
Abstract
We investigated the impact of a diabetic state with hyperglycemia on morphometry of β cell mitochondria and modifying influence of a K+-ATP channel opener and we related in vivo findings with glucose effects in vitro. For in vivo experiments islets from syngeneic rats were transplanted under the kidney capsule to neonatally streptozotocin-diabetic or non-diabetic recipients. Diabetic recipients received vehicle, or tifenazoxide (NN414), intragastrically for 9 weeks. Non-diabetic rats received vehicle. Transplants were excised 7 d after cessation of treatment (wash-out) and prepared for electron microscopy. Morphological parameters were measured from approx. 25,000 mitochondria. Rat islets were cultured in vitro for 2–3 weeks at 27 or 11 (control) mmol/l glucose. Transplants to diabetic rats displayed decreased numbers of mitochondria (-31%, p < 0.05), increased mitochondrial volume and increased mitochondrial outer surface area, p < 0.001. Diabetes increased variability in mitochondrial size with frequent appearance of mega-mitochondria. Tifenazoxide partly normalized diabetes-induced effects, and mega-mitochondria disappeared. Long-term culture of islets at 27 mmol/l glucose reproduced the in vivo morphological abnormalities. High-glucose culture was also associated with reduced ATP and ADP contents, reduced oxygen consumption, reduced signaling by MitoTracker Red and reduction of mitochondrial proteins (complexes I–IV), OPA 1 and glucose-induced insulin release.
We conclude that (1) a long-term diabetic state leads to a reduced number of mitochondria and to distinct morphological abnormalities which are replicated by high glucose in vitro; (2) the morphological abnormalities are coupled to dysfunction; (3) K+-ATP channel openers may have potential to partly reverse glucose-induced effects.
Introduction
Glucose-induced insulin secretion depends on oxidative metabolism.Citation1,Citation2 The crucial importance of mitochondrial function has been highlighted by investigations in mitochondrial DNA-depleted INS-1 cells.Citation3,Citation4 In humans rare mutations of mitochondrial DNA reduce glucose-induced insulin secretion and lead to “mitochondrial diabetes.”Citation5 In type 2 diabetes—the most common form of the disease—polymorphisms of mitochondrial genes such as TFB1M, associate with decreased insulin secretion and risk of type 2 diabetes.Citation6 Furthermore, mitochondrial abnormalities have been observed in islets from type 2 diabetic subjects.Citation7
Not only a genetic background but also a diabetic milieu with chronic hyperglycemia may adversely affect mitochondria. Such influence could be responsible for the progression of insulin-independent diabetes with declining capacity for insulin release during the course of the disease. Indeed, a report demonstrates that deterioration of β-cell function with time can occur in parallel with mitochondrial dysfunction and morphometric abnormalities.Citation8
However, evidence is equivocal for an effect of the diabetic state and specifically for chronically elevated glucose. Most previous studies in animal models of diabetes did not distinguish between a role of the genetic background of the experimental model used vis-à-vis an effect of the diabetic state. Also few studies, whether in vivo or in vitro, have reported results in quantitative terms. Quantitative morphometry from in vivo studies are important since they yield robust data that are apt to reflect mitochondrial function.Citation9 Qualitative evaluations are not sufficient, since they fail to take into account the increasingly acknowledged large variability and plasticity of normal mitochondria.Citation10 Further, quantitative data are needed in order to evaluate any modifying effect of pharmacological agents that interact with β-cells.
Our primary aim was to assess the impact of a diabetic milieu with hyperglycemia on quantitative mitochondrial morphometry. To this end we transplanted rat pancreatic islets to diabetic recipients and measured mitochondrial numbers and morphometry (see for in vivo study design and Materials and Methods). We then tested for and found reproducibility of morphological effects by elevated glucose in an in vitro system. Upon finding reproducibility we next tested for associations with mitochondrial dysfunction.
Figure 1. In vivo study design.
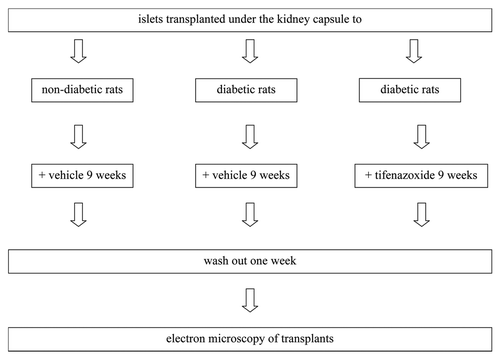
A second aim was to investigate possible corrective effects of K+-ATP channel openers (tifenazoxide). The rationale for this part of the study was the protective effects by such agents on glucose-induced insulin secretion during chronic hyperglycemia that we have previously documented.Citation11 We demonstrate moderate but significant corrective effects on morphometry in the transplantation model.
Results
Transplantation study
Blood glucose
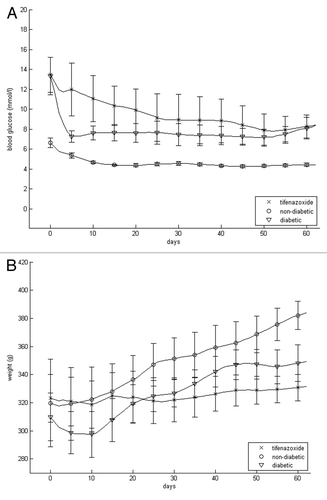
Trans-plantation moderately but swiftly decreased blood glucose in the diabetic-vehicle treated rats, presumably because of the added β-cell mass (). Such a rapid decline was not evident in tifenazoxide-treated rats, probably because of the blocking effect of this compound on insulin secretion. All in all, moderate hyperglycemia prevailed. The mean blood glucose during the treatment period in diabetic rats treated with vehicle was 7.7 ± 1.0, in the tifenazoxide group 9.5 ± 1.4 mmol/l and in the non-diabetic control group 4.6 ± 0.5 mmol/l. Blood glucose did not change significantly as a result of the 7 d of wash out, being 8.3 ± 1.1 in diabetic rats treated with vehicle, 9.7 ± 1.9 in tifenazoxide-treated and 4.1 ± 0.1 mmol/l in non-diabetic controls.
Figure 2. Blood glucose (A) and body weight (B) of transplanted rats that were non-diabetic and vehicle-treated, diabetic and vehicle-treated or diabetic and tifenazoxide-treated. There were eight rats in each group. Treatment was removed after 9 weeks i.e., after day 63.
Body weight
Body weight increased less in the vehicle-treated diabetic rats than in non-diabetic recipients (). Body weight did not increase appreciably in tifenazoxide-treated diabetic rats. After the 7 d wash out period body weight had increased 2% in the vehicle-treated diabetic rats, 5% in tifenazoxide-treated diabetic rats and 3% in the vehicle-treated non-diabetic rats.
Quantitative morphometric analysis: diabetes induces fewer, larger and more variably sized mitochondria
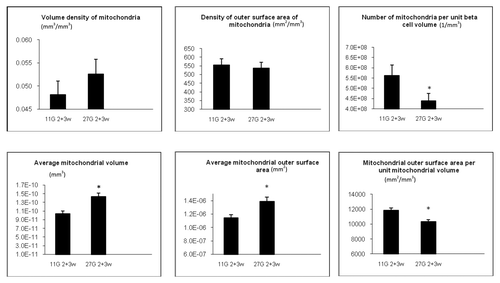
Quantitative morphometric analysis was performed by electron microscopy and primary and secondary parameters were calculated ( and Materials and Methods). The volume of mitochondria per unit β-cell volume (VVM) was not significantly affected in vehicle-treated diabetic rats vs. non-diabetic rats (, upper left graph). The outer surface area of mitochondria per unit β-cell volume (SVM) was however decreased by 12 percent by the diabetic state (, upper middle graph).
Table 1. Morphometric parameters of islets cell mitochondria
Figure 3. Quantitative morphology of primary (upper panel) and secondary (lower panel) mitochondrial parameters. Data were collected from 22 sections per islet graft. There were eight transplants per group. Totally 528 EM sections were evaluated (176 from each treatment group i.e., approx. 25,000 mitochondria altogether). *p < 0.05 vs. mitochondria from transplants to non-diabetic animals, †p < 0.05 vs. mitochondria from transplants to vehicle-treated diabetic animals. NN414 = tifenazoxide.
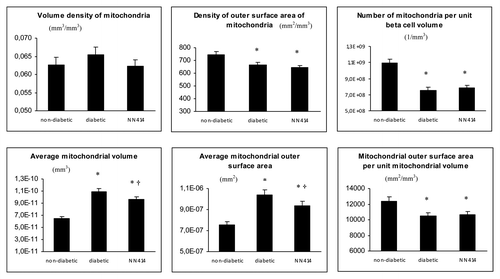
Most importantly the number of mitochondria per unit β-cell volume (NVM) was decreased (by 31%) in vehicle-treated diabetic rats compared with non-diabetic rats, p < 0.05 (, upper right graph).
We considered the possibility that the nucleus can occupy variable amounts of the cytoplasm and thereby influence the afore-mentioned results. We therefore calculated the cytoplasmic area of the cells. The percent of cytoplasmic to nuclear area was increased in transplants to diabetic recipients vs. transplants to non-diabetic recipients (92.5 ± 2.0 vs. 85.5 ± 2.9%, p < 0.05) whereas transplants to tifenazoxide-treated rats seemed intermediate (89.4 ± 2.7% not significant). Expressing our data per cytoplasmic area would therefore enhance rather than decrease the difference described above.
From the above given primary parameters secondary ones were calculated. The diabetic state led to larger mitochondria as demonstrated by an increase in average mitochondrial volume (VVM/NVM), p < 0.05, and average mitochondrial outer surface area (SVM/NVM), p < 0.05 (, lower left and lower middle graphs). Conversely, the mitochondrial outer surface area per unit mitochondrial volume (SVM/VVM) was smaller in diabetic vs. non-diabetic rats (, lower right graph). This indicates that there was a change in shape from elongated to more round mitochondria in diabetic vs. non-diabetic vehicle-treated animals. Examples of round, very large mitochondria (megamitochondria) are given in .
Figure 4. Electron microscopy showing representative sections of β-cells from transplanted islets to non-diabetic, vehicle-treated (A), diabetic, vehicle-treated (B and D) and diabetic, tifenazoxide-treated (C) rats. (E) shows an example of a likely fusion event in a section from a diabetic vehicle-treated rat. In (B) black arrows = mitochondria. In (C) black arrows = mature granule and white arrows = immature granule.
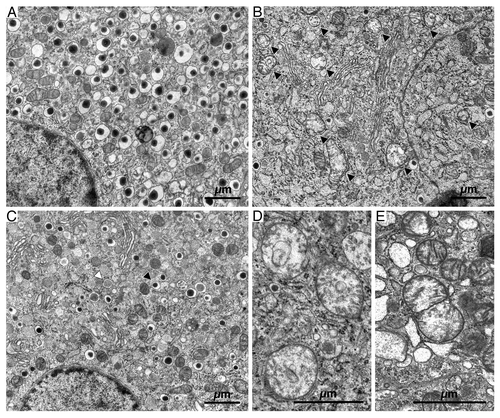
As evidenced from we observed mitochondria of at least 2–3 fold increased size and with reduced density in many sections of grafts in vehicle-treated diabetic animals. These observations (as well as the influence of tifenazoxide, see below) are quantified in . The variation in average mitochondrial volume and outer surface area was larger in grafts from vehicle-treated diabetic vs. non-diabetic-treated animals.
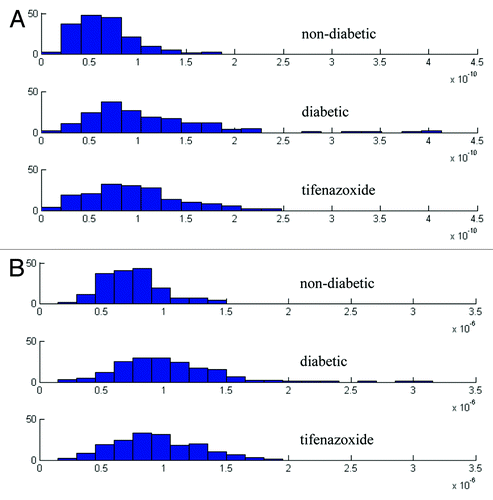
Figure 5. Frequency distribution of average mitochondrial volume (A) and average mitochondrial outer surface area (B).
Treatment with tifenazoxide partially corrects effects by the diabetic milieu
Previous exposure to tifenazoxide, (i.e., long-term effect remaining after wash-out), did not significantly affect any of the primary parameters. Of secondary parameters, tifenazoxide-treatment of the diabetic rats partly but significantly normalized the diabetes-induced differences in average mitochondrial volume and average mitochondrial outer surface area, p < 0.05 ( vs. 4C and , left and middle lower graphs). Tifenazoxide did not correct the decrease in average mitochondrial outer surface area per unit mitochondrial volume (SVM/VVM) that was seen in vehicle-treated diabetic rats (, lower right graph). Tifenazoxide reduced the variability of mitochondrial size by eliminating megamitochondria ().
Morphometrics and number of insulin granules
Insulin release and synthesis is dependent on mitochondrial function. Hence it was of interest to assess here insulin granules in relation to the diabetic state and its effects on mitochondria. Insulin granules (immature and mature) were quantified according to volume of granules per unit β-cell volume (VVM), number of granules per unit β-cell volume (NVM) and average volume (VVM/NVM). All parameters were decreased in β-cells from grafts in diabetic animals (mature granules, Fig. S2A–C). There was a significant negative correlation between the number of mitochondria on one hand and mature or immature insulin granules on the other with correlation coefficients -0.37 and -0.27 respectively, p < 0.05 for both correlations.
Qualitative ultrastructural findings
The β-cells of the transplants from vehicle-treated non-diabetic rats had moderately developed ER and Golgi apparatus. The mitochondrial profiles were round or elongated and the mitochondria had matrix of moderate density. The internal membrane formed distinct cristae ().
The morphology of β-cells of the vehicle-treated diabetic rats displayed striking differences. The mitochondrial matrix had an extremely low density and cristae were usually disrupted forming an irregular pattern, (). Often circular membranous profiles were found in the mitochondria (). Occasionally mitochondrial profiles were seen where in the middle of the mitochondrion the inner mitochondrial membrane was double folded over the entire width of the organelle (). This is a view often associated with mitochondrial fusion or fission.Citation12 We captured a couple of usually rare events of fusion/fission. One is shown in . Tifenazoxide-treatment of the diabetic rats resulted in a β-cell morphology that seemed reminiscent of that in transplants to non-diabetic recipients. ER and Golgi seemed less elaborated than in the transplants to vehicle-treated diabetic rats (). The mitochondrial morphology resembled that found in transplants from vehicle-treated non-diabetic rats ().
Long-term in vitro culture in high glucose
Culture reproduces changes in mitochondrial morphology obtained in vivo
After culture in vitro for 2 and 3 weeks islets of Langerhans were examined by electron microscopy. All changes registered in vivo due to the diabetic state were reproduced after high glucose culture in vitro both after two and three weeks (compare and ).
Figure 6. Quantitative morphology of primary (upper panel) and secondary (lower panel) mitochondrial parameters of islets cultured in vitro. Data were collected from 22 sections per rat. Data from 2 weeks (n = 2) and 3 weeks (n = 1) culture in 11 or 27 mmol/l glucose were pooled since quantitative morphology was similar after 2 and 3 weeks culture. *p < 0.05 vs. mitochondria from islets cultured in 11 mmol/l glucose.
Abnormal morphometry associates with mitochondrial dysfunction
DNA content did not differ between islets cultured in 11 vs. 27 mmol/l glucose for three weeks (6.11 ± 0.1 and 6.10 ± 0.2 ng/islet, n = 4). There was an increase by 1.53 ± 0.3 fold in the mtDNA/nDNA ratio after culture in 27 mmol/l vs. 11 mmol/l glucose, n = 8, p < 0.01. Yet, culture in 27 vs. 11 mmol/l glucose decreased insulin secretion in final incubations in response to 16.7 mmol/l glucose (Table S1). Furthermore, long-term culture in high glucose reduced constituents of mitochondrial complexes I-IV (). Complex V was not affected. Culture in high glucose significantly decreased ATP and ADP levels ().
Figure 7. Effects of 3 weeks culture in vitro in 11 or 27 mmol/l glucose on sub-units of mitochondrial complexes I-V (A and quantified in B), OPA 1 (A and quantified in C) and on ATP and ADP levels (D). Immunoblotting was performed on islets cultured either at 27 or 11 mmol/l glucose. In (A), (B) and (C) mean ± SEM of four individual experiments expressed as percentage of results in islets cultured in 11 mmol/l glucose (100%). *p < 0.05 or less vs. 11 mmol/l glucose culture. ATP and ADP levels: Mean ± SEM of four individual experiments. *p < 0.001 or less vs. 11 mmol/l glucose culture.
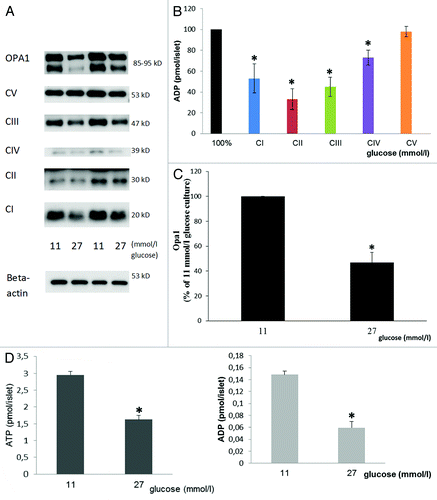
OPA 1 has been implicated as a key protein involved in the fusion process of mitochondria.Citation13 It was therefore of interest to test for in the present context. Culture in 27 vs. 11 mmol/l glucose for three weeks significantly decreased OPA1 ().
Single β-cells from two weeks cultured islets in 11 or 27 mmol/l glucose were studied for uptake of MTR fluorescence. Fluorescence in relation to cell area (ratio) was decreased from 1.1 ± 0.04 to 0.8 ± 0.04, n = 73, p < 0.001.
Basal oxygen consumption and oligomycin-induced reduction therof was measured in islets after three weeks culture in 27 vs. 11 mmol/l glucose at the same glucose concentration that was present during culture. The mean of 2–3 measurements was for basal consumption 6.6 pmol/s/400 islets cultured at 11 and 5.5 pmol/s/400 islets after 27 mmol/l glucose culture. The mean reduction after oligomycin was 78% after 11 and 57% after 27 mmol/l glucose.
Discussion
Our study documents that a chronic diabetic state with only moderate hyperglycemia induces fewer, larger, swollen and more variably sized mitochondria. These morphological aberrations can be replicated by long-term exposure to elevated glucose in vitro and are then associated with dysfunction of oxidative phosphorylation. Further, we provide evidence that abnormal morphology can be influenced, i.e., partly corrected, by the in vivo presence of the K+-ATP channel opener tifenazoxide.
Our in vitro data strongly support the notion of linkage between the morphological alterations in vivo and long-term exposure to elevated glucose. Hence, the pattern of morphological abnormalities that we document in vivo was closely reproduced during 2–3 weeks of tissue culture with 27 vs. 11 mmol/l glucose. We used 11 mmol/l rather than a lower glucose concentration as “control” because of the documented adverse effects during culture at a lower glucose concentration.Citation14 Furthermore, the in vitro data give evidence for the pathological nature of the altered morphology. Hence, after culture in long-term elevated glucose we demonstrate overall insufficiency of energy production. Hence there was reduction in levels of ATP, of oxygen consumption tied to ATP-production, signaling by MitoTracker Red (a marker of active respiring mitochondria) as well as of key proteins of oxidative phosphorylation i.e., complexes I–IV. Interestingly, complex V, ATP synthase which is not part of the oxidative phosphorylation per se, was not affected, and mitochondrial DNA in relation to nuclear DNA was increased and not decreased after high glucose culture. This indicates selectivity for effects of long-term elevated glucose on mitochondrial function.
Our morphological findings differ from those in the spontaneously diabetic GK rats where an increased number of mitochondria have been reported, coupled with a decrease in size.Citation15 Our diabetes model tested specifically the influence of the diabetic milieu with moderate hyperglycemia whereas in the GK rat genetically determined perturbations of β-cell function and structure cause and precede diabetes. It seems possible that the genetic factors, known to be responsible for β-cell inadequacy in the GK rats, could affect mitochondrial structure in a way distinct from influences by the diabetic milieu per se. In this context, it is interesting to note that the challenge to the β-cells in GK rat by giving sucroseCitation16 resulted in similar morphological alterations that we describe here.
The pathological morphology that we find (including the decrease in number of mitochondria) is in line with a reportCitation8 from diabetic MKR mice that only harbor genetically determined and muscle-specific insulin resistance. It seems possible that long-term excessive demands on β-cell function is a common denominator for the negative effects exerted both by elevated glucose and by insulin resistance.
We find that a diabetic state increases variability of mitochondrial morphometry (). Our finding is in line with an in vitro study reporting that prolonged exposure to elevated glucose increases the variability of functional and structural characteristics of β-cell mitochondria.Citation17 These observations highlight the fact that morphological descriptions of mitochondria only in qualitative terms can be misleading.
In the transplantation study we looked for associations between mitochondrial morphometry and the occurrence of newly formed insulin granules (termed immature) as well as older ones (termed mature). As expected, we found lesser amounts of granules in grafts to diabetic recipients. This probably reflects, in part at least, increased demand for insulin secretion in the diabetic state. Interestingly, in these grafts we found a negative correlation between the number of mitochondria and the amount of insulin granules. This suggests, but does not prove, that the degree of mitochondrial dysfunction that we presume to be present (as indicated by the in vitro results) had decreased the synthesis of insulin. Such a notion is compatible with the known requirement of energy for insulin biosynthesis. At first glance the notion disagrees with observations from mitochondrial DNA-depleted β-cellsCitation3 in which glucose-induced insulin secretion is abolished but islet insulin content retained. The more chronic conditions of mitochondrial dysfunction in our experimental protocols could be important for our results. Hence long-term increased demands for secretion and thereby synthesis as dictated by hyperglycemia could have unraveled attenuation of insulin synthesizing capacity.
In light of our observations on megamitochondria it was of interest to test for perturbation of the dynamin-related GTPase OPA1 protein. OPA1 is localized in the inter-mitochondrial space or attached to the inner mitochondrial membrane.Citation18 OPA1 has been identified as a key—but not the only—protein necessary for the fusion process of mitochondria and lack of OPA1 results in fragmentation of mitochondria. The decrease in OPA1 that we observe clearly negates OPA 1 as a driver in the process that results in megamitochondria. One may speculate that the reduction of OPA1 is an adaption to other, presently unclarified, factors that induce fusion and/or inhibit fission.
Of note, OPA1 may also exert effects on β-cell metabolism and was recently shown to affect glucose-induced oxidative metabolism, such an effect being caused primarily by an impact on complex 4.Citation19 We cannot exclude that downregulation of OPA1 is of importance for the decrease in oxidation that we observe. However, our findings are not completely compatible with those of Zhang et al. Hence, our results indicate that the long-term adverse effects of elevated glucose associate with a generalized rather than selective attenuation of components (complex 1–4) of oxidative phosphorylation.
The beneficial effects of tifenazoxide in our in vivo diabetes model are in line with beneficial ones on insulin secretion using a very similar protocol.Citation11 Tifenazoxide and another (diazoxide) K+-ATP-channel opener were administered in vivo to diabetic rats during 8 weeks. Glucose-induced insulin secretion from transplant holding kidneys ex vivo was markedly increased by the previous exposure whether to tifenazoxide or to diazoxide. Taken together the results suggest that K+-ATP channel openers can protect glucose-induced insulin secretion by alleviating mitochondrial dysfunction. Further studies on the molecular mechanisms behind the beneficial effect of tifenazoxide have been hampered by unavailability of the drug for supplemental in vitro investigations. Also our experiences with diazoxide, indicate that this drug is unsuitable for longer term culture in vitro (results not shown). Hence, the molecular mechanisms behind the drug-induced beneficial effects remain to be investigated.
What is the possible significance of the present findings for human type 2 diabetes? Progression of type 2 diabetes, i.e., reduced β-cell function with duration of the disease is the one aspect that relates to our findings. The diabetic milieu with “glucotoxicity” and “lipotoxicity” is widely thought to be important for the decline of β-cell function in type 2 diabetes. The present findings indicate that a diabetic milieu with moderate hyperglycemia (resembling the situation in many patients) exerts a marked influence on mitochondria and that elevated glucose per se can be a sufficient factor for negative effects.
Materials and Methods
Animals
Female and male Wistar-Furth (WF) rats were obtained from Scanbur (Sollentuna, Sweden). Female rats served as donors and male syngeneic rats as recipients in the transplantation experiments. Male rats were made diabetic by an intraperitoneal injection of 90 mg/kg streptozotocin (Sigma) at day 1 after birth. Diabetes was confirmed at day 3 by blood glucose > 10.0 mmol/l. For in vitro experiments islets from male Sprague-Dawley (SD) rats (200–300 g) were used.
The animal studies were approved by the northern Stockholm or by the northern Swedish Ethical Committee on Experimental Animal Care. The studies were performed in accordance with guidelines from the Swedish National Board for Laboratory Animals.
Transplantation protocol
Rat islets of Langerhans were isolated by collagenase from 16–20 week old female WF rats. Islets were cultured overnight in RPMI 1640 (Statens Veterinärmedicinska Anstalt, Uppsala, Sweden; for details see Sup. Materials). Fifty to one hundred 50 islets were transplanted under the left kidney capsule as previously described in references Citation11 and Citation20. To minimize inter-donor variations, islets isolated from two to three rats were mixed and then divided into three equal portions; one-third being transplanted to a non-diabetic rat, one-third to a diabetic vehicle-treated and one-third to a diabetic tifenazoxide-treated rat (see for the in vivo study design). The number of islets transplanted to each rat was purposely kept low enough not to reverse diabetes in the recipients.
Following transplantation, rats were administered vehicle (non-diabetic and diabetic recipients) or tifenazoxide (3 mg/kg, diabetic recipients) once daily between 08.00 and 10.00 h. The vehicle component consisted of glycerol 5 vol%, gelatine 0.5%, 40 vol% and carboxy-methyl-cellulose 2%, 55 vol%. Tifenazoxide was dissolved in 2% NaOH. The treatment period lasted for 63 d. It was followed by exactly 7 d of wash-out, i.e., the cessation of vehicle or tifenazoxide administration. At all times the animals had free access to water and a standard laboratory chow for rats (Scanbur). Blood glucose levels were measured non-fasting once every week just before medication throughout the experimental period.
Ultrastructural morphometry
The transplants were cut out from under the renal capsule and fixed by immersion in an ice-cold mixture of 2.5% glutaraldehyde in 0.05 M phosphate buffer, pH 7.2, with an osmolarity of 390 mOsmol/kg. After dehydration the transplants were embedded in an epoxy resin. Ultra-thin sections giving a silver interference color were prepared on an LKB ultramicrotome and picked up on form-coated one-hole grids. The sections were stained with uranyl acetate and lead citrate. The electron microscopy was performed with a Hitachi H-700 with 75 kV acceleration tension.
After fixation and embedding, eight transplants from each experimental group were subsequently prepared for electron microscopy. Twenty-two electron micrographs at a primary enlargement of 7,200x were made from each transplant. The micrographs were distributed over the section areas by systematic random sampling of β-cell-rich areas.
The negative film was scanned (Epson Perfection 4990 Photo) and analyzed with Adobe Photoshop C5 extended with The Image Processing Tool Kit version 3.0. In the stereological analysis, β-cells were identified by their typical appearance of secretory granules. The parameters used for evaluation of mitochondria are summarized in . Totally 528 EM sections were evaluated (176 from each treatment group). All together approximately 25,000 mitochondria were evaluated. Non-β-cell data points were subtracted when number of points on mitochondria per number of test points (Pp), number of intersections of mitochondria per unit length of test line (LL) and number of interceptions of mitochondria per unit test area (NA) were calculated. Figure S1 shows how Pp, LL and NA were calculated in practice. The volume of mitochondria per unit β-cell volume (volume density, VVM), the outer surface area of mitochondria per unit β-cell volume (density of outer surface area, SVM), and the number of mitochondria per unit β-cell volume (numerical density, NVM) were determined according to methods described by WeibelCitation21 and Borg.Citation22 Secondary morphometric parameters, such as average mitochondrial volume (VVM/NVM), average mitochondrial outer surface area (SVM/NVM) and average mitochondrial surface area per unit mitochondrial volume (SVM/VVM), were also calculated. Insulin granules were classified as either mature from the appearance of a dense core and a wide halo or immature from the appearance of pale content and a narrow halo. Insulin granules (mature, immature and total) were quantified according to volume density, number of granules and average volume.
Single islets of Langerhans cultured in vitro as described were prepared for electron microscopy and evaluated in a similar way as above.
Insulin release measurements including batch-type incubations. Following culture and preincubation as described in Supplemental Materials equal sized islets were incubated in groups of three for 60 min at 37°C in 300 μl KRB containing 3.3 or 16.7 mmol/l glucose. Each experimental condition consisted of three or four individual groups of three islets. The insulin accumulated in the incubation medium was measured as described.Citation23 Islet insulin content was measured after acid-ethanol extractionCitation24 of islets retrieved from batch incubations.
Western blot
Protein extracts were separated on SDS-PAGE gels, transferred to nitrocellulose filters and probed with mitoprofile Total OXPHOS Rodent WB antibody Cocktail (cat. no. MS604, MitoSciences, 1:5,000) and for OPA-1, an antibody with cat. no. 612606 BD from Transduction Laboratories, 1:1,000. For further details see Supplemental Materials.
ATP and ADP measurements
ATP was determined using a bioluminescence kit (HS II; cat. no. 11 699 709 001, Roche Diagnostics). ADP was measured after enzymatic removal of ATP, as described.Citation25
DNA measurements of cultured islets
After culture islets were washed twice with cold PBS and 1 ∝l PBS/islet was added followed by sonication. DNA was measured by fluorescent DNA Quantitation Kit (170–2480, Bio-Rad) according to manufactures’ instructions.
Mitochondrial DNA (mtDNA) amounts by real time PCR
After culture, islet DNA was extracted by DNeasy Blood and Tissue kit (Qiagen, Sweden). Mitochondrial relative DNA copy number was determined by calculation of the mtDNA/nDNA to the control ratio (i.e., 11 mmol/l glucose).Citation26 mtDNA-encoded NADH dehydrogenase 2 gene (ND-2) was used as a marker for total mtDNA and 18S rDNA for nDNA. All primers and real-time PCR kit were from Applied Biosystems.
Imaging of live cell mitochondria
After culture plated cells from SD rats (for details see Sup. Materials) were rinsed and positioned in new Petri dishes for 60 min preincubation at 37°C with 3.3 mmol/l glucose and 70 nmol/l Red CMXRos, MitoTracker Red (MTR) from Molecular probes (Eugen Oregon) was added for 10 min. Live cell images were collected using a Leica SP2 spectral laser scanning confocal microscopy system (Leica Microsystems, Mannheim, Germany) equipped with a 250 mW argon/krypton laser (Omnichrome, Inc.). For further details see Supplemental Materials.
Oxygen consumption
The oxygen consumption was measured by Clark-type polarographic oxygen sensors and high-resolution respirometry (Oxygraph-2k, OROBOROS). Samples of 400 islets in cell culture medium were added to a chamber with magnetic stirring and allowed to equilibrate with air for 2 min before closing the chamber and recording oxygen uptake at basal respiration during 20 min of stable oxygen consumption. Consumption rates were calculated as the negative time derivate followed by the addition of 2 mg/ml of oligomycin of the oxygen concentration present in the chamber (pmol/s/400 islets).
Statistical analysis
Data in Tables and figures are expressed as means ± SEM. For multiple comparisons, one-way ANOVA was performed. When normality test failed then Kruskal-Wallis ANOVA on ranks followed by Student-Newman-Keuls post-hoc test was performed. A p value of less than 0.05 was considered significant.
| Abbreviations: | ||
| K+-ATP channel | = | ATP sensitive potassium channel |
| SD | = | Sprague Dawley, WF, Wistar Furth |
Additional material
Download Zip (19.3 MB)Acknowledgments
We thank Prof Erol Cerasi for helpful comments and discussions during the preparation of this manuscript. This study was supported by NovoNordisk, Denmark, Swedish Society of Medicine, Swedish Medical Research Council (grant 2003–6289) and an EFSD/Merck grant to Valdemar Grill.
Disclosure of Potential Conflicts of Interest
During the performance of the in vivo transplantation experiments in this manuscript John Bondo Hansen was employed by Novo-Nordisk A/S. He holds stock in Novo-Nordisk.
Supplemental Material
Supplemental materials may be found here: http://www.landesbioscience.com/journals/islets/article/20516/
References
- Wiederkehr A, Wollheim CB. Minireview: implication of mitochondria in insulin secretion and action. Endocrinology 2006; 147:2643 - 9; http://dx.doi.org/10.1210/en.2006-0057; PMID: 16556766
- Fujimoto S, Nabe K, Takehiro M, Shimodahira M, Kajikawa M, Takeda T, et al. Impaired metabolism-secretion coupling in pancreatic beta-cells: role of determinants of mitochondrial ATP production. Diabetes Res Clin Pract 2007; 77:Suppl 1 S2 - 10; http://dx.doi.org/10.1016/j.diabres.2007.01.026; PMID: 17449130
- Kennedy ED, Maechler P, Wollheim CB. Effects of depletion of mitochondrial DNA in metabolism secretion coupling in INS-1 cells. Diabetes 1998; 47:374 - 80; http://dx.doi.org/10.2337/diabetes.47.3.374; PMID: 9519742
- Soejima A, Inoue K, Takai D, Kaneko M, Ishihara H, Oka Y, et al. Mitochondrial DNA is required for regulation of glucose-stimulated insulin secretion in a mouse pancreatic beta cell line, MIN6. J Biol Chem 1996; 271:26194 - 9; http://dx.doi.org/10.1074/jbc.271.42.26194; PMID: 8824267
- Maassen JA, Janssen GM, ’t Hart LM. Molecular mechanisms of mitochondrial diabetes (MIDD). Ann Med 2005; 37:213 - 21; http://dx.doi.org/10.1080/07853890510007188; PMID: 16019720
- Koeck T, Olsson AH, Nitert MD, Sharoyko VV, Ladenvall C, Kotova O, et al. A common variant in TFB1M is associated with reduced insulin secretion and increased future risk of type 2 diabetes. Cell Metab 2011; 13:80 - 91; http://dx.doi.org/10.1016/j.cmet.2010.12.007; PMID: 21195351
- Anello M, Lupi R, Spampinato D, Piro S, Masini M, Boggi U, et al. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 2005; 48:282 - 9; http://dx.doi.org/10.1007/s00125-004-1627-9; PMID: 15654602
- Lu H, Koshkin V, Allister EM, Gyulkhandanyan AV, Wheeler MB. Molecular and metabolic evidence for mitochondrial defects associated with beta-cell dysfunction in a mouse model of type 2 diabetes. Diabetes 2010; 59:448 - 59; http://dx.doi.org/10.2337/db09-0129; PMID: 19903739
- Hackenbrock CR. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J Cell Biol 1966; 30:269 - 97; http://dx.doi.org/10.1083/jcb.30.2.269; PMID: 5968972
- Dimmer KS, Scorrano L. (De)constructing mitochondria: what for?. Physiology (Bethesda) 2006; 21:233 - 41; http://dx.doi.org/10.1152/physiol.00010.2006; PMID: 16868312
- Björklund A, Bondo Hansen J, Falkmer S, Grill V. Openers of ATP-dependent K+-channels protect against a signal-transduction-linked and not freely reversible defect of insulin secretion in a rat islet transplantation model of Type 2 diabetes. Diabetologia 2004; 47:885 - 91; http://dx.doi.org/10.1007/s00125-004-1386-7; PMID: 15088085
- Choi SY, Huang P, Jenkins GM, Chan DC, Schiller J, Frohman MA. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol 2006; 8:1255 - 62; http://dx.doi.org/10.1038/ncb1487; PMID: 17028579
- Bossy-Wetzel E, Barsoum MJ, Godzik A, Schwarzenbacher R, Lipton SA. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr Opin Cell Biol 2003; 15:706 - 16; http://dx.doi.org/10.1016/j.ceb.2003.10.015; PMID: 14644195
- Bensellam M, Van Lommel L, Overbergh L, Schuit FC, Jonas JC. Cluster analysis of rat pancreatic islet gene mRNA levels after culture in low-, intermediate- and high-glucose concentrations. Diabetologia 2009; 52:463 - 76; http://dx.doi.org/10.1007/s00125-008-1245-z; PMID: 19165461
- Serradas P, Giroix MH, Saulnier C, Gangnerau MN, Borg LA, Welsh M, et al. Mitochondrial deoxyribonucleic acid content is specifically decreased in adult, but not fetal, pancreatic islets of the Goto-Kakizaki rat, a genetic model of noninsulin-dependent diabetes. Endocrinology 1995; 136:5623 - 31; http://dx.doi.org/10.1210/en.136.12.5623; PMID: 7588317
- Mizukami H, Wada R, Koyama M, Takeo T, Suga S, Wakui M, et al. Augmented beta cell loss and mitochondrial abnormalities in sucrose-fed GK rats. Virchows Arch 2008; 452:383 - 92; http://dx.doi.org/10.1007/s00428-007-0508-2; PMID: 18236074
- Wikstrom JD, Katzman SM, Mohamed H, Twig G, Graf SA, Heart E, et al. beta-Cell mitochondria exhibit membrane potential heterogeneity that can be altered by stimulatory or toxic fuel levels. Diabetes 2007; 56:2569 - 78; http://dx.doi.org/10.2337/db06-0757; PMID: 17686943
- Chen H, Chan DC. Physiological functions of mitochondrial fusion. Ann N Y Acad Sci 2010; 1201:21 - 5; http://dx.doi.org/10.1111/j.1749-6632.2010.05615.x; PMID: 20649534
- Zhang Z, Wakabayashi N, Wakabayashi J, Tamura Y, Song WJ, Sereda S, et al. The dynamin-related GTPase Opa1 is required for glucose-stimulated ATP production in pancreatic beta cells. Mol Biol Cell 2011; 22:2235 - 45; http://dx.doi.org/10.1091/mbc.E10-12-0933; PMID: 21551073
- Korsgren O, Jansson L, Andersson A. Effects of hyperglycemia on function of isolated mouse pancreatic islets transplanted under kidney capsule. Diabetes 1989; 38:510 - 5; http://dx.doi.org/10.2337/diabetes.38.4.510; PMID: 2494093
- Weibel ER, Gomez DM. A principle for counting tissue structures on random sections. J Appl Physiol 1962; 17:343 - 8; PMID: 14005589
- Borg LA, Andersson A, Berne C, Westman J. Glucose dependent alterations of mitochondrial ultrastructure and enzyme content in mouse pancreatic islets maintained in tissue culture: a morphometrical and biochemical study. Cell Tissue Res 1975; 162:313 - 21; http://dx.doi.org/10.1007/BF00220177; PMID: 1102103
- Herbert V, Lau KS, Gottlieb CW, Bleicher SJ. Coated charcoal immunoassay of insulin. J Clin Endocrinol Metab 1965; 25:1375 - 84; http://dx.doi.org/10.1210/jcem-25-10-1375; PMID: 5320561
- Grill V, Rundfeldt M, Efendić S. Previous exposure to glucose enhances somatostatin secretion from the isolated perfused rat pancreas. Diabetologia 1981; 20:495 - 500; http://dx.doi.org/10.1007/BF00253414; PMID: 6113182
- Schultz V, Sussman I, Bokvist K, Tornheim K. Bioluminometric assay of ADP and ATP at high ATP/ADP ratios: assay of ADP after enzymatic removal of ATP. Anal Biochem 1993; 215:302 - 4; http://dx.doi.org/10.1006/abio.1993.1591; PMID: 8122794
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Δ Δ C(T)) Method. Methods 2001; 25:402 - 8; http://dx.doi.org/10.1006/meth.2001.1262; PMID: 11846609

