that develop in the thymus. T cells possessing receptors that strongly bind to one of the body's own peptides bound to an endogenous MHC molecule are triggered to undergo apoptosis. Because most B cells require the support of helper T cells for their reactions with antigens, the elimination of self-reacting helper T cells also insures that self-reacting B cells are harmless. The repertoire of T cells thus consists of a combination of positive and negative selection processes.
During ontogeny, the potentially immunogenic embryonic antigens are expressed by the cells of the early embryo for a short period of time. Early embryonic antigens in the fetus disappear long before the development and maturation of the immune system of a new individual. The maturation of the immune system is associated with the appearance of the clones of immunocompetent lymphocytes, including memory lymphocytes. As a result, the new individual gains the ability to produce a normal immune response (i.e., antibody production and/or cell-mediated immunity) following exposure to different antigens, except antigens of the early embryo, since such antigens are absent at the time of immune system development. During pregnancy, when antigens of semi-alien embryos are expressed, the maternal memory lymphocytes are unable to initiate the cascade of immune responses against the early embryo because memory lymphocytes have nothing to remember about antigens of blastocyst-stage embryos. That is why we named this selective immune tolerance to the embryonic antigens a “phylogenetic immune tolerance.”
CSCs, which express embryonic antigens, usurp the mechanism of “phylogenetic immune tolerance” for their own, unhampered, in vivo growth.
The first shift of host immune reactivity results in the establishment of transient selective tolerance either to cells of the implanted blastocyst or to cells of the invasive tumor spheroid. Such shifts in host immune reactivity occur due to graft-vs.-host interactions, where the graft is represented either by a blastocyst embryo or by a malignant tumor. The graft-vs.-host interaction results in the development of host specific syndromes: paraembryonic syndrome, known as pregnancy, in the case of embryonic development, and paraneoplastic syndrome in the case of cancer development. These syndromes are mediated by humoral factors (hormones, cytokines, polyamines, embryonic antigens, and others) secreted by either embryonic or cancer cells. In mammals, development of such phylogenetically determined syndromes during pregnancy underlies the transient and selective immune tolerance of a host to a semi-allograft fetus, and secures its development. A malignant tumor, as a pseudo-embryo, usurps the mechanism of protective tolerance that phylogeny creates for embryo development.
Thus, through mammalian evolution were elaborated phylogenetic immune mechanisms, which are repeated in ontogeny, that secure the development of semi-allograft embryos. A malignant tumor as a mimic embryo, usurps these secure mechanisms for its own benefit to protect itself against host immunity.
Stage III: The second shift of maternal immunity in the postpartum period: immune rejection of the remaining embryonic cells The second shift in maternal immune reactivity occurs in the postpartum period when selective immune tolerance is replaced by an active immune response against remaining embryonic cells, presumably to cells of the semi-allograft placenta. In mammals, the resuming of a maternal immune response to the remaining embryonic cells in the postpartum period is phylogenetically determined and designed to protect the host against the development of trophoblastic and embryonal tumors. In the case of cancer development, the second shift of host immune reactivity is absent. Host immune tolerance to cells of growing malignant tumors is continuously maintained. Due to this, the host is unable to use its natural immune defense mechanisms against embryonic antigens of cancer cells, which can have fatal consequences for the host. Thus, from the standpoint of developmental biology, to have cancer is tantamount to be gestating with a “bad embryo.”
Mechanisms of the Overcoming of Immune Tolerance to Trophoblastic Cells are Also Effective in the Overcoming of Immune Tolerance to Malignant Cells
Embryo and cancer share similar defense mechanisms that protect them against host-vs.-graft immune responses. In the case of the embryo, the defense mechanism is transient and is restricted by the term of pregnancy. Unlike the embryo during pregnancy, a cancer is protected by such mechanisms permanently.
Nevertheless, hundreds of cases of spontaneous regression of malignant tumors and metastases have been recorded.Citation142-Citation144 To date, this phenomenon remains unexplained. We believe that, in cases of spontaneous regression of cancer, there is a switch from a program of “preserving foreign” tissue in a host to a program of “rejecting foreign” tissue, similar to what occurs in the postpartum period. Understanding this phenomenon would lead to a new direction in future cancer-remission research. It would allow us to move from a passive statement of the self-healing capabilities of a cancer patient to a meaningful modeling of “rejecting foreign” scenarios in patients with cancer in order to radically cure cancer. Ideally that means the removal of the “critical mass” of tumor cells combined with the modeling of the second shift of host immune reactivity against any remaining cancer cells. That is why the encoding of the immune events that occur in the postpartum period has a great importance for the development of new biological anti-cancer therapies of high efficiency.
It is particularly important that the mechanisms of the overcoming of immune tolerance to trophoblastic cells are also effective in the overcoming of immune tolerance to malignant cells. Ingegerd Hellstrom and Karl Erik HellstromCitation145 showed that lymph-node and spleen cells from pregnant BALB/c mice are selectively cytotoxic to cultivated syngeneic tumor cells from BALB/c 3T3 cells (3T3-MSV, 3T3-SV40, and 3T12), as well as to two lines of tumors induced in vivo. Skin fibroblasts and non-transformed 3T3 cells were not similarly affected. The cytotoxic lymphocyte effect could be blocked by serum taken from pregnant mice and from mice carrying methyl-cholanthrene-induced sarcomas. Both the cytotoxic lymphocyte effect and the blocking serum activity were seen already during the first pregnancy and lasted for at least 1 mo subsequent to delivery. The authors have assumed that serum from pregnant mice could “arm” control lymph-node cells and produce a lymphocyte-dependent cytotoxic effect on neoplastic, but not on control, target cells.Citation145 It was found that animals that have given birth, unlike virgin animals, had cytotoxic antibodies against the cells of the tumor induced by SV40 virus. The frequency of SV40 virus-induced tumors was substantially lower in female hamsters that had several previous pregnancies than in virgin females of the same age or in males.Citation146 Mice that had gone through a single pregnancy showed a decreased rate of tumor growth and tumor metastases (Lewis T241 fibrosarcoma in C57BL/6J mice).Citation147 The authors showed that the tumor inhibitory response evoked by fetal cell immunization is due to fetal antigens, and the male specific HY antigen is not responsible for antitumor responses.
Neonatal Vaccination with Embryonic Antigens: A New Way to Overcome “Phylogenetic Immune Tolerance” and Significantly Reduce Cancer Risk
Immunization against cancer using embryonic material has a history of more than 100 years. Oncodevelopmental antigens were reported to have been identified by transplantation as early as 1906 when SchoneCitation148 found that tumor transplants that would kill normal mice would be rejected by mice that had previously been immunized with fetal tissue; immunization with adult tissue was ineffective.Citation149 In the following decades, many reports affirmed the general idea that immunization of an individual with fetal tissue, including placental tissue, can result in immunologic rejection of transplantable tumors, as well as prevention of the development of chemically-induced tumors.Citation150
Pottathil R and Meier H (1977) described the antitumor effects of certain RNAs isolated from murine tumors and embryos. A single i.v. injection of 10 to 30 µg of these RNAs induced necrosis, hemorrhages, and regression of solid tumors in the strain of origin; in pregnant mice it was embryotoxic, causing resorption of the embryos.Citation151 The effects of RNAs were specific, since there were no toxic actions against pregnant mice or mice with tumors.
Khare P, Singh O, Jain, at al. (2012) showed the inhibitory effect of antibodies against human chorionic gonadotropin on the growth of colorectal tumor cells. Anti-hCG antiserum specifically reduced the viability of colorectal cancer cells (CCL-253) and the addition of complement increased in vitro anti-tumor effects. In nude mice implanted with CCL-253 cells, administration of anti-hCG antiserum caused a significant reduction in tumor volume; all treated animals survived, while mortality was observed in control animals.Citation152
Our experiments showed that preventive immunization of C57BL/6 mice with carcinoma 3LL by human chorionic gonadotropin (hCG) as well as by syngeneic placental homogenates significantly decreased the number and volume of lung metastases and increased the number of metastases-free mice. Preliminary immunization of mice with homogenates of syngeneic adult tissues did not affect tumor development.Citation21,Citation153-Citation155 Placental vaccination may function as a multi-epitope vaccine; the body recognizes the placental antigens of this vaccine as foreign, and thus stimulates a cross-reactive humoral and cell-mediated immune response targeting cancer tumor-associated antigens (TAA), as well as proteins that aid in cancer development.Citation156 Zhao B, Wang Y, Wu B, et al. (2013) used placenta-derived heat shock protein gp96, which induces prophylactic anti-tumor T cell responses. Immunization with placental gp96 provided partial protection; long-term (>3 months) anti-tumor immunity against growth of transplantable melanoma or breast tumors in mice, elicited total protection against 7, 12-dimethylbenz(a)-anthracene (DMBA)-induced mammary tumors in rats, and significantly reduced the occurrence and growth of autochthonous breast tumors in HER2 transgenic mice. Placental gp96 activated HER2- and MUC1-specific T cell responses through binding to tumor-associated antigens.Citation157
There have been promising attempts to develop vaccines against the development of cancers of all kinds, the so-called “universal” vaccine, using embryonic stem cells as immunogens.Citation158
Based on the concept of “phylogenetic immune tolerance” to early embryonic antigens, which is due to absence in the host of long-lived immunological memory to these antigens, we anticipated that the immunization of newborns with early embryonic antigens would induce the appearance of the corresponding long-lived immunological memory cells, which in turn would provide lifetime immunity against cancer.
Our anticipation of the enhancing protective efficacy of neonatal vaccination against cancer was supported by data showing high anti-cancer efficacy of neonatal immunization with tumor tissue homogenates. It has been shown that immunization of white inbred mice 1–3 d-old with homogenate of Ehrlich ascites carcinoma (EAC) cells result in the development of strong, long-lasting immunity against EAC cells, sarcoma 37 ascites (S37) cells, and cells in which sarcoma was induced by 20-methylcholanthrene.Citation159,Citation160 After neonatally immunized mice reached 3 mo of age, they were inoculated (i.p.) with EAC cells or S37 cells. The cell-dose threshold, or minimum number of cells required for successful i.p. transplantation was determined for immunized and control (intact) mice. The EAC cell-dose threshold for control mice was 25 cells and the rate of successful inoculations was 100%. In contrast, the EAC cell-dose threshold for immunized mice was 10 000 cells and the successful inoculation rate was 50%. The S37 cell-dose threshold for control mice was 50 cells and the successful inoculation rate was 100%. In contrast the S37 cell-dose threshold for immunized mice was 2500 cells and the successful inoculation rate was 50%. The neonatal immunized mice also displayed significant resistance against methylcholanthrene-induced sarcoma: the number of tumor free mice in the treated group was two times higher than in the control group. Immunization of 3-mo-old mice with homogenates of EAC cells was ineffective against transplantation of ascites tumors and 20-methylcholantrene induced sarcomas.
We consider the development of effective cancer vaccines using tumor-embryo cross-reacting antigens for early-life immunization as a new research strategy aimed at creation of life-long immunity against cancer. Immune responses of human newborns are special and cannot be predicted from those of human adults or animal models. Therefore, understanding and modeling age-specific human immune responses will be vital to the rational design and development of safe and effective cancer prophylactic vaccines for newborns.
Conclusions
There are seven innovative ideas that the Oncogerminative Theory of Tumorigenesis adds to our understanding of cancer biology.
First, it states that the essence of the malignant transformation of the somatic cell is the awakening of the germline cell’s phenotypic properties in this cell. This means that the transformation of a somatic cell into a CSC (an oncogerminative cell) is driven exclusively by those genomic mutations and epigenomic alterations that result in “germinalization” of somatic cells, i.e., that result in their being reprogrammed into pseudo-germline cells. Mimicking the behavior of a fertilized germline cell, the oncogerminative cell achieves its potential immortality by passing through the stages of its life-cycle and developing into a pseudo-blastula-stage embryo, which manifests in the body as a malignant tumor.
Second, it considers tumor formation as a dynamic self-organizing process that mimics a self-organizing process of early embryo development. The theory explains the initiating of cancer as a phenomenon of developmental biology, which we have called a “desperate asexual self-cloning event.” In mammals this phenomenon manifests as cancer development, which represents a desperate attempt of the oncogerminative cell to implement its potential immortality through pseudo-embryogenesis by creation of an endless line of intermediate generations of pseudo-clones that are malignant tumors.
Third, it postulates that the interaction between a malignant tumor and a host should be considered in the context of the interaction between a pseudo-embryo and a host. This means that the diagnostic and therapeutic value of new findings in malignant cells and malignant tumors should be evaluated by a comparative study of similar findings in embryo cells and embryo tissues.
Fourth, it postulates that in order to initiate the development of a metastatic tumor, a circulating CSC must first create a multicellular tumor spheroid, just as a fertilized germ cell creates a multicellular blastocyst-stage embryo before its implantation.
Fifth, the theory explains the body’s immune tolerance to cancer embryonic antigens as a natural “phylogenetic immune tolerance” that underlies maternal immune tolerance to the antigens of an early embryo. We believe that the reason of “phylogenetic immune tolerance” to early embryonic antigens is the absence in the host of long-lived memory cells that can give rise to effector cells against these antigens. Cancer, as a pseudo-embryo that produces embryonic antigens, usurps the phylogenetic immune tolerance for its own benefit.
Sixth, it considers a postnatal immunization with tumor-associated embryonic antigens as a promising approach to overcome immune tolerance to cancer cells by inducing the appearance of the corresponding long-lived immunological memory cells, which in turn will provide lifetime immunity against cancer and significantly reduce cancer risk. Thus, the theory suggests new strategies for effective cancer prevention: the development of effective cancer vaccines using tumor-embryo cross-reacting antigens for early-life immunization aimed at creation of life-long immunity against cancer.
Seventh, the oncogerminative theory of tumorigenesis proposes a new oncogerminative model of cancer development. This model explains the 5 basic stages of tumorigenesis: (1) the reprogramming (i.e., germinalization) of a somatic cell into a pseudo-germline cell that is a CSC; (2) parthenogenetic-like multiplication of CSCs and formation of a multicellular tumor spheroid with a heterogeneous cell population, which is a pseudo-blastocyst-stage-embryo; (3) vascularization of the tumor spheroid and its further invasive growth as a vascularized tumor that mimics the implantation and invasive behavior of the blastocyst-stage embryo; (4) disaggregation of the CSC compartment of the tumor into individual CSCs and their migration into body tissues, which mimics primordial cell migration; (5) development of new generations of metastatic tumors with a modified ratio of malignant to non-malignant cells that underlie tumor progression. All these events occur in the context of natural selective immune tolerance to the embryonic antigens of the cancer, which we have termed “phylogenetic immune tolerance.”
We believe that proposed theory will have a substantial practical impact. It already organizes numerous findings collected to date that directly or indirectly support the theory that tumorigenesis mimics a self-organizing process of early embryo development. If the Oncogerminative Theory of Tumorigenesis is validated, it would help focus the attention and efforts of researchers on the following tasks: (1) Explore the genetic and proteomic patterns of the reprogramming (i.e., germinalization) of a somatic cell into a mimic germline cell and develop gene therapies targeting somatic cells to prevent this reprogramming. (2) Develop targeted therapies to destroy oncogerminative CSCs, the only cells that are able to give rise to malignant tumors. (3) Identify tumor markers indicative of the development of tumor spheroids and their implantation and invasive properties, and develop advanced anti-cancer therapies targeting the formation of tumor spheroids. (4) Explore natural defense mechanisms associated with embryo development that control invasiveness, immune tolerance, and the phenotypic profile of cells, and use these natural defense mechanisms in the development of targeted anti-cancer therapies. (5) Develop advanced anti-cancer vaccines based on embryonic-antigens for neonatal use as preventive vaccines, which are intended to prevent cancer from developing in healthy people. Develop fetal tissue vaccines that are intended to treat an existing cancer by restoring the body’s natural defenses against the cancer. (6) Develop advanced combined therapies that include the removal of a “critical mass” of CSCs followed by the restoration of host immune reactivity against the remaining cancer cells, similar to a maternal host restoring her immunity to the remaining embryonic cells in the postpartum period.
We believe that the Oncogerminative Theory of Tumorigenesis may serve as a road map in cancer research and that using that road map to develop targeted anti-cancer therapies should allow us to prevent, control or eradicate cancer.
Disclosure of Potential Conflicts of Interest
No conflict of interest was disclosed.
Acknowledgments
We thank Dr Karen Bullock, Dr Susan Chubinskaya, and Mr. Earle Martin for their productive comments that helped us improve the contents of the manuscript. We thank Jeremy Fields for helpful suggestions regarding the writing of the manuscript. Additionally we thank Val Treshchov for preparation of the figures.
References
- TakebeN, IvySP. Controversies in cancer stem cells: targeting embryonic signaling pathways. Clin Cancer Res2010; 16:3106 - 12; http://dx.doi.org/10.1158/1078-0432.CCR-09-2934; PMID: 20530695
- MaYL, ZhangP, WangF, MoyerMP, YangJJ, LiuZH, PengJY, ChenHQ, ZhouYK, LiuWJ, et al. Human embryonic stem cells and metastatic colorectal cancer cells shared the common endogenous human microRNA-26b. J Cell Mol Med2011; 15:1941 - 54; http://dx.doi.org/10.1111/j.1582-4934.2010.01170.x; PMID: 20831567
- Virchow R. Die Cellularpathologie in ihrer Begrundung auf physiologische und pathologische Gewebelehre. Berlin: August Hirschwald, (1858).
- Cohnheim J. Vorlesungen uber ellgemine Pathologie. Hirschwald, Berlin, (1877).
- BignoldLP, CoghlanB, JersmannH. Biography on David Paul von Hansemann: Contributions to Oncology. Oncology2007; 159:371
- BeardJ. Embryological aspects and etiology of carcinoma. Lancet1902; 1:1758 - 61; http://dx.doi.org/10.1016/S0140-6736(01)89936-0
- ChanGL. On the nature of oncogenic transformation of cells. Int Rev Cytol1981; 70:101 - 37; http://dx.doi.org/10.1016/S0074-7696(08)61131-6; PMID: 7014504
- ConwayFJ. A proposed model of cancer as the inappropriate expression of non-body introns. J Theor Biol1983; 100:1 - 24; http://dx.doi.org/10.1016/0022-5193(83)90090-5; PMID: 6220182
- FeketeB. An idea on the genesis of the first cancer cell. Med Hypotheses1987; 23:109 - 12; http://dx.doi.org/10.1016/0306-9877(87)90187-3; PMID: 3613999
- ErenpreissJG. Gametogenesis as a molecular model of cancerogenesis: A current view of the embryological theory of cancer. . Proc. Latv. Acad.Sci., Part B.1992; 3:55 - 63
- KnudsonAGJr.. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A1971; 68:820 - 3; http://dx.doi.org/10.1073/pnas.68.4.820; PMID: 5279523
- RenanMJ. How many mutations are required for tumorigenesis? Implications from human cancer data. Mol Carcinog1993; 7:139 - 46; http://dx.doi.org/10.1002/mc.2940070303; PMID: 8489711
- HahnWC, CounterCM, LundbergAS, BeijersbergenRL, BrooksMW, WeinbergRA. Creation of human tumour cells with defined genetic elements. Nature1999; 400:464 - 8; http://dx.doi.org/10.1038/22780; PMID: 10440377
- HanahanD, WeinbergRA. The hallmarks of cancer. Cell2000; 100:57 - 70; http://dx.doi.org/10.1016/S0092-8674(00)81683-9; PMID: 10647931
- HanahanD, WeinbergRA. Hallmarks of cancer: the next generation. Cell2011; 144:646 - 74; http://dx.doi.org/10.1016/j.cell.2011.02.013; PMID: 21376230
- LoboNA, ShimonoY, QianD, ClarkeMF. The biology of cancer stem cells. Annu Rev Cell Dev Biol2007; 23:675 - 99; http://dx.doi.org/10.1146/annurev.cellbio.22.010305.104154; PMID: 17645413
- GhiaurG, GerberJ, JonesRJ. Concise review: Cancer stem cells and minimal residual disease. Stem Cells2012; 30:89 - 93; http://dx.doi.org/10.1002/stem.769; PMID: 22045578
- BaccelliI, TrumppA. The evolving concept of cancer and metastasis stem cells. J Cell Biol2012; 198:281 - 93; http://dx.doi.org/10.1083/jcb.201202014; PMID: 22869594
- VinnitskyVB. [On the nature of host tolerance to cancer] [Russian.]. Exp Oncol1981; 3:3 - 12
- VinnitskyVB. [Oncogerminative Hypothesis of Tumor Growth] [Russian.]. Exp Oncol1989; 21:239 - 42
- Vinnitsky VB, ed. Mosienko MD, Glinsky GV, Surgova TM, Sidorenko MV, Tarakhovsky AM, Ivanova AB, Kartavova NS, Polishchuk LA. Biologiia markerov raka i beremennosti [Biology of markers of cancer and pregnancy] Kiev: Naukova Dumka; 1990. 252 p. Russian.
- VinnitskyVB. Oncogerminative hypothesis of tumor formation. Med Hypotheses1993; 40:19 - 27; http://dx.doi.org/10.1016/0306-9877(93)90191-R; PMID: 8455462
- BonnetD, DickJE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med1997; 3:730 - 7; http://dx.doi.org/10.1038/nm0797-730; PMID: 9212098
- Al-HajjM, WichaMS, Benito-HernandezA, MorrisonSJ, ClarkeMF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A2003; 100:3983 - 8; http://dx.doi.org/10.1073/pnas.0530291100; PMID: 12629218
- ClarkeMF, DickJE, DirksPB, EavesCJ, JamiesonCH, JonesDL, VisvaderJ, WeissmanIL, WahlGM. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res2006; 66:9339 - 44; http://dx.doi.org/10.1158/0008-5472.CAN-06-3126; PMID: 16990346
- SprouffskeK, Athena AktipisC, RadichJP, CarrollM, NedelcuAM, MaleyCC. An evolutionary explanation for the presence of cancer nonstem cells in neoplasms. Evol Appl2013; 6:92 - 101; http://dx.doi.org/10.1111/eva.12030; PMID: 23397439
- JanM, SnyderTM, Corces-ZimmermanMR, VyasP, WeissmanIL, QuakeSR, MajetiR. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med2012; 4:ra118; http://dx.doi.org/10.1126/scitranslmed.3004315; PMID: 22932223
- JanicA, MendizabalL, LlamazaresS, RossellD, GonzalezC. Ectopic expression of germline genes drives malignant brain tumor growth in Drosophila. Science2010; 330:1824 - 7; http://dx.doi.org/10.1126/science.1195481; PMID: 21205669
- MonkM, HoldingC. Human embryonic genes re-expressed in cancer cells. Oncogene2001; 20:8085 - 91; http://dx.doi.org/10.1038/sj.onc.1205088; PMID: 11781821
- Van DorenM. The cell biology of the germ cell life cycle. Curr Opin Cell Biol2010; 22:707 - 8; http://dx.doi.org/10.1016/j.ceb.2010.10.003; PMID: 21093242
- NakamuraA, SeydouxG. Less is more: specification of the germline by transcriptional repression. Development2008; 135:3817 - 27; http://dx.doi.org/10.1242/dev.022434; PMID: 18997110
- BaoS, LeitchHG, GillichA, NicholsJ, TangF, KimS, LeeC, ZwakaT, LiX, SuraniMA. The germ cell determinant Blimp1 is not required for derivation of pluripotent stem cells. Cell Stem Cell2012; 11:110 - 7; http://dx.doi.org/10.1016/j.stem.2012.02.023; PMID: 22770244
- LeitchHG, TangWW, SuraniMA. Primordial germ-cell development and epigenetic reprogramming in mammals. Curr Top Dev Biol2013; 104:149 - 87; http://dx.doi.org/10.1016/B978-0-12-416027-9.00005-X; PMID: 23587241
- SuraniMA. Germ cells: the eternal link between generations. C R Biol2007; 330:474 - 8; http://dx.doi.org/10.1016/j.crvi.2007.03.009; PMID: 17631440
- ErenpreisaJ, CraggMS. Three steps to the immortality of cancer cells: senescence, polyploidy and self-renewal. Cancer Cell Int2013; 13:92; http://dx.doi.org/10.1186/1475-2867-13-92; PMID: 24025698
- ErenpreisaJA, CraggMS, FringesB, SharakhovI, IllidgeTM. Release of mitotic descendants by giant cells from irradiated Burkitt’s lymphoma cell line. Cell Biol Int2000; 24:635 - 48; http://dx.doi.org/10.1006/cbir.2000.0558; PMID: 10964453
- IanziniF, KosmacekEA, NelsonES, NapoliE, ErenpreisaJ, KalejsM, MackeyMA. Activation of meiosis-specific genes is associated with depolyploidization of human tumor cells following radiation-induced mitotic catastrophe. Cancer Res2009; 69:2296 - 304; http://dx.doi.org/10.1158/0008-5472.CAN-08-3364; PMID: 19258501
- ErenpreisaJ, CraggMS. MOS, aneuploidy and the ploidy cycle of cancer cells. Oncogene2010; 29:5447 - 51; http://dx.doi.org/10.1038/onc.2010.310; PMID: 20676137
- GorgoulisVG, ZacharatosP, MariatosG, LiloglouT, KokotasS, KastrinakisN, KotsinasA, AthanasiouA, FoukasP, ZoumpourlisV, et al. Deregulated expression of c-mos in non-small cell lung carcinomas: relationship with p53 status, genomic instability, and tumor kinetics. Cancer Res2001; 61:538 - 49; PMID: 11212247
- LindseySF, ByrnesDM, EllerMS, RosaAM, DabasN, EscandonJ, GrichnikJM. Potential role of meiosis proteins in melanoma chromosomal instability. J Skin Cancer2013; 2013:190109; http://dx.doi.org/10.1155/2013/190109; PMID: 23840955
- TakahashiK, TanabeK, OhnukiM, NaritaM, IchisakaT, TomodaK, YamanakaS. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell2007; 131:861 - 72; http://dx.doi.org/10.1016/j.cell.2007.11.019; PMID: 18035408
- TakahashiK, YamanakaS. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell2006; 126:663 - 76; http://dx.doi.org/10.1016/j.cell.2006.07.024; PMID: 16904174
- HannaJH, SahaK, JaenischR. Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell2010; 143:508 - 25; http://dx.doi.org/10.1016/j.cell.2010.10.008; PMID: 21074044
- TanabeBK, TakahashiK, YamanakaS. Induction of pluripotency by defined factors. Proc. Jpn. Acad. Ser. B2014; 90:83 - 96
- LinSL, ChangDC, Chang-LinS, LinCH, WuDTS, ChenDT, YingSY. Mir-302 reprograms human skin cancer cells into a pluripotent ES-cell-like state. RNA2008; 14:2115 - 24; http://dx.doi.org/10.1261/rna.1162708; PMID: 18755840
- SpergerJM, ChenX, DraperJS, AntosiewiczJE, ChonCH, JonesSB, BrooksJD, AndrewsPW, BrownPO, ThomsonJA. Gene expression patterns in human embryonic stem cells and human pluripotent germ cell tumors. Proc Natl Acad Sci U S A2003; 100:13350 - 5; http://dx.doi.org/10.1073/pnas.2235735100; PMID: 14595015
- LeebM, WalkerR, MansfieldB, NicholsJ, SmithA, WutzA. Germline potential of parthenogenetic haploid mouse embryonic stem cells. Development2012; 139:3301 - 5; http://dx.doi.org/10.1242/dev.083675; PMID: 22912412
- LinSL, ChangDC, LinCH, YingSY, LeuD, WuDT. Regulation of somatic cell reprogramming through inducible mir-302 expression. Nucleic Acids Res2011; 39:1054 - 65; http://dx.doi.org/10.1093/nar/gkq850; PMID: 20870751
- OnderTT, DaleyGQ. microRNAs become macro players in somatic cell reprogramming. Genome Med2011; 3:40; http://dx.doi.org/10.1186/gm256; PMID: 21699744
- FarehM, TurchiL, VirolleV, DebruyneD, AlmairacF, de-la-Forest DivonneS, PaquisP, Preynat-SeauveO, KrauseKH, ChneiweissH, et al. The miR 302-367 cluster drastically affects self-renewal and infiltration properties of glioma-initiating cells through CXCR4 repression and consequent disruption of the SHH-GLI-NANOG network. Cell Death Differ2012; 19:232 - 44; http://dx.doi.org/10.1038/cdd.2011.89; PMID: 21720384
- PeterME. Let-7 and miR-200 microRNAs: guardians against pluripotency and cancer progression. Cell Cycle2009; 8:843 - 52; http://dx.doi.org/10.4161/cc.8.6.7907; PMID: 19221491
- WangX, BelguiseK, O’NeillCF, Sánchez-MorganN, RomagnoliM, EddySF, MinevaND, YuZ, MinC, Trinkaus-RandallV, et al. RelB NF-kappaB represses estrogen receptor alpha expression via induction of the zinc finger protein Blimp1. Mol Cell Biol2009; 29:3832 - 44; http://dx.doi.org/10.1128/MCB.00032-09; PMID: 19433448
- ZwakaTP, ThomsonJA. A germ cell origin of embryonic stem cells?. Development2005; 132:227 - 33; http://dx.doi.org/10.1242/dev.01586; PMID: 15623802
- FujimoriH, ShikanaiM, TeraokaH, MasutaniM, YoshiokaK. Induction of cancerous stem cells during embryonic stem cell differentiation. J Biol Chem2012; 287:36777 - 91; http://dx.doi.org/10.1074/jbc.M112.372557; PMID: 22961983
- Raff RA. The shape of life: genes, development, and the evolution of animal form. University of Chicago Press; 1996. 544 p.
- VasudevanS, SeliE, SteitzJA. Metazoan oocyte and early embryo development program: a progression through translation regulatory cascades. Genes Dev2006; 20:138 - 46; http://dx.doi.org/10.1101/gad.1398906; PMID: 16418480
- NyströmT. Aging in bacteria. Curr Opin Microbiol2002; 5:596 - 601; http://dx.doi.org/10.1016/S1369-5274(02)00367-3; PMID: 12457704
- AckermannM, StearnsSC, JenalU. Senescence in a bacterium with asymmetric division. Science2003; 300:1920 - 1920; http://dx.doi.org/10.1126/science.1083532; PMID: 12817142
- MartínezDE. Mortality patterns suggest lack of senescence in hydra. Exp Gerontol1998; 33:217 - 25; http://dx.doi.org/10.1016/S0531-5565(97)00113-7; PMID: 9615920
- JoplingC, BoueS, Izpisua BelmonteJC. Dedifferentiation, transdifferentiation and reprogramming: three routes to regeneration. Nat Rev Mol Cell Biol2011; 12:79 - 89; http://dx.doi.org/10.1038/nrm3043; PMID: 21252997
- Kirkwood TBL, Holliday R. The evolution of ageing and longevity. Proc R Soc Lond B Biol Sci Series B 1979; 205: 531–46.
- SundaramM, GuernseyDL, RajaramanMM, RajaramanR. Neosis: a novel type of cell division in cancer. Cancer Biol Ther2004; 3:207 - 18; http://dx.doi.org/10.4161/cbt.3.2.663; PMID: 14726689
- RajaramanR, RajaramanMM, RajaramanSR, GuernseyDL. Neosis--a paradigm of self-renewal in cancer. Cell Biol Int2005; 29:1084 - 97; http://dx.doi.org/10.1016/j.cellbi.2005.10.003; PMID: 16316756
- ErenpreisaJ, CraggMS. Cancer: a matter of life cycle?. Cell Biol Int2007; 31:1507 - 10; http://dx.doi.org/10.1016/j.cellbi.2007.08.013; PMID: 17936649
- LokkiJ, SuomalainenE, SauraA, LankinenP. Genetic polymorphism and evolution in parthenogenetic animals. II. Diploid and polyploid Solenobia triquetrella (Lepidoptera: Psychidae). Genetics1975; 79:513 - 25; PMID: 1126629
- SuomalainenE, SauraA, LokkiJ, TeeriT. Genetic polymorphism and evolution in parthenogenetic animals : Part 9: Absence of variation within parthenogenetic aphid clones. Theor Appl Genet1980; 57:129 - 32; http://dx.doi.org/10.1007/BF00253887; PMID: 24302494
- White MJD. Animal Cytology and Evolution. 3rd ed. University Press, Cambridge. 1973. 468 p.
- VranaKE, HippJD, GossAM, McCoolBA, RiddleDR, WalkerSJ, WettsteinPJ, StuderLP, TabarV, CunniffK, et al. Nonhuman primate parthenogenetic stem cells. Proc Natl Acad Sci U S A2003; 100:Suppl 111911 - 6; http://dx.doi.org/10.1073/pnas.2034195100; PMID: 14504386
- RevazovaES, TurovetsNA, KochetkovaOD, KindarovaLB, KuzmichevLN, JanusJD, PryzhkovaMV. Patient-specific stem cell lines derived from human parthenogenetic blastocysts. Cloning Stem Cells2007; 9:432 - 49; http://dx.doi.org/10.1089/clo.2007.0033; PMID: 17594198
- RevazovaES, TurovetsNA, KochetkovaOD, AgapovaLS, SebastianJL, PryzhkovaMV, SmolnikovaVI, KuzmichevLN, JanusJD. HLA homozygous stem cell lines derived from human parthenogenetic blastocysts. Cloning Stem Cells2008; 10:11 - 24; http://dx.doi.org/10.1089/clo.2007.0063; PMID: 18092905
- PeytonA. Tumeurs des glandes genitales. Bull Assoc Fr Etud Cancer1922; 11:215 - 74
- BeutnerR. Ober die Ursache der Neoplasie. Ztschr. Krebsforsch.1927; 24:99 - 116; http://dx.doi.org/10.1007/BF02176093
- Erenpreiss JG. Current concepts of malignant growth. Part A. From a normal cell to cancer. Riga: Zvaigzne Publishers; 1993. 192 p.
- OliveiraFG, DozortsevD, DiamondMP, FracassoA, AbdelmassihS, AbdelmassihV, GonçalvesSP, AbdelmassihR, NagyZP. Evidence of parthenogenetic origin of ovarian teratoma: case report. Hum Reprod2004; 19:1867 - 70; http://dx.doi.org/10.1093/humrep/deh345; PMID: 15192066
- StevensLC. Teratocarcinogenesis and spontaneous parthenogenesis in mice. Results Probl Cell Differ1980; 11:265 - 74; http://dx.doi.org/10.1007/978-3-540-38267-6_34; PMID: 7444199
- HiraoY, EppigJJ. Parthenogenetic development of Mos-deficient mouse oocytes. Mol Reprod Dev1997; 48:391 - 6; http://dx.doi.org/10.1002/(SICI)1098-2795(199711)48:3<391::AID-MRD13>3.0.CO;2-Z; PMID: 9322252
- MostofiFK. Histological Typing of Testis Tumours. International Histological Classification of Tumours. Br J Cancer1977; 15 - 31
- BotchanA, HauserR, YogevL, GamzuR, PazG, LessingJB, YavetzH. Testicular cancer and spermatogenesis. Hum Reprod1997; 12:755 - 8; http://dx.doi.org/10.1093/humrep/12.4.755; PMID: 9159438
- Damajanov I. Overview. In Oosterhuis, JW, Walt H, and Damajanov I. (eds), Pathobiology of Human Germ Cell Neoplasia. Recent Results in Cancer Research. Springer Verlag, Berlin-Heidelberg, (1991): 1–21.
- KimJB, ZaehresH, WuG, GentileL, KoK, SebastianoV, Araúzo-BravoMJ, RuauD, HanDW, ZenkeM, et al. Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature2008; 454:646 - 50; http://dx.doi.org/10.1038/nature07061; PMID: 18594515
- KimJB, ZaehresH, WuG, GentileL, KoK, SebastianoV, Araúzo-BravoMJ, RuauD, HanDW, ZenkeM, et al. Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature2008; 454:646 - 50; http://dx.doi.org/10.1038/nature07061; PMID: 18594515
- LiuC, MaZ, HouJ, ZhangH, LiuR, WuW, LiuW, LuY. Germline traits of human hepatoblastoma cells associated with growth and metastasis. Biochem Biophys Res Commun2013; 437:120 - 6; http://dx.doi.org/10.1016/j.bbrc.2013.06.050; PMID: 23800414
- SauraA, LokkiJ, SuomalainenE. Origin of polyploidy in parthenogenetic weevils. J Theor Biol1993; 163:449 - 56; http://dx.doi.org/10.1006/jtbi.1993.1130
- MosieniakG, SikoraE. Polyploidy: the link between senescence and cancer. Curr Pharm Des2010; 16:734 - 40; http://dx.doi.org/10.2174/138161210790883714; PMID: 20388083
- LiQ, DangCV. c-Myc overexpression uncouples DNA replication from mitosis. Mol Cell Biol1999; 19:5339 - 51; PMID: 10409725
- MintzB. Genetic Mosaicism in Adult Mice of Quadriparental Lineage. Science1965; 148:1232 - 3; http://dx.doi.org/10.1126/science.148.3674.1232; PMID: 14280005
- EakinGS, HadjantonakisAK. Production of chimeras by aggregation of embryonic stem cells with diploid or tetraploid mouse embryos. Nat Protoc2006; 1:1145 - 53; http://dx.doi.org/10.1038/nprot.2006.173; PMID: 17406396
- SutherlandRM, McCredieJA, InchWR. Growth of multicell spheroids in tissue culture as a model of nodular carcinomas. J Natl Cancer Inst1971; 46:113 - 20; PMID: 5101993
- MaHL, JiangQ, HanS, WuY, Cui TomshineJ, WangD, GanY, ZouG, LiangXJ. Multicellular tumor spheroids as an in vivo-like tumor model for three-dimensional imaging of chemotherapeutic and nano material cellular penetration. [My paper]Mol Imaging2012; 11:487 - 98; PMID: 23084249
- CrawfordS, DiamondD, BrustolonL, PenarretaR. Effect of increased extracellular ca on microvesicle production and tumor spheroid formation. Cancer Microenviron2010; 4:93 - 103; http://dx.doi.org/10.1007/s12307-010-0049-0; PMID: 21505564
- WuLY, Di CarloD, LeeLP. Microfluidic self-assembly of tumor spheroids for anticancer drug discovery. Biomed Microdevices2008; 10:197 - 202; http://dx.doi.org/10.1007/s10544-007-9125-8; PMID: 17965938
- Mueller-KlieserW. Tumor biology and experimental therapeutics. Crit Rev Oncol Hematol2000; 36:123 - 39; http://dx.doi.org/10.1016/S1040-8428(00)00082-2; PMID: 11033302
- AbramovitchR, MeirG, NeemanM. Neovascularization induced growth of implanted C6 glioma multicellular spheroids: magnetic resonance microimaging. Cancer Res1995; 55:1956 - 62; PMID: 7537176
- ZabierowskiSE, HerlynM. Melanoma stem cells: the dark seed of melanoma. J Clin Oncol2008; 26:2890 - 4; http://dx.doi.org/10.1200/JCO.2007.15.5465; PMID: 18539969
- AillesLE, WeissmanIL. Cancer stem cells in solid tumors. Curr Opin Biotechnol2007; 18:460 - 6; http://dx.doi.org/10.1016/j.copbio.2007.10.007; PMID: 18023337
- Pierce GB. Cellular heterogeneity of cancers. In Chemical Carcinogenesis: Part B, P.O.P. Ts'o and J.A. Di Paolo (eds.) New York: Marcel Dekker Inc. 1974; 463-472.
- FrostP, KerbelRS. On a possible epigenetic mechanism(s) of tumor cell heterogeneity. The role of DNA methylation. Cancer Metastasis Rev1983; 2:375 - 8; http://dx.doi.org/10.1007/BF00048568; PMID: 6375860
- Savre-TrainI, GollahonLS, HoltSE. Clonal heterogeneity in telomerase activity and telomere length in tumor-derived cell lines. Proc Soc Exp Biol Med2000; 223:379 - 88; http://dx.doi.org/10.1046/j.1525-1373.2000.22354.x; PMID: 10721008
- PierceGB, WallaceC. Differentiation of malignant to benign cells. Cancer Res1971; 31:127 - 34; PMID: 5545265
- DotsikasG, KonowalchukT, MajorPP, KovacPE, WardGK, StewartSS, PriceGB, ElhilaliMM, MackillopWJ. Cellular heterogeneity in normal and neoplastic human urothelium: a study using murine monoclonal antibodies. Br J Cancer1987; 56:439 - 44; http://dx.doi.org/10.1038/bjc.1987.220; PMID: 3689660
- MottlaGL, AdelmanMR, HallJL, GindoffPR, StillmanRJ, JohnsonKE. Lineage tracing demonstrates that blastomeres of early cleavage-stage human pre-embryos contribute to both trophectoderm and inner cell mass. Hum Reprod1995; 10:384 - 91; PMID: 7769068
- FolkmanJ. Influence of geometry on growth of normal and malignant cells. Adv Pathobiol1976; 4:12 - 28; PMID: 1023766
- StevensLC, VarnumDS. The development of teratomas from parthenogenetically activated ovarian mouse eggs. Dev Biol1974; 37:369 - 80; http://dx.doi.org/10.1016/0012-1606(74)90155-9; PMID: 4826282
- AllanRW, AlgoodCB, ShihIeM. Metastatic epithelioid trophoblastic tumor in a male patient with mixed germ-cell tumor of the testis. Am J Surg Pathol2009; 33:1902 - 5; http://dx.doi.org/10.1097/PAS.0b013e3181c03792; PMID: 19898219
- Lee P, Pincus MR, McPherson RA. Diagnosis and management of cancer using serologic tumor markers. In: McPherson RA, Pincus MR, eds. Henry’s Clinical Diagnosis and Management by Laboratory Methods. 21st ed. Philadelphia, Pa: Saunders Elsevier; 2007: 1353-1366.
- SurgovaTM, VinnitskyVB, SidorenkoMV. Protein markers of pregnancy and cancer. Advances in Modern Biology1989; 107:418 - 34
- YagelS, ParharRS, JeffreyJJ, LalaPK. Normal nonmetastatic human trophoblast cells share in vitro invasive properties of malignant cells. J Cell Physiol1988; 136:455 - 62; http://dx.doi.org/10.1002/jcp.1041360309; PMID: 3170642
- KhoshyomnS, PenarPL, McBrideWJ, TaatjesDJ. Four-dimensional analysis of human brain tumor spheroid invasion into fetal rat brain aggregates using confocal scanning laser microscopy. J Neurooncol1998; 38:1 - 10; http://dx.doi.org/10.1023/A:1005758626348; PMID: 9540052
- BasogluA, SengulAT, BuyukkarabacakYB, YetimTD, YildizL. Pure yolk-sac tumor of the lung. Asian Cardiovasc Thorac Ann2008; 16:410 - 1; http://dx.doi.org/10.1177/021849230801600514; PMID: 18812352
- FlamantF, SchwartzL, DelonsE, CaillaudJM, HartmannO, LemerleJ. Nonseminomatous malignant germ cell tumors in children. Multidrug therapy in Stages III and IV. Cancer1984; 54:1687 - 91; http://dx.doi.org/10.1002/1097-0142(19841015)54:8<1687::AID-CNCR2820540833>3.0.CO;2-U; PMID: 6089996
- WeidnerN. Germ-cell tumors of the mediastinum. Semin Diagn Pathol1999; 16:42 - 50; PMID: 10355653
- KaufmanMH, RobertsonEJ, HandysideAH, EvansMJ. Establishment of pluripotential cell lines from haploid mouse embryos. Embryol. exp. Morph.1983; 73:249 - 61
- GeijsenN, HoroschakM, KimK, GribnauJ, EgganK, DaleyGQ. Derivation of embryonic germ cells and male gametes from embryonic stem cells. Nature2004; 427:148 - 54; http://dx.doi.org/10.1038/nature02247; PMID: 14668819
- ChambersI, SmithA. Self-renewal of teratocarcinoma and embryonic stem cells. Oncogene2004; 23:7150 - 60; http://dx.doi.org/10.1038/sj.onc.1207930; PMID: 15378075
- Dyban AP. Rannee razvitie mlekopitayushchikh [Early Development in Mammals], Leningrad: Nauka; 1988. 228 p. Russian.
- LehmanJM, SpeersWC, SwartzendruberDE, PierceGB. Neoplastic differentiation: characteristics of cell lines derived from a murine teratocarcinoma. J Cell Physiol1974; 84:13 - 27; http://dx.doi.org/10.1002/jcp.1040840103; PMID: 4858519
- Logothetou-RellaH. Description of primordial germ cells, oogonia, oocytes and embryo-like growth in squash preparations of tissues from hematological malignancies. Histol Histopathol1996; 11:965 - 84; PMID: 8930640
- AubertG, LansdorpPM. Telomeres and aging. Physiol Rev2008; 88:557 - 79; http://dx.doi.org/10.1152/physrev.00026.2007; PMID: 18391173
- KimNW, PiatyszekMA, ProwseKR, HarleyCB, WestMD, HoPL, CovielloGM, WrightWE, WeinrichSL, ShayJW. Specific association of human telomerase activity with immortal cells and cancer. Science1994; 266:2011 - 5; http://dx.doi.org/10.1126/science.7605428; PMID: 7605428
- SimpsonAJG, CaballeroOL, JungbluthA, ChenYT, OldLJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer2005; 5:615 - 25; http://dx.doi.org/10.1038/nrc1669; PMID: 16034368
- MorrisonSJ, KimbleJ. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature2006; 441:1068 - 74; http://dx.doi.org/10.1038/nature04956; PMID: 16810241
- ScaffidiP, MisteliT. In vitro generation of human cells with cancer stem cell properties. Nat Cell Biol2011; 13:1051 - 61; http://dx.doi.org/10.1038/ncb2308; PMID: 21857669
- ZhangS, Mercado-UribeI, LiuJ. Tumor stroma and differentiated cancer cells can be originated directly from polyploid giant cancer cells induced by paclitaxel. Int J Cancer2014; 134:508 - 18; http://dx.doi.org/10.1002/ijc.28319; PMID: 23754740
- LacroixM, LeclercqG. Relevance of breast cancer cell lines as models for breast tumours: an update. Breast Cancer Res Treat2004; 83:249 - 89; http://dx.doi.org/10.1023/B:BREA.0000014042.54925.cc; PMID: 14758095
- LiJ, WangK, JensenTD, LiS, BolundL, WiufC. Tumor heterogeneity in neoplasms of breast, colon, and skin. BMC Res Notes2010; 3:321; http://dx.doi.org/10.1186/1756-0500-3-321; PMID: 21108813
- KongD, LiY, WangZ, SarkarFH. Cancer Stem Cells and Epithelial-to-Mesenchymal Transition (EMT)-Phenotypic Cells: Are They Cousins or Twins?. Cancers (Basel)2011; 3:716 - 29; http://dx.doi.org/10.3390/cancers30100716; PMID: 21643534
- ThieryJP, SleemanJP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol2006; 7:131 - 42; http://dx.doi.org/10.1038/nrm1835; PMID: 16493418
- ThieryJP, AcloqueH, HuangRY, NietoMA. Epithelial-mesenchymal transitions in development and disease. Cell2009; 139:871 - 90; http://dx.doi.org/10.1016/j.cell.2009.11.007; PMID: 19945376
- KalluriR, WeinbergRA. The basics of epithelial-mesenchymal transition. J Clin Invest2009; 119:1420 - 8; http://dx.doi.org/10.1172/JCI39104; PMID: 19487818
- LiR, LiangJ, NiS, ZhouT, QingX, LiH, HeW, ChenJ, LiF, ZhuangQ, et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell2010; 7:51 - 63; http://dx.doi.org/10.1016/j.stem.2010.04.014; PMID: 20621050
- RichardsonBE, LehmannR. Mechanisms guiding primordial germ cell migration: strategies from different organisms. Nat Rev Mol Cell Biol2010; 11:37 - 49; http://dx.doi.org/10.1038/nrm2815; PMID: 20027186
- MamsenLS, BrøchnerCB, ByskovAG, MøllgardK. The migration and loss of human primordial germ stem cells from the hind gut epithelium towards the gonadal ridge. Int J Dev Biol2012; 56:771 - 8; http://dx.doi.org/10.1387/ijdb.120202lm; PMID: 23417399
- ManiSA, GuoW, LiaoMJ, EatonEN, AyyananA, ZhouAY, BrooksM, ReinhardF, ZhangCC, ShipitsinM, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell2008; 133:704 - 15; http://dx.doi.org/10.1016/j.cell.2008.03.027; PMID: 18485877
- ChafferCL, WeinbergRA. A perspective on cancer cell metastasis. Science2011; 331:1559 - 64; http://dx.doi.org/10.1126/science.1203543; PMID: 21436443
- HofmannMC, MillánJL. Developmental expression of alkaline phosphatase genes; reexpression in germ cell tumours and in vitro immortalized germ cells. Eur Urol1993; 23:38 - 44, discussion 45; PMID: 8386657
- ChangTC, WangJK, HungMW, ChiaoCH, TsaiLC, ChangGG. Regulation of the expression of alkaline phosphatase in a human breast-cancer cell line. Biochem J1994; 303:199 - 205; PMID: 7945240
- LiottaLA, StrackeML. Tumor invasion and metastases: biochemical mechanisms. Cancer Treat Res1988; 40:223 - 38; http://dx.doi.org/10.1007/978-1-4613-1733-3_10; PMID: 2908652
- ZhangJ, TamWL, TongGQ, WuQ, ChanHY, SohBS, LouY, YangJ, MaY, ChaiL, et al. Sall4 modulates embryonic stem cell pluripotency and early embryonic development by the transcriptional regulation of Pou5f1. Nat Cell Biol2006; 8:1114 - 23; http://dx.doi.org/10.1038/ncb1481; PMID: 16980957
- CaoD, HumphreyPA, AllanRW. SALL4 is a novel sensitive and specific marker for metastatic germ cell tumors, with particular utility in detection of metastatic yolk sac tumors. Cancer2009; 115:2640 - 51; http://dx.doi.org/10.1002/cncr.24308; PMID: 19365862
- CalcagnoAM, SalcidoCD, GilletJP, WuCP, FostelJM, MumauMD, GottesmanM. M, Varticovski L, Ambudkar SV. Prolonged Drug Selection of Breast Cancer Cells and Enrichment of Cancer Stem Cell Characteristics. JNCI J. Natl. Cancer Inst.2010; 102:1637 - 52
- MedawarP. Some immunological and endocrinological problems raised by the evolution of viviparity in vertebrates. Symp Soc Exp Biol1953; 7:320 - 38
- ColeWH. Spontaneous regression of cancer: the metabolic triumph of the host?. Ann N Y Acad Sci1974; 230:111 - 41; http://dx.doi.org/10.1111/j.1749-6632.1974.tb14441.x; PMID: 4362919
- StewartFW. Experiences in spontaneous regression of neoplastic disease in man. Tex Rep Biol Med1952; 10:239 - 53; PMID: 14922277
- ChallisGB, StamHJ. The spontaneous regression of cancer. A review of cases from 1900 to 1987. Acta Oncol1990; 29:545 - 50; http://dx.doi.org/10.3109/02841869009090048; PMID: 2206563
- HellströmI, HellströmKE. Cytotoxic effect of lymphocytes from pregnant mice on cultivated tumor cells. I. Specificity, nature of effector cells and blocking by serum. Int J Cancer1975; 15:1 - 16; http://dx.doi.org/10.1002/ijc.2910150102; PMID: 1168624
- VendrovEL. Effect of single and repeated pregnancy on frequency of origin of primary tumors induced by SV40 virus in syrian hamsters. Bull Exp Biol Med1977; 84:1620 - 2
- GautamS, DeodharSD. T-cell-mediated antitumor immune response induced by oncofetal antigens. J Natl Cancer Inst1981; 67:939 - 45; PMID: 6974275
- SchöneG. Untersuchungen uber Kakzinomimmunitat bei Mausen. Munch Med Wochenschr1906; 53:2517 - 9
- SellS, BeckerFF, LeffertHL, WatabeL. Expression of an oncodevelopmental gene product (alpha-fetoprotein) during fetal development and adult oncogenesis. Cancer Res1976; 36:4239 - 49; PMID: 61804
- TingCC, GrantJP. Humoral antibody response and tumor transplantation resistance elicited by fetal tissues in mice. J Natl Cancer Inst1976; 56:401 - 4; PMID: 1255770
- PottathilR, MeierH. Antitumor effects of RNA isolated from murine tumors and embryos. Cancer Res1977; 37:3280 - 6; PMID: 577892
- KhareP, SinghO, JainSK, JavedS, PalR. Inhibitory effect of antibodies against human chorionic gonadotropin on the growth of colorectal tumour cells. Indian J Biochem Biophys2012; 49:92 - 6; PMID: 22650005
- VinnitskyVB, MosienkoMD, RyabukhaVN. [Effect of antiserum to human chorionic gonadotropin on the viability and tumorigenicity of malignant cells and oncospheroids] [Russian.]. Exp Oncol1993; 15:51 - 6
- VinnitskyVB, MosienkoMD, RyabukhaVN, PyaskovskayaON. [Decrease in tumorigenic potency of malignant cells after the treatment with antiserum against human chorionic gonadotropin and its beta-subunit] [Russian.]. Exp Oncol1997; 19:44 - 9
- MosienkoMD, RyabukhaVN, PyaskovskayaON, VinnitskyVB. [Anti-tumor activity of preventive immunization of mice C57BL/6 with carcinoma Lewis against placental tissue] [Russian.]. Exp Oncol2001; 23:278 - 81
- HarandiA. Immunoplacental therapy, a potential multi-epitope cancer vaccine. Med Hypotheses2006; 66:1182 - 7; http://dx.doi.org/10.1016/j.mehy.2005.12.011; PMID: 16439066
- ZhaoB, WangY, WuB, LiuS, WuE, FanH, GuiM, ChenL, LiC, JuY, et al. Placenta-derived gp96 as a multivalent prophylactic cancer vaccine. Sci Rep2013; 3:1947; http://dx.doi.org/10.1038/srep01947; PMID: 23739295
- BrewerBG, MitchellRA, HarandiA, EatonJW. Embryonic vaccines against cancer: an early history. Exp Mol Pathol2009; 86:192 - 7; http://dx.doi.org/10.1016/j.yexmp.2008.12.002; PMID: 19171137
- KazminSD, TodorIN. Effect of postnatal immunization on the ascetic tumors transplantation and chemical cancerogenesis induced by 20-methylcholantrene. Intl. J. Med. Biol. Environ.2001; 29:159 - 64
- KazminSD, TodorIN, ChekhunVE. Antitumor resistance formation in animals in early neonatal period. J Exp Clin Cancer Res2007; 26:83 - 90; PMID: 17550136