
Abstract
The incidence of type 1 diabetes (T1D), as with several other autoimmune diseases and conditions, began to notably rise in the latter half of the last century. Most cases of T1D are not solely attributable to genetics and therefore, environmental influences are proposed to account for the difference. Humans live today in general under much more hygienic conditions than their ancestors. Although human enteroviruses (HEV) have been strongly implicated as causative environmental agents of T1D, recent work has shown that the bacterial genera in the gut of diabetics compared with non-diabetics, can vary significantly. Here, we consider these data in light of our non-hygienic human past in order to discuss a possible relationship between the resident bacterial biome and acute infectious events by HEV, suggesting how this may have influenced T1D incidences in the past and the risk for developing T1D today.
Introduction
The rapid global increase in type 1 diabetes (T1D) incidence since the middle of the 20th century, coupled with the realization that most cases of T1D are not attributable solely to individual genetics, have led to the inescapable conclusion that one or more powerful (and recently emerged) environmental factors are influencing the disease etiology. This rise in T1D cases is recent because T1D was rare in the past: although symptoms consistent with T1D were described in ancient Greece,Citation1 T1D seldom occurred until about the mid-20th century when incidences began noticeably to increase.Citation2-Citation4 Although human genetics form a risk factor for T1D development,Citation5-Citation8 genetics alone are not responsible for the rapid expansion of the number of cases occurring in the last 70–100 y.Citation6,Citation9-Citation11 Fewer than 10% of people with HLA-determined susceptibility to T1D develop the diseaseCitation9,Citation12 while monozygotic twins have a less than 40% concordance of developing T1D early in life (although concordance rises with ageCitation13). Type 1 diabetes incidences are commonly higher in developed countries and in wealthy and uncrowded urban environments than elsewhere.Citation14-Citation21 Several studies have noted a trend of a decreasing proportion of T1D patients with high risk HLA in recent than in previous cohorts, a trend matched by an increase in the proportion of T1D with less high risk HLA genotypes.Citation22-Citation25 A study of the genetics of medieval Poles noted that at-risk genotypes were present more frequently 700 y ago than in the current Polish population despite increasing T1D incidence in modern Europe.Citation26 As noted by RookCitation27, “…any explanation (for increased T1D incidences) that omits environmental change is incomplete.”
What then is/are the “environmental factors” causing this modern increase in T1D cases? Viral infections currently represent the most likely candidate, with overwhelming supporting data for being the primary, perhaps the sole significant, environmental cause of T1D onset. The increasing incidence of T1D in many countries also has shown geographical localization of increasesCitation28,Citation29 as one might expect of an infectious association. The role of human enteroviral (HEV) infections has been supported by the detection of HEV antigens, RNA and occasionally isolation of virus from T1D patient samples.Citation30-Citation33 The presence of HEV RNA in cases of myocarditis and cardiomyopathy supported a role for these viruses in inflammatory cardiac disease, although detection of virus in hearts can be difficult due to the focal nature and the low level of infection and the difficulty of sampling so essential an organ.Citation34,Citation35 These difficulties are even greater in T1D as the pancreas is nearly impossible to biopsy and patients fortunately survive their disease due to the use of insulin. As HEV have been shown to leave a signature persistence of low level defective enteroviruses in cases of myocarditis,Citation36-Citation38 this mechanism may account for the late term presence of enterovirus antigens and RNA in T1D as well. Although this article focuses on the HEV, other viruses (e.g., mumps and rubellaCitation39) have been associated with human T1D, indicating that the HEV may well not be the only players.Citation40 But it's not just viruses anymore: bacteria are increasingly seen as playing a part in T1D etiology.Citation41-Citation46
Another question derives from the observation that T1D has been a rare disease throughout the majority of humans' time on earth: what were the environmental factors in our long history which so effectively suppressed T1D that the disease was uncommon? From studies in the NOD mouse model of T1D, we know that 'dirty' mice (i.e., those in conventional housing) are healthier mice whereas if animals are kept in pristine conditions, most develop T1D.Citation47 In an interesting parallel, human history reveals that until quite recently, our ancestors were largely unaware of the importance of hygiene in preventing many diseases and thus they lived in, ate and drank their own filth to varying degrees. This, of course, dramatically changed in developed societies of the modern era within the past 100 y or so. The “hygiene hypothesis,” proposed by StrachanCitation48 to account for rising asthma in developed societies, is a working approachCitation49,Citation50 to considering the rapid rise in T1D cases beginning in the 20th century as well as other autoimmune diseases and disorders.Citation4,Citation40 A commonality to the story emerging from mice and men is exposure to infectious agents that indicates pervasive viral and/or bacterial exposure as a potentially protective mechanism. Given the significant change regarding personal hygiene in the human environment from the past to the present, it is difficult to ignore the possibility that human hygiene levels have been continually linked to previously low, and currently rising, incidences of T1D.
Hygiene, Life and Human Disease
From a 21st century perspective, one might wish to hold one's breath while considering the generally abysmal human living standards in the past. Flush toilets, enclosed sewers and sewage treatment plants became commonplace only in the 20th century and are still rare/nonexistent in many highly populated parts of the world today. Sewage was certainly recognized as foul but relatively little was done about it: when diseases came, the awful smell was blamed (the 'miasma' theoryCitation51). It was not until the mid-1800s that the perception of the key issue changed from smell to the quality of fresh water itself as key. As late as the mid-1800s, for example, the Thames River in England was a running sewer. The stench was so bad in the summer of 1858, it was called “The Big Stink,”Citation52 an occurrence that initiated the construction of enclosed sewers. It is important to realize that well beyond this time and throughout the world, sewage in streets and feces-contaminated waterways remained (and still remain to this day in many areas) part of the daily fabric of human life. Cholera (a marker of fecal pollution) outbreaks were devastating: in one case, cholera reportedly killed 20,000 people in Paris in 1832,Citation53 about 3% of the city's population. It can be strongly inferred that nearly every significant stream and river system that bordered any significant human habitation harbored a significant load of microorganisms and viruses of human fecal origin. This situation existed well into the 20th century and has waned only in areas where societies progressively “cleaned up.”
Personal hygiene was also at a level far below that which one would consider acceptable today. A simple example suffices: the cleansing of one's hands following a visit to a privy, even if one were scrupulous, was problematic. Running water was generally uncommon, but even if present, the microbial contents may well have been significant, having been drawn from a nearby polluted water supply. If one rinsed one's hands in a water container used by others for the same purpose, it would have scarcely been an improvement over not washing at all. Wells were (and still are in many places) easily polluted from privies located nearby. Lacking toilet paper or a substitute, a hand would be used; in many cultures still, the left hand is not used for eating for this very reason or out of respect for cultural memory. Even now, human sewage remains a rich source for viruses, including a large array of untyped viruses.Citation54
Hygiene and Polio: A Case Study
Poliomyelitis (polio), an example of a HEV-caused disease that incorporates the concepts of human environment and hygiene, can be used to help understand aspects of T1D etiology. Prior to the 20th century, polio—the most thoroughly characterized HEV disease—was rare in outbreak or epidemic form.Citation55,Citation56 PaulCitation57 suggests that had polio been epidemic in ancient times, the astute Hippocrates would surely have recognized its presence and noted it in his 'Of The Epidemics'. Instead, Hippocrates described rare cases of 'clubfoot', presumably the dropped foot common to many paralytic polio cases. Beginning around the end of the 1800s, annual polio outbreaksCitation58 were startling occurrences in themselves, for polio had previously been rare.Citation59,Citation60 The regular appearance of polio epidemics has been linked to the increased recognition, and progressive adaptation, of the germ theory and the importance of societal and personal hygiene, sewage containment and treatment, and microbiologically clean(er) water supplies.Citation55,Citation61 As discussed above, sewage containment was generally poor or did not exist, and hygienic concepts were dimly understood. Passive (through breast feeding) transfer of maternal anti-PV protective antibodiesCitation62,Citation63 would protect children against polio contracted from exposure to excrement-contaminated, PV-containing water supplies and poor hygiene as long as the mother had previously developed immunity to the same PV serotypes. As children aged, robust protective anti-PV immunity would develop through repetitive infectious contact with the viruses in the environment. As hygienic practices improved,Citation64,Citation65 people were less frequently immunized via naturally occurring, sub-clinical infections in childhood and survived to adulthood without immunity to PV. As a result, whenever PV circulated in the community, a large group of people became infected (and were sources themselves of infection) producing more cases of polio. Cohorts without anti-PV immunity set the stage for epidemics of polio.
Polioviruses and other HEV are quite similar in terms of how they are spreadCitation66. Just as natural PV exposure began to be less common, it is reasonably certain that the numbers of other non-PV HEV exposures also started to decrease in many societies by mid-20th century (certainly those which recognized the advantages of increasing hygienic standardsCitation3,Citation64). Symptoms consistent with T1D were described in antiquityCitation1 but the disease was rare until the mid 20th century.Citation4,Citation21,Citation67,Citation68 Interestingly, increasing Finnish T1D incidencesCitation4,Citation69 have been coincident with decreasing HEV infection rates, an observation consistent with improving hygienic conditions.Citation69 StrachanCitation48 originally proposed that increased asthma incidenceCitation40 in industrialized or more hygienic societies, is related to increased hygiene. This fundamental concept forms the basis for a working hypothesis to consider how widespread anti-HEV immunity as well as bacterial exposure, obtained through constant close environmental exposure to feces, may once have had importance greater than hitherto supposed in the suppression of T1D.
How T1D in NOD Mice Responds to HEV Infections: Lessons for Us
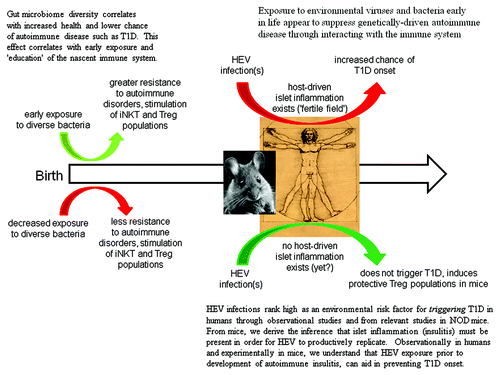
Non-obese diabetic (NOD) mice, which spontaneously develop autoimmune T1D starting around week 12 of life, have been used to model a variety of aspects of T1DCitation47. Although the model is criticized as incomplete and/or of limited translational value,Citation70 the NOD mouse is an excellent system for studying the impact which HEV infections have upon T1D development. A female NOD mouse population will develop T1D in 70–100% of the animals by week 15–25 of life if kept in a clean environment but if the mice are not often transferred to fresh sterile cages, water and food, incidences can drop to as low as 20–30%. After inoculating 4 week old NOD mice with group B coxsackievirus (CVB), mice were well protected from developing host-driven autoimmune T1D with of just 0–20%.Citation71 But when older NOD mice were used, which in this system of genetically closely identical mice corresponds to the extent of autoimmune insulitis, CVB rapidly induced T1D onset.Citation72 The age of the mouse (extent of islet inflammation) was key: virus was only demonstrable in islets of older mice, not in young mice. It was shown that as NOD mice aged and insulitis increased, the mice became susceptible—not to protection from T1D onset—but to rapid induction of T1D. This volte face—protection from, to induction of, disease—occurred as a function of the extent of the host's own autoimmune islet inflammation. The strain of the virus used to inoculate the mice, defined in this case by how rapidly it replicates, was also importantCitation73: a more rapidly replicating CVB strain required logs fewer infectious particles to induce T1D than a more slowly replicating strain. Thus, virus strain and infectious dose were linked, as is the case for many infectious disease agents. From these observations with a highly defined system, a link between exposure to HEV in general and how it might affect human T1D was proposed.Citation10
A concern regarding the studies mentioned above is that NOD (or any) mice lack a naturally-occurring infection that triggers T1D onset, in contrast to accumulating evidence that supports a role for HEV as a trigger of human T1D. In this context, it must be recalled that mouse models of various and diverse human diseases have been extremely useful to aid in the understanding of pathogenesis. Two excellent models exist, for example, for human HEV diseases. Though PV is not an infection of mice, mouse-adapted strainsCitation74,Citation75 exist and have been used to understand the mechanisms underlying PV-induced paralysis. The construction of mice expressing the human poliovirus receptor (CD155) facilitated these studies further.Citation76,Citation77 Similarly, the CVB replicate well in all mice and have been used in numerous studies to elucidate mechanisms underlying HEV myocarditis, for example.Citation38,Citation78,Citation79 In each of these cases, despite the lack of a natural enterovirus that induces these diseases in mice, the models have proven exceptionally useful and capable of recapitulating observations from humans.
But viruses alone do not comprise the entire cast of T1D-linked infectious agents. Acknowledging the above arguments, one should consider that a failure to develop a sufficiently robust protective immune response against fecal microbiota—bacterial and viral—beginning early in life, may be a primary influence on whether or not a genetically predisposed individual develops T1D. This lack would fail to suppress, either significantly or totally, the destructive naturally-occurring autoimmune response in a genetically prone individual, thereby leading to T1D onset. Moreover, recent insights attribute a major role to intestinal microbiota in the post-thymic functional education of various T cell subsets.Citation80,Citation81 Specifically, it appears that reduced intestinal bacterial diversity is associated with a propensity to develop inappropriate T cell responses against self antigens and development of autoimmune disease. The emerging thought is that intestinal microbiota, as well as viruses, are instrumental in shaping the functionality of T cells in the periphery. Evidence is mounting to suggest that a diverse gut microbiome associates with protection from T1D onsetCitation82,Citation83 in genetically at-risk individuals. It is not surprising, then, that a better name for the hygiene hypothesis has been suggested to be the “microbial deprivation hypothesis”Citation84. Consequently, an HEV infection which meets key criteriaCitation49 to trigger T1D in an at-risk individual, might better be understood as an example of an infectious disease, much like a PV infection might induce polio. Because for polio, fewer than 1% of PV infections led to paralytic disease,Citation60 it is reasonable to assume that similar infection/disease ratios hold for other HEV. In the case of a modern human population genetically at risk for developing T1D, only the rare HEV infection might trigger T1D onset. The genetic susceptibility factor supplied by the potential T1D patient may well be modulated by the impact of having a stimulatory diverse gut microbiome from birth onward.
We have argued for the necessity of active insulitis as a key part of the mechanism of HEV-induced destruction of islet β cells and subsequent T1D onset.Citation49 We suggest broadening this to considering that any case of T1D may involve a significant lack of prior infectious agent-induced immune stimulation. The genetic landscape of the host is pivotal in deciding whether viruses and/or bacteria will be effectively cleared or ignored (in the gut), or whether deleterious responses are initiated that lead to T1D. Lacking autoimmune infiltration of islets, humans (based on work in the NOD mouse systemCitation71) might be more resistant to a productive HEV infection of the islets. Were this not the case, we would expect the incidence of T1D to be higher simply based on the large number of HEV infections occurring annually in any population.Citation66,Citation85 Of course, insulitis patterns in NOD mice differ significantly from those seen in humansCitation86 and as little is known about insulitis in individuals without overt disease,Citation87 we must infer more than is desirable. It is, however, worth noting that a recent study suggests that detection of HEV in human islets may be dependent upon insulitis,Citation88 in line with results from the NOD mouse system.Citation72,Citation89
The “fertile field hypothesis”Citation90 originally proposed that environmental infections can prepare the way for subsequent autoimmune process that initiate the disease. However, it has been arguedCitation49 that at least in the specific case of T1D, HEV may turn this approach on its head, by taking advantage of a pre-existing fertile field (islets assaulted by the host's own autoimmune disorder) in which to replicate and cause disease. This was based on two experimental results. Experimental infection of young NOD mice without insulitis by CVB show neither islet infections nor can be induced into developing T1D.Citation71 In contrast and in older NOD mice with active autoimmune islet inflammation, CVB readily replicate within islet cells and can rapidly cause T1D onset.Citation72 Seen in the light of a necessity for a pre-existing robust gut microbiome which helps to prevent T1D onset, this model can be theoretically modified to depend on the history of the individual's microbiological exposure as well. Importantly, the host participates in its own protection or demise in either case.
It is interesting to speculate that purely genetically-driven T1D, as well as virus-induced T1D, might now be more common than it was in our past because of a lowered chance of exposure to diverse fecal associated infectious agents early in life. Routine exposure, which surely was a constant part of life in our human past, has now been, at least in part, interdicted by higher levels of hygiene. By this argument, it may be no coincidence that T1D incidences are increasing. While HEV infections in early life may indeed help to lower the risk for T1D development later on, we must assume that either this has (1) enormous importance for the developing immune system in at-risk people, out of proportion to HEV infection frequency, or (2) that it is contributory toward health but not the sole arbiter of protection from T1D. It would therefore seem that another key arbiter early in life in at-risk individuals, must be the nature of one’s gut bacterial population.
The Gut Biome: Another Factor Linking Changes in Human Hygiene to T1D Frequency
When microbiologically clean water was not widely available in the past, humans were routinely exposed to diverse microbes. Both bacterial contamination due to nearly ubiquitous contamination and HEV exposure due to the well-established fecal-oral route of infection for HEV, were common. One may then hypothesize a causal link between microbial exposure and the suppression of T1D in a human population that itself has not significantly changed genetically over centuries. The insertion of hygienic cleanliness into this basic equation must therefore be seen as disruptive of a historically constant fecal-oral transmission of fecal microorganisms and viruses.
The picture that emerges is an anethema to modern sensibilities: T1D was rare in history most likely due to the constant microbial stimulation which humans encountered from day one of life, a protective microbial stimulation that commonly was fecal in origin. We know that hygienic cleanliness not only means we appear and smell much better but from a clinical perspective, we are also less commonly exposed to potential disease organisms. Nonetheless, it should be noted that we remain exposed to diverse viruses and microorganisms as, for example, in food.Citation91 The lettuce, sprouts and other fresh vegetables and fruit that are obtained from sparklingly clean supermarkets and generally eaten raw, for example, contain variable levels of bacteria despite being rinsed prior to ingestion.Citation92-Citation94 Salmonella outbreaks are often linked to raw vegetable and fruit consumption. Meats are often contaminated with intestinal bacteria during the butchering process. It is interesting that Matricardi and Bonini suggested that food-borne infections may impact allergic disease, and wondered whether certain diets, perhaps lacking in fresh foods, might be sufficiently deficient in bacteria to fail to continually stimulate the immune system.Citation95 Diet as a source for diverse bacterial populations, must also be factored into this story.Citation96
Of course, infants are not usually fed such things but are nursed or fed formula. Mothers impart important nutrition through nursing as well as passive immunization against microbes which she already endured and against which she raised a protective immune response. The infant in a modern industrialized society has a lower chance of being exposed to microbial contamination through the mother's care to keep her child neat and clean than would have been a similar child in the past. We can with reason assume that in the past, infants were exposed to bacterial and viral contamination as a matter of course when the only source of water was often contaminated and when a mother's (and anyone else's) hands and body were also contaminated due to inadequate or no sanitation. Thus from day 1 of life, the child would have been continually exposed to the fecal microbial population which existed in its immediate environment. To this point, the incidence of HEV infection in infants is decreased by breast feeding and increased by the presence of siblings (perhaps carrying infection from community sources to the family environmentCitation97). It is also established that infants delivered by caesarian show different bacterial colonization patterns than infants born vaginallyCitation98 as do children who are breast fed vs. fed with formula.Citation99
Here, then, is an interesting concept: aspects of microbial “filth” from which humans have endeavored to remove themselves, may actually be a significant component for long-term human health. Numerous studies have characterized gut bacterial populations in diabetic and non-diabetic humans, generally finding that a greater bacterial diversity is a commonality in the non-diabetic individuals,Citation41-Citation43,Citation46,Citation82,Citation83 findings that have also been confirmed with experimental studies in mice and rats.Citation44,Citation100-Citation103 For example, Bacteroides fragilis, a human intestinal bacterial denizen, can influence Treg developmentCitation104 through secretion of polysaccharide A. And when used to colonize mice, B. fragilis induced Tregs which ablated experimentally-induced colitis.Citation102 The intestinal flora of children who became diabetic showed a higher Bacteroidetes/Firmicutes species ratio than did control children.Citation83 Controls, in contrast, showed a decreasing level of Bacteroidetes species while Firmicutes species increased. A large metagenomic studyCitation41 showed that the genus Bacteroides was somewhat less than 2-fold more common in diabetics than in control patients, whereas the genera Faecalibacterium and Prevotella were significantly diminished in diabetics vs. well represented in controls. Taken together, this is a strong argument to support a role for intestinal bacterial populations in human T1D etiology. Yet, there is an underlying question: are less diverse (as well as different) bacterial populations in diabetic patients the cause of disease onset through an active mechanism or do such populations reflect that which was previously present in the individual and which in the early time of infancy, failed to sufficiently activate key immune regulators.Citation105 It is worth considering that it is still early in our understanding which of the overwhelmingly numerous bacterial genera and species may be the key players in T1D etiology. One might wish to recall the earlier (and misguided) eagerness to adopt CVB4 as the primary HEV pathogen of T1D and so keep an open mind.
It is likely that the age when microbial diversity is established may be significant for helping suppress T1D development. The adaptive immune response develops early in life,Citation99 reacting to the stimulation of diverse infectious agents: failure to be exposed or an inadequate exposure to a wide array of fecal flora, for example, could predispose the host to the development of inappropriate immune reactions (autoimmunity) to self or environmental antigensCitation10,Citation27 and/or lessen the chance of developing protective immunity.Citation80,Citation106,Citation107 Preceding discussions suggest that with sufficient exposure to fecal microbial diversity early in life, T1D may be suppressed even in those who are genetically predisposed to T1D development. Indeed, infections in the first year of life are linked to lower chances of T1D.Citation108 Fewer—not more—HEV infections correlated with a higher T1D risk: despite a higher incidence of HEV infections than a Finnish group, the incidence of T1D in an Estonian study group was five times lower than in neighboring Finland (7 vs. 36/100,000). This recalls results from experimental CVB infections of NOD mice, which demonstrated that infections early in life protect mice better from T1D than infections later in life. Young NOD mice without active islet inflammation, inoculated with any of several diverse strains of CVBs, enjoy considerably lowered risk of T1D development than do control counterparts.Citation71 While inoculation of older, prediabetic (with active islet inflammation) mice also slightly reduces the risk of T1D,Citation72 the reduction is much less than that in younger mice. These observations suggest while the same mechanism is triggered in both young and older mice by exposure to CVB, the impact of that protective mechanism on the suppression of deleterious islet inflammation is much more robust in younger than in older mice. This suppressive mechanism, the generation of protective Tregs,Citation109 may not be able to overcome the widespread autoimmune insulitis in older mice and thus its effect is much less pronounced or mooted entirely. In toto, these results are consistent with a beneficial effect of early, rather than later, exposure to more infectious diseases for the suppression of T1D incidence.
These findings support a general hypothesis that constant exposure to environmental bacteria from birth onward could lessen a risk for a variety of autoimmune diseases including T1D onset. It seems reasonable that both bacterial species diversity and bacterial load would be maintained/stimulated with constant fecal-oral contamination. Our predecessors successfully evolved over eons for many reasons, one of which may well have been the ability to live successfully in the midst of microbial contamination. Over evolutionary time, human immune responses to frequent microbial exposures may have allowed individuals with genes promoting autoimmunity to avoid the expression of that pathogenic immune response and survive sufficiently long to have children. As we have sought to avoid illness by improving hygiene, we may have unintentionally revealed these pathogenic immune responses.
Gut Bacteria and Enteroviruses: A Synergy Affecting T1D Development
It now appears—based upon both historical reference and modern research observations—that the environmental impact upon potential disease outcome (protection or onset) begins very early in life for those individuals who are genetically predisposed to developing T1D. This impact is positive, deterring or preventing the destructive, pathogenic attack of autoimmune T cells on β cells. But failure to experience sufficient environmental immune stimulation early in life when the immune system is developing may more readily permit development of the pathogenic autoimmune response that leads to T1D onset over time. We hypothesize based upon available evidence, that diverse bacteria and viruses (particularly the HEV), will be found to be the prime environmental players.
We can understand the impacts of HEV on human T1D specifically by drawing strong inferences from both NOD mouse studies and from human observational studies. These tell us that early in life and prior to development of the (yet to happen and host determined) autoimmune islet inflammation, bacterial or HEV exposures significantly reduce the chance of T1D onset later in life. It is in the first few years of life that the human immune system matures and in this time period when infectious agents interact in a significant (once in a lifetime?) way with the adaptive immune system. It may also be important that the host's autoimmune insulitis be absent or minimal during this time of exposure and immunological education, in order to permit the rise in and establishment of what we believe at present to be a dominant and protective population(s) of Tregs.Citation109,Citation110 The generation of neutralizing antibody producing B cell populations against specific HEV serotypes also protects the host against subsequent infection by the same HEV serotype, effectively mooting those serotypes' potential for causing T1D later on. Further, this exposure to diverse HEV can prompt immune memory to respond more rapidly to an infection by related but as yet inexperienced HEV serotypes, through recognition of evolutionarily well-conserved viral antigenic epitopes. A recently discovered mechanism by which HEV can persist in the host well beyond the acute infection period of a few weeks,Citation36-Citation38 suggests the possibility that immune stimulation by such HEV infections could be a benefit for the host.
Should a genetically predisposed individual not be exposed at some rate (the extent of which we do not know at present) to HEV infections or be exposed only to few or far apart infections, protective Tregs may not proliferate, letting insulitis develop determined by the individual's genetic constellation. From murine studies, we know that without islet inflammation, CVB do not productively infect β cells and other islet cell typesCitation49,Citation71 although as stated above, this is fundamentally unknown territory in humans at present. The key is “productive infection”: there is ample evidence from explanted islet studies and NOD mice, that diverse HEV can infect islets and β cells and that various HEV receptors are expressed in islet cells.Citation72,Citation111,Citation112 But in NOD mice—though we cannot say in humans—this does not proceed to widespread islet cell destruction in vivo in the absence of islet inflammation.Citation71 But once insulitis has begun, however, HEV such as the CVB can and do infect murine islets, destroying β cells and triggering T1D onset.Citation72,Citation73 Thus, the impact of HEV alone can be either protective or destructive, depending upon factors determined both by the host (state of insulitis and previous antiviral immunities) and the infecting virus (strain and dose).
Exposure to human waste entails exposure to the various gut flora including (predominantly) diverse bacterial species. An important correlary as well as consequence, to this argument is the fact that a human being carries approximately 10-fold more bacterial than human cellsCitation113 (a human-bacterial “hybrid”Citation114 and superorganismCitation115). It is too early at present to know whether the primary impact of the gut biome is upon protection from, or induction of, T1D, or both: this has been discussed in terms of other diseases as well.Citation105 However, strong evidence indicates that at least protection is a benefit from a diverse bacterial population in the host.Citation107 Though different bacterial population populations appear to exist in diabetic vs. healthy individuals, this may represent a de facto situation, not a causative one. Exposure to diverse bacteria and subsequent colonization of the host's digestive system, would most beneficially occur during very early life as well, influencing the host's immune development long-term.Citation106 Lack of a sufficiently diverse bacterial load in the intestine, might similarly fail to appropriately direct development of the immune system (discussed in ref. Citation107). This supposition is consistent with NOD mouse T1D incidences based on conditions of housing as well as observational determinations of differences in bacterial populations in diabetic vs. healthy individuals. As suggested by observations linking antibiotic exposure very early in life and increased risk of inflammatory bowel diseaseCitation116 and obesity,Citation117 it is interesting to speculate that repeated and/or long-term exposure to antibiotics early in life could significantly suppress bacterial diversity. This in turn might act as an exacerbating and difficult to trace influence on T1D development later in life.
Perhaps the most mechanistically convincing data linking T1D to bacterial gut populations derive from the study by Wen et al. in the NOD mouseCitation118: this work showed that genetic deletion of an innate signaling molecule (MyD88) changed certain ratios between gut microbiota, an activity that resulted in protection against T1D. However, administration of antibiotics or transfer of normal gut microbiota, restored this balance and as a consequence, reinstated diabetes development. As in the case with viruses, the host's own genetics seems to govern the manner in which the immune system ‘sees’ bacteria. Of note, rat and human studies have identified a ‘leaky’ gut phenotype in T1D,Citation43,Citation46 a feature that may allow immune cells to interact more closely with gut bacteria than is good for the host. Strategies aimed at generating a robust microbial diversity and restoring microbial balance may aid in preventing T1D development by induction of regulatory pathways ().Citation119
Figure 1. Microbiota in the environment can deleteriously or beneficially affect the progress of T1D.
Summary
A key societal impact of understanding the germ theory has been to move the human population to a much more hygienic way of life. This very significant change has in great part removed humanity from a vicious microbiological environment in which life-threatening infectious diseases were far too common. But paradoxically, living a 'cleaner' life may now mean that humans are threatened by diseases which were previously kept in check by a much less hygienic daily environment. Though somewhat non-intuitive, this realization may be the breakthrough needed to successfully designing preventative measures to reduce the chance of developing T1D. Potentially protective immunostimulatory approaches that can be readily tested include characterizing specific probiotic treatments as well as vaccination against common HEV antigens.
| Abbreviations: | ||
| T1D | = | type 1 diabetes |
| HEV | = | human enteroviruses |
| NOD | = | nonobese diabetic |
| CVB | = | group B coxsackievirus |
| Tregs | = | regulatory T cells |
Acknowledgments
We thank Mark Atkinson and Alberto Pugliesi for graciously reviewing this paper prior to submission. This work was supported in part by a grant from the NIH (N.M.C., S.T.), NIH/NIAID grant P01 AI089624-02 (M. Kronenberg, PI; M.vH. as leader of 'Project #3–Treg stability in viral infection and autoimmunity' and a Return Grant from the Belgian Federal Science Policy Office and a Marie Curie Career Reintegration Grant from the European Commission (K.C.).
References
- Sanders LJ. From Thebes to Toronto and the 21st century: An incredible journey. Diabetes Spectrum 2002; 15:56 - 60; http://dx.doi.org/10.2337/diaspect.15.1.56
- Harjutsalo V, Sjöberg L, Tuomilehto J. Time trends in the incidence of type 1 diabetes in Finnish children: a cohort study. Lancet 2008; 371:1777 - 82; http://dx.doi.org/10.1016/S0140-6736(08)60765-5; PMID: 18502302
- Viskari HR, Koskela P, Lönnrot M, Luonuansuu S, Reunanen A, Baer M, et al. Can enterovirus infections explain the increasing incidence of type 1 diabetes?. Diabetes Care 2000; 23:414 - 6; http://dx.doi.org/10.2337/diacare.23.3.414; PMID: 10868874
- Gale EA. The rise of childhood type 1 diabetes in the 20th century. Diabetes 2002; 51:3353 - 61; http://dx.doi.org/10.2337/diabetes.51.12.3353; PMID: 12453886
- Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al, Type 1 Diabetes Genetics Consortium. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 2009; 41:703 - 7; http://dx.doi.org/10.1038/ng.381; PMID: 19430480
- Todd JA. Etiology of type 1 diabetes. Immunity 2010; 32:457 - 67; http://dx.doi.org/10.1016/j.immuni.2010.04.001; PMID: 20412756
- Ziegler AG, Nepom GT. Prediction and pathogenesis in type 1 diabetes. Immunity 2010; 32:468 - 78; http://dx.doi.org/10.1016/j.immuni.2010.03.018; PMID: 20412757
- Nejentsev S, Howson JM, Walker NM, Szeszko J, Field SF, Stevens HE, et al, Wellcome Trust Case Control Consortium. Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature 2007; 450:887 - 92; http://dx.doi.org/10.1038/nature06406; PMID: 18004301
- Knip M. Pathogenesis of type 1 diabetes: implications for incidence trends. Horm Res Paediatr 2011; 76:Suppl 1 57 - 64; http://dx.doi.org/10.1159/000329169; PMID: 21778751
- Drescher KM, Tracy SM. The CVB and etiology of type 1 diabetes. Curr Top Microbiol Immunol 2008; 323:259 - 74; http://dx.doi.org/10.1007/978-3-540-75546-3_12; PMID: 18357774
- Renz H, Autenrieth IB, Brandtzæg P, Cookson WO, Holgate S, von Mutius E, et al. Gene-environment interaction in chronic disease: a European Science Foundation Forward Look. J Allergy Clin Immunol 2011; 128:Suppl S27 - 49; http://dx.doi.org/10.1016/j.jaci.2011.09.039; PMID: 22118218
- Haller MJ, Atkinson MA, Schatz D. Type 1 diabetes mellitus: etiology, presentation, and management. Pediatr Clin North Am 2005; 52:1553 - 78; http://dx.doi.org/10.1016/j.pcl.2005.07.006; PMID: 16301083
- Redondo MJ, Yu L, Hawa M, Mackenzie T, Pyke DA, Eisenbarth GS, et al. Heterogeneity of type I diabetes: analysis of monozygotic twins in Great Britain and the United States. Diabetologia 2001; 44:354 - 62; http://dx.doi.org/10.1007/s001250051626; PMID: 11317668
- Staines A, Bodansky HJ, McKinney PA, Alexander FE, McNally RJ, Law GR, et al. Small area variation in the incidence of childhood insulin-dependent diabetes mellitus in Yorkshire, UK: links with overcrowding and population density. Int J Epidemiol 1997; 26:1307 - 13; http://dx.doi.org/10.1093/ije/26.6.1307; PMID: 9447411
- Patterson CC, Carson DJ, Hadden DR, Northern Ireland Diabetes Study Group. Epidemiology of childhood IDDM in Northern Ireland 1989-1994: low incidence in areas with highest population density and most household crowding. Diabetologia 1996; 39:1063 - 9; http://dx.doi.org/10.1007/BF00400655; PMID: 8877290
- LaPorte RE, Fishbein HA, Drash AL, Kuller LH, Schneider BB, Orchard TJ, et al. The Pittsburgh insulin-dependent diabetes mellitus (IDDM) registry. The incidence of insulin-dependent diabetes mellitus in Allegheny County, Pennsylvania (1965-1976). Diabetes 1981; 30:279 - 84; http://dx.doi.org/10.2337/diabetes.30.4.279; PMID: 7202862
- Karvonen M, Tuomilehto J, Libman I, LaPorte R, World Health Organization DIAMOND Project Group. A review of the recent epidemiological data on the worldwide incidence of type 1 (insulin-dependent) diabetes mellitus. Diabetologia 1993; 36:883 - 92; http://dx.doi.org/10.1007/BF02374468; PMID: 8243865
- Colle E, Siemiatychi J, West R, Belmonte M, Crepeau M, Poirier R, et al. Incidence of juvenile onset diabetes in Montreal-demonstration of ethnic and socio-economic class differences. J Chronic Dis 1981; 34:611 - 6; http://dx.doi.org/10.1016/0021-9681(81)90060-6; PMID: 7309825
- Cherubini V, Carle F, Gesuita R, Iannilli A, Tuomilehto J, Prisco F, et al. Large incidence variation of Type I diabetes in central-southern Italy 1990-1995: lower risk in rural areas. Diabetologia 1999; 42:789 - 92; http://dx.doi.org/10.1007/s001250051228; PMID: 10440119
- D’Angeli MA, Merzon E, Valbuena LF, Tirschwell D, Paris CA, Mueller BA. Environmental factors associated with childhood-onset type 1 diabetes mellitus: an exploration of the hygiene and overload hypotheses. Arch Pediatr Adolesc Med 2010; 164:732 - 8; http://dx.doi.org/10.1001/archpediatrics.2010.115; PMID: 20679164
- Karvonen M, Tuomilehto J, Libman I, LaPorte R, World Health Organization DIAMOND Project Group. A review of the recent epidemiological data on the worldwide incidence of type 1 (insulin-dependent) diabetes mellitus. Diabetologia 1993; 36:883 - 92; http://dx.doi.org/10.1007/BF02374468; PMID: 8243865
- Steck AK, Armstrong TK, Babu SR, Eisenbarth GS, Type 1 Diabetes Genetics Consortium. Stepwise or linear decrease in penetrance of type 1 diabetes with lower-risk HLA genotypes over the past 40 years. Diabetes 2011; 60:1045 - 9; http://dx.doi.org/10.2337/db10-1419; PMID: 21307077
- Fourlanos S, Varney MD, Tait BD, Morahan G, Honeyman MC, Colman PG, et al. The rising incidence of type 1 diabetes is accounted for by cases with lower-risk human leukocyte antigen genotypes. Diabetes Care 2008; 31:1546 - 9; http://dx.doi.org/10.2337/dc08-0239; PMID: 18487476
- Vehik K, Hamman RF, Lezotte D, Norris JM, Klingensmith GJ, Rewers M, et al. Trends in high-risk HLA susceptibility genes among Colorado youth with type 1 diabetes. Diabetes Care 2008; 31:1392 - 6; http://dx.doi.org/10.2337/dc07-2210; PMID: 18356404
- Hermann R, Knip M, Veijola R, Simell O, Laine AP, Akerblom HK, et al, FinnDiane Study Group. Temporal changes in the frequencies of HLA genotypes in patients with Type 1 diabetes--indication of an increased environmental pressure?. Diabetologia 2003; 46:420 - 5; PMID: 12687342
- Witas HW, Jedrychowska-Dańska K, Zawicki P. Changes in frequency of IDDM-associated HLA DQB, CTLA4 and INS alleles. Int J Immunogenet 2010; 37:155 - 8; http://dx.doi.org/10.1111/j.1744-313X.2010.00896.x; PMID: 20345871
- Rook GA. Hygiene hypothesis and autoimmune diseases. Clin Rev Allergy Immunol 2012; 42:5 - 15; http://dx.doi.org/10.1007/s12016-011-8285-8; PMID: 22090147
- Bruno G, Maule M, Merletti F, Novelli G, Falorni A, Iannilli A, et al, RIDI Study Group. Age-period-cohort analysis of 1990-2003 incidence time trends of childhood diabetes in Italy: the RIDI study. Diabetes 2010; 59:2281 - 7; http://dx.doi.org/10.2337/db10-0151; PMID: 20566665
- Aamodt G, Stene LC, Njølstad PR, Søvik O, Joner G, Norwegian Childhood Diabetes Study Group. Spatiotemporal trends and age-period-cohort modeling of the incidence of type 1 diabetes among children aged <15 years in Norway 1973-1982 and 1989-2003. Diabetes Care 2007; 30:884 - 9; http://dx.doi.org/10.2337/dc06-1568; PMID: 17392550
- Lönnrot M, Korpela K, Knip M, Ilonen J, Simell O, Korhonen S, et al. Enterovirus infection as a risk factor for beta-cell autoimmunity in a prospectively observed birth cohort: the Finnish Diabetes Prediction and Prevention Study. Diabetes 2000; 49:1314 - 8; http://dx.doi.org/10.2337/diabetes.49.8.1314; PMID: 10923631
- Andréoletti L, Hober D, Hober-Vandenberghe C, Belaich S, Vantyghem MC, Lefebvre J, et al. Detection of coxsackie B virus RNA sequences in whole blood samples from adult patients at the onset of type I diabetes mellitus. J Med Virol 1997; 52:121 - 7; http://dx.doi.org/10.1002/(SICI)1096-9071(199706)52:2<121::AID-JMV1>3.0.CO;2-5; PMID: 9179756
- Sarmiento L, Cabrera-Rode E, Lekuleni L, Cuba I, Molina G, Fonseca M, et al. Occurrence of enterovirus RNA in serum of children with newly diagnosed type 1 diabetes and islet cell autoantibody-positive subjects in a population with a low incidence of type 1 diabetes. Autoimmunity 2007; 40:540 - 5; http://dx.doi.org/10.1080/08916930701523429; PMID: 17966045
- Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia 2009; 52:1143 - 51; http://dx.doi.org/10.1007/s00125-009-1276-0; PMID: 19266182
- Chapman NM, Kim KS. Persistent coxsackievirus infection: enterovirus persistence in chronic myocarditis and dilated cardiomyopathy. Curr Top Microbiol Immunol 2008; 323:275 - 92; http://dx.doi.org/10.1007/978-3-540-75546-3_13; PMID: 18357775
- Tracy S, Chapman NM, McManus BM, Pallansch MA, Beck MA, Carstens J. A molecular and serologic evaluation of enteroviral involvement in human myocarditis. J Mol Cell Cardiol 1990; 22:403 - 14; http://dx.doi.org/10.1016/0022-2828(90)91476-N; PMID: 2167382
- Chapman NM, Kim KS, Drescher KM, Oka K, Tracy S. 5′ terminal deletions in the genome of a coxsackievirus B2 strain occurred naturally in human heart. Virology 2008; 375:480 - 91; http://dx.doi.org/10.1016/j.virol.2008.02.030; PMID: 18378272
- Kim KS, Chapman NM, Tracy S. Replication of coxsackievirus B3 in primary cell cultures generates novel viral genome deletions. J Virol 2008; 82:2033 - 7; http://dx.doi.org/10.1128/JVI.01774-07; PMID: 18057248
- Kim KS, Tracy S, Tapprich W, Bailey J, Lee CK, Kim K, et al. 5′-Terminal deletions occur in coxsackievirus B3 during replication in murine hearts and cardiac myocyte cultures and correlate with encapsidation of negative-strand viral RNA. J Virol 2005; 79:7024 - 41; http://dx.doi.org/10.1128/JVI.79.11.7024-7041.2005; PMID: 15890942
- van der Werf N, Kroese FG, Rozing J, Hillebrands JL. Viral infections as potential triggers of type 1 diabetes. Diabetes Metab Res Rev 2007; 23:169 - 83; http://dx.doi.org/10.1002/dmrr.695; PMID: 17103489
- Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med 2002; 347:911 - 20; http://dx.doi.org/10.1056/NEJMra020100; PMID: 12239261
- Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS One 2011; 6:e25792; http://dx.doi.org/10.1371/journal.pone.0025792; PMID: 22043294
- Valladares R, Sankar D, Li N, Williams E, Lai KK, Abdelgeliel AS, et al. Lactobacillus johnsonii N6.2 mitigates the development of type 1 diabetes in BB-DP rats. PLoS One 2010; 5:e10507; http://dx.doi.org/10.1371/journal.pone.0010507; PMID: 20463897
- Neu J, Lorca G, Kingma SD, Triplett EW. The intestinal microbiome: relationship to type 1 diabetes. Endocrinol Metab Clin North Am 2010; 39:563 - 71; http://dx.doi.org/10.1016/j.ecl.2010.05.008; PMID: 20723820
- Roesch LF, Lorca GL, Casella G, Giongo A, Naranjo A, Pionzio AM, et al. Culture-independent identification of gut bacteria correlated with the onset of diabetes in a rat model. ISME J 2009; 3:536 - 48; http://dx.doi.org/10.1038/ismej.2009.5; PMID: 19225551
- King C, Sarvetnick N. The incidence of type-1 diabetes in NOD mice is modulated by restricted flora not germ-free conditions. PLoS One 2011; 6:e17049; http://dx.doi.org/10.1371/journal.pone.0017049; PMID: 21364875
- Vaarala O, Atkinson MA, Neu J. The “perfect storm” for type 1 diabetes: the complex interplay between intestinal microbiota, gut permeability, and mucosal immunity. Diabetes 2008; 57:2555 - 62; http://dx.doi.org/10.2337/db08-0331; PMID: 18820210
- Atkinson MA, Leiter EH. The NOD mouse model of type 1 diabetes: as good as it gets?. Nat Med 1999; 5:601 - 4; http://dx.doi.org/10.1038/9442; PMID: 10371488
- Strachan DP. Hay fever, hygiene, and household size. BMJ 1989; 299:1259 - 60; http://dx.doi.org/10.1136/bmj.299.6710.1259; PMID: 2513902
- Tracy S, Drescher KM, Jackson JD, Kim K, Kono K. Enteroviruses, type 1 diabetes and hygiene: a complex relationship. Rev Med Virol 2010; 20:106 - 16; http://dx.doi.org/10.1002/rmv.639; PMID: 20049905
- Okada H, Kuhn C, Feillet H, Bach JF. The ‘hygiene hypothesis’ for autoimmune and allergic diseases: an update. Clin Exp Immunol 2010; 160:1 - 9; http://dx.doi.org/10.1111/j.1365-2249.2010.04139.x; PMID: 20415844
- An PG. Constructing and dismantling frameworks of disease etiology: the rise and fall of sewer gas in America, 1870-1910. Yale J Biol Med 2004; 77:75 - 100; PMID: 15829149
- Halliday S. Death and miasma in Victorian London: an obstinate belief. BMJ 2001; 323:1469 - 71; http://dx.doi.org/10.1136/bmj.323.7327.1469; PMID: 11751359
- Delaporte F. Disease and Civilization: The Cholera in Paris, 1832. Cambridge, MA: The MIT Press, 1989.
- Cantalupo PG, Calgua B, Zhao G, Hundesa A, Wier AD, Katz JP, et al. Raw sewage harbors diverse viral populations. MBio 2011; 2; http://dx.doi.org/10.1128/mBio.00180-11; PMID: 21972239
- Nathanson N, Martin JR. The epidemiology of poliomyelitis: enigmas surrounding its appearance, epidemicity, and disappearance. Am J Epidemiol 1979; 110:672 - 92; PMID: 400274
- Bunimovich-Mendrazitsky S, Stone L. Modeling polio as a disease of development. J Theor Biol 2005; 237:302 - 15; http://dx.doi.org/10.1016/j.jtbi.2005.04.017; PMID: 15975604
- Paul JR. A History of Poliomyelitis. Yale University Press, 1978.
- Horstmann DM. The poliomyelitis story: a scientific hegira. Yale J Biol Med 1985; 58:79 - 90; PMID: 2994307
- Modlin JF. Poliomyelitis and poliovirus immunization. In: Rotbart HA, ed. Human Enterovirus Infections. Washington, D.C.: ASM Press, 1995:195-220.
- Minor PD. Poliovirus. In: Nathanson N, ed. Viral Pathogenesis. Philadelphia: Lippincott-Raven, 1996:555-74.
- Nathanson N, Kew OM. From emergence to eradication: the epidemiology of poliomyelitis deconstructed. Am J Epidemiol 2010; 172:1213 - 29; http://dx.doi.org/10.1093/aje/kwq320; PMID: 20978089
- Horstmann DM. Poliomyelitis: severity and type of disease in different age groups. Ann N Y Acad Sci 1955; 61:956 - 67; http://dx.doi.org/10.1111/j.1749-6632.1955.tb42554.x; PMID: 13340604
- Fales WT, Taback M. Observations on the epidemiology of poliomyelitis. Public Health Rep 1952; 67:47 - 52; http://dx.doi.org/10.2307/4587979; PMID: 14883288
- Nathanson N, McGann KA, Wilesmith J, Desrosiers RC, Brookmeyer R. The evolution of virus diseases: their emergence, epidemicity, and control. Virus Res 1993; 29:3 - 20; http://dx.doi.org/10.1016/0168-1702(93)90122-4; PMID: 8212850
- Nathanson N, Murphy FA. Evolution of viral diseases. In: Nathanson N, ed. Viral Pathogenesis. Philadelphia: Lippincott-Raven, 1996:353-70.
- Pallansch M, Roos R. Enteroviruses: Polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses. In: Knipe DM, ed. Fields Virology. Philadelphia: Wolters Kluwer, 2007:839-94.
- Karvonen M, Viik-Kajander M, Moltchanova E, Libman I, LaPorte R, Tuomilehto J. Incidence of childhood type 1 diabetes worldwide. Diabetes Mondiale (DiaMond) Project Group. Diabetes Care 2000; 23:1516 - 26; http://dx.doi.org/10.2337/diacare.23.10.1516; PMID: 11023146
- Padaiga Z, Tuomilehto J, Karvonen M, Podar T, Brigis G, Urbonaite B, et al. Incidence trends in childhood onset IDDM in four countries around the Baltic sea during 1983-1992. Diabetologia 1997; 40:187 - 92; http://dx.doi.org/10.1007/s001250050661; PMID: 9049479
- Viskari H, Ludvigsson J, Uibo R, Salur L, Marciulionyte D, Hermann R, et al. Relationship between the incidence of type 1 diabetes and maternal enterovirus antibodies: time trends and geographical variation. Diabetologia 2005; 48:1280 - 7; http://dx.doi.org/10.1007/s00125-005-1780-9; PMID: 15902401
- Roep BO, Atkinson M, von Herrath M. Satisfaction (not) guaranteed: re-evaluating the use of animal models of type 1 diabetes. Nat Rev Immunol 2004; 4:989 - 97; http://dx.doi.org/10.1038/nri1502; PMID: 15573133
- Tracy S, Drescher KM, Chapman NM, Kim KS, Carson SD, Pirruccello S, et al. Toward testing the hypothesis that group B coxsackieviruses (CVB) trigger insulin-dependent diabetes: inoculating nonobese diabetic mice with CVB markedly lowers diabetes incidence. J Virol 2002; 76:12097 - 111; http://dx.doi.org/10.1128/JVI.76.23.12097-12111.2002; PMID: 12414951
- Drescher KM, Kono K, Bopegamage S, Carson SD, Tracy S. Coxsackievirus B3 infection and type 1 diabetes development in NOD mice: insulitis determines susceptibility of pancreatic islets to virus infection. Virology 2004; 329:381 - 94; http://dx.doi.org/10.1016/j.virol.2004.06.049; PMID: 15518817
- Kanno T, Kim K, Kono K, Drescher KM, Chapman NM, Tracy S. Group B coxsackievirus diabetogenic phenotype correlates with replication efficiency. J Virol 2006; 80:5637 - 43; http://dx.doi.org/10.1128/JVI.02361-05; PMID: 16699045
- Pevear DC, Oh CK, Cunningham LL, Calenoff M, Jubelt B. Localization of genomic regions specific for the attenuated, mouse-adapted poliovirus type 2 strain W-2. J Gen Virol 1990; 71:43 - 52; http://dx.doi.org/10.1099/0022-1317-71-1-43; PMID: 2154539
- Couderc T, Guinguene B, Horaud F, Aubert-Combiescu A, Crainic R. Molecular pathogenesis of type 2 poliovirus in mice. Eur J Epidemiol 1989; 5:270 - 4; http://dx.doi.org/10.1007/BF00144825; PMID: 2551721
- Racaniello VR. One hundred years of poliovirus pathogenesis. Virology 2006; 344:9 - 16; http://dx.doi.org/10.1016/j.virol.2005.09.015; PMID: 16364730
- Racaniello VR, Ren R. Transgenic mice and the pathogenesis of poliomyelitis. Arch Virol Suppl 1994; 9:79 - 86; PMID: 8032284
- Liu W, Huber SA. Cross-talk between cd1d-restricted nkt cells and γδ cells in t regulatory cell response. Virol J 2011; 8:32; http://dx.doi.org/10.1186/1743-422X-8-32; PMID: 21255407
- Gauntt C, Huber S. Coxsackievirus experimental heart diseases. Front Biosci 2003; 8:e23 - 35; http://dx.doi.org/10.2741/928; PMID: 12456330
- Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, et al. Microbial Exposure During Early Life Has Persistent Effects on Natural Killer T Cell Function. Science (New York, NY) 2012.
- Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, et al. Peripheral education of the immune system by colonic commensal microbiota. Nature 2011; 478:250 - 4; http://dx.doi.org/10.1038/nature10434; PMID: 21937990
- Kriegel MA, Sefik E, Hill JA, Wu HJ, Benoist C, Mathis D. Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc Natl Acad Sci U S A 2011; 108:11548 - 53; http://dx.doi.org/10.1073/pnas.1108924108; PMID: 21709219
- Giongo A, Gano KA, Crabb DB, Mukherjee N, Novelo LL, Casella G, et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J 2011; 5:82 - 91; http://dx.doi.org/10.1038/ismej.2010.92; PMID: 20613793
- Björkstén B. The hygiene hypothesis: do we still believe in it?. Nestle Nutr Workshop Ser Pediatr Program 2009; 64:11 - 8, discussion 18-22, 251-7; http://dx.doi.org/10.1159/000235780; PMID: 19710512
- Froeschle JE, Feorino PM, Gelfand HM. A continuing surveillance of enterovirus infection in healthy children in six United States cities. II. Surveillance enterovirus isolates 1960-1963 and comparison with enterovirus isolates from cases of acute central nervous system disease. Am J Epidemiol 1966; 83:455 - 69; PMID: 5932697
- Roep BO, Atkinson M. Animal models have little to teach us about type 1 diabetes: 1. In support of this proposal. Diabetologia 2004; 47:1650 - 6; http://dx.doi.org/10.1007/s00125-004-1517-1; PMID: 15490110
- In’t Veld P. Insulitis in human type 1 diabetes: The quest for an elusive lesion. Islets 2011; 3:131 - 8; http://dx.doi.org/10.4161/isl.3.4.15728; PMID: 21606672
- Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Immunohistochemical analysis of the relationship between islet cell proliferation and the production of the enteroviral capsid protein, VP1, in the islets of patients with recent-onset type 1 diabetes. Diabetologia 2011; 54:2417 - 20; http://dx.doi.org/10.1007/s00125-011-2192-7; PMID: 21597997
- Tracy S, Drescher KM. Coxsackievirus infections and NOD mice: relevant models of protection from, and induction of, type 1 diabetes. Ann N Y Acad Sci 2007; 1103:143 - 51; http://dx.doi.org/10.1196/annals.1394.009; PMID: 17376828
- von Herrath MG, Fujinami RS, Whitton JL. Microorganisms and autoimmunity: making the barren field fertile?. Nat Rev Microbiol 2003; 1:151 - 7; http://dx.doi.org/10.1038/nrmicro754; PMID: 15035044
- Berger CN, Sodha SV, Shaw RK, Griffin PM, Pink D, Hand P, et al. Fresh fruit and vegetables as vehicles for the transmission of human pathogens. Environ Microbiol 2010; 12:2385 - 97; http://dx.doi.org/10.1111/j.1462-2920.2010.02297.x; PMID: 20636374
- Lynch MF, Tauxe RV, Hedberg CW. The growing burden of foodborne outbreaks due to contaminated fresh produce: risks and opportunities. Epidemiol Infect 2009; 137:307 - 15; http://dx.doi.org/10.1017/S0950268808001969; PMID: 19200406
- Heaton JC, Jones K. Microbial contamination of fruit and vegetables and the behaviour of enteropathogens in the phyllosphere: a review. J Appl Microbiol 2008; 104:613 - 26; http://dx.doi.org/10.1111/j.1365-2672.2007.03587.x; PMID: 17927745
- Sivapalasingam S, Friedman CR, Cohen L, Tauxe RV. Fresh produce: a growing cause of outbreaks of foodborne illness in the United States, 1973 through 1997. J Food Prot 2004; 67:2342 - 53; PMID: 15508656
- Matricardi PM, Bonini S. Mimicking microbial ‘education’ of the immune system: a strategy to revert the epidemic trend of atopy and allergic asthma?. Respir Res 2000; 1:129 - 32; http://dx.doi.org/10.1186/rr22; PMID: 11667975
- Muegge BD, Kuczynski J, Knights D, Clemente JC, González A, Fontana L, et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 2011; 332:970 - 4; http://dx.doi.org/10.1126/science.1198719; PMID: 21596990
- Witsø E, Cinek O, Aldrin M, Grinde B, Rasmussen T, Wetlesen T, et al. Predictors of sub-clinical enterovirus infections in infants: a prospective cohort study. Int J Epidemiol 2010; 39:459 - 68; http://dx.doi.org/10.1093/ije/dyp333; PMID: 19939809
- Penders J, Thijs C, Vink C, Stelma FF, Snijders B, Kummeling I, et al. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 2006; 118:511 - 21; http://dx.doi.org/10.1542/peds.2005-2824; PMID: 16882802
- Kelly D, King T, Aminov R. Importance of microbial colonization of the gut in early life to the development of immunity. Mutat Res 2007; 622:58 - 69; http://dx.doi.org/10.1016/j.mrfmmm.2007.03.011; PMID: 17612575
- Schwartz RF, Neu J, Schatz D, Atkinson MA, Wasserfall C. Comment on: Brugman S et al. (2006) Antibiotic treatment partially protects against type 1 diabetes in the Bio-Breeding diabetes-prone rat. Is the gut flora involved in the development of type 1 diabetes? Diabetologia 49:2105-2108. Diabetologia 2007; 50:220 - 1; http://dx.doi.org/10.1007/s00125-006-0526-7; PMID: 17119915
- Brugman S, Klatter FA, Visser JT, Wildeboer-Veloo AC, Harmsen HJ, Rozing J, et al. Antibiotic treatment partially protects against type 1 diabetes in the Bio-Breeding diabetes-prone rat. Is the gut flora involved in the development of type 1 diabetes?. Diabetologia 2006; 49:2105 - 8; http://dx.doi.org/10.1007/s00125-006-0334-0; PMID: 16816951
- Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A 2010; 107:12204 - 9; http://dx.doi.org/10.1073/pnas.0909122107; PMID: 20566854
- Wingender G, Stepniak D, Krebs P, Lin L, McBride S, Wei B, et al. Intestinal microbes affect phenotypes and functions of invariant natural killer T cells in mice. Gastroenterology 2012; 143:418 - 28; http://dx.doi.org/10.1053/j.gastro.2012.04.017; PMID: 22522092
- Romano-Keeler J, Weitkamp JH, Moore DJ. Regulatory properties of the intestinal microbiome effecting the development and treatment of diabetes. Curr Opin Endocrinol Diabetes Obes 2012; 19:73 - 80; PMID: 22357099
- Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet 2012; 13:260 - 70; PMID: 22411464
- Petrovsky N. Immunomodulation with microbial vaccines to prevent type 1 diabetes mellitus. Nat Rev Endocrinol 2010; 6:131 - 8; http://dx.doi.org/10.1038/nrendo.2009.273; PMID: 20173774
- Boerner BP, Sarvetnick NE. Type 1 diabetes: role of intestinal microbiome in humans and mice. Ann N Y Acad Sci 2011; 1243:103 - 18; http://dx.doi.org/10.1111/j.1749-6632.2011.06340.x; PMID: 22211896
- Juhela S, Hyöty H, Roivainen M, Härkönen T, Putto-Laurila A, Simell O, et al. T-cell responses to enterovirus antigens in children with type 1 diabetes. Diabetes 2000; 49:1308 - 13; http://dx.doi.org/10.2337/diabetes.49.8.1308; PMID: 10923630
- Filippi CM, Estes EA, Oldham JE, von Herrath MG. Immunoregulatory mechanisms triggered by viral infections protect from type 1 diabetes in mice. J Clin Invest 2009; 119:1515 - 23; PMID: 19478458
- Strauch UG, Obermeier F, Grunwald N, Gürster S, Dunger N, Schultz M, et al. Influence of intestinal bacteria on induction of regulatory T cells: lessons from a transfer model of colitis. Gut 2005; 54:1546 - 52; http://dx.doi.org/10.1136/gut.2004.059451; PMID: 15987795
- Ylipaasto P, Klingel K, Lindberg AM, Otonkoski T, Kandolf R, Hovi T, et al. Enterovirus infection in human pancreatic islet cells, islet tropism in vivo and receptor involvement in cultured islet beta cells. Diabetologia 2004; 47:225 - 39; http://dx.doi.org/10.1007/s00125-003-1297-z; PMID: 14727023
- Smura T, Ylipaasto P, Klemola P, Kaijalainen S, Kyllönen L, Sordi V, et al. Cellular tropism of human enterovirus D species serotypes EV-94, EV-70, and EV-68 in vitro: implications for pathogenesis. J Med Virol 2010; 82:1940 - 9; http://dx.doi.org/10.1002/jmv.21894; PMID: 20872722
- Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, et al. Metagenomic analysis of the human distal gut microbiome. Science 2006; 312:1355 - 9; http://dx.doi.org/10.1126/science.1124234; PMID: 16741115
- Sekirov I, Finlay BB. Human and microbe: united we stand. Nat Med 2006; 12:736 - 7; http://dx.doi.org/10.1038/nm0706-736; PMID: 16829917
- Goodacre R. Metabolomics of a superorganism. J Nutr 2007; 137:Suppl 259S - 66S; PMID: 17182837
- Shaw SY, Blanchard JF, Bernstein CN. Association between the use of antibiotics in the first year of life and pediatric inflammatory bowel disease. Am J Gastroenterol 2010; 105:2687 - 92; http://dx.doi.org/10.1038/ajg.2010.398; PMID: 20940708
- Ajslev TA, Andersen CS, Gamborg M, Sørensen TI, Jess T. Childhood overweight after establishment of the gut microbiota: the role of delivery mode, pre-pregnancy weight and early administration of antibiotics. Int J Obes (Lond) 2011; 35:522 - 9; http://dx.doi.org/10.1038/ijo.2011.27; PMID: 21386800
- Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 2008; 455:1109 - 13; http://dx.doi.org/10.1038/nature07336; PMID: 18806780
- Calcinaro F, Dionisi S, Marinaro M, Candeloro P, Bonato V, Marzotti S, et al. Oral probiotic administration induces interleukin-10 production and prevents spontaneous autoimmune diabetes in the non-obese diabetic mouse. Diabetologia 2005; 48:1565 - 75; http://dx.doi.org/10.1007/s00125-005-1831-2; PMID: 15986236

