
Abstract
The aim of this study was to assess multifactorial β-cell responses to metabolic perturbations in primary rat and human islets. Treatment of dispersed rat islet cells with elevated glucose and free fatty acids (FFAs, oleate:palmitate = 1:1 v/v) resulted in increases in the size and the number of lipid droplets in β-cells in a time- and concentration-dependent manner. Glucose and FFAs synergistically stimulated the nutrient sensor mammalian target of rapamycin complex 1 (mTORC1). A potent mTORC1 inhibitor, rapamycin (25 nM), significantly reduced triglyceride accumulation in rat islets. Importantly, lipid droplets accumulated only in β-cells but not in α-cells in an mTORC1-dependent manner. Nutrient activation of mTORC1 upregulated the expression of adipose differentiation related protein (ADRP), known to stabilize lipid droplets. Rat islet size and new DNA synthesis also increased under nutrient overload. Insulin secretion into the culture medium increased steadily over a 4-day period without any significant difference between glucose (10 mM) alone and the combination of glucose (10 mM) and FFAs (240 μM). Insulin content and insulin biosynthesis, however, were significantly reduced under the combination of nutrients compared with glucose alone. Elevated nutrients also stimulated lipid droplet formation in human islets in an mTORC1-dependent manner. Unlike rat islets, however, human islets did not increase in size under nutrient overload despite a normal response to nutrients in releasing insulin. The different responses of islet cell growth under nutrient overload appear to impact insulin biosynthesis and storage differently in rat and human islets.
Introduction
Type 2 diabetes mellitus (T2DM) is characterized by insulin resistance and defects in β-cell function, growth, and survival. Chronic nutrient overload associated with obesity is implicated in both insulin resistance and β-cell defects. Adaptive responses of β-cells under conditions of insulin resistance and nutrient overload, including lipid accumulation and β-cell mass expansion, have been widely observed in various experimental models.Citation1-Citation5 However, the specific molecular mechanisms by which nutrients promote lipid accumulation and β-cell mass expansion and the causal relationship between these adaptive responses and β-cell defects are not clearly understood. Furthermore, metabolic and morphological changes of human islets under nutrient overload are poorly understood, despite their potential impact on understanding the etiology of T2DM.
The storage of TG in lipid droplets requires the stimulation of the two pathways: TG biosynthesis through the esterification pathwayCitation6 and expression of lipid droplet-associated proteins that stabilize lipid droplets by preventing lipolysis. Lipid droplets contain neutral lipids (TG, cholesterol ester) in the core, surrounded by a phospholipid monolayer and a coat of specific proteins.Citation7 These lipid droplet-associated proteins are designated as the PAT proteins [perilipin, adipophilin/adipose differentiation related protein (ADRP), TIP47 and other related proteins], which play structural and functional roles in cellular lipid metabolism.Citation8 Both perilipin and ADRP have been found in the rat and human islets.Citation9,Citation10
The expansion of β-cell mass has been observed in genetically modified obese rodents,Citation4,Citation11 normal mice fed a high-fat dietCitation5,Citation12 and mice infused with elevated levels of glucose or FFAs.Citation13,Citation14 The increase in β-cell mass associated with insulin resistance and nutrient overload has been widely accepted to correlate with hypersecretion of insulin, providing a compensatory mechanism to overcome insulin resistance. However, recent evidence that an increase in β-cell mass under conditions such as a chronic high fat diet or 60% pancreatectomy was not associated with a corresponding improvement of β-cell functionCitation12,Citation15-Citation17 suggests that an increase in β-cell mass is not always correlated with enhanced β-cell function. The causal relationship between β-cell mass expansion and β-cell function under nutrient overload, therefore, remains to be elucidated.
Under chronic nutrient overload, rodent β-cells undergo characteristic metabolic adaptations including lipid accumulation and β-cell mass expansion. We propose that mammalian target of rapamycin complex 1 (mTORC1), a conserved serine/threonine kinase that functions as a nutrient sensor, is a common mediator that regulates both responses. mTORC1 is known to integrate signals from growth factors and nutrients to regulate protein translation, DNA synthesis, cell size, and proliferation.Citation18-Citation23 Two prominent downstream targets of mTORC1 are the 70-kDa ribosomal protein S6 kinase (S6K1) and eukaryotic initiation factor 4E-binding protein 1 (4EBP1), which regulate protein translation.Citation20,Citation24 The mTORC1 signaling pathway has been implicated as a link between nutrient excess and development of both obesity and insulin resistance.Citation25,Citation26 Recent evidence indicates that mTORC1 signaling is also implicated in β-cell regenerative processes.Citation27-Citation30 Furthermore, the present study suggests that mTORC1 plays a pivotal role in lipid accumulation in β-cells.
Preservation of β-cell function and mass under chronic nutrient overload is of great interest as a potential strategy for therapeutic intervention for type 2 diabetes. To this end, we studied the complex β-cell metabolic responses to nutrient overload in rat and human islets using a combination of microscopy and biochemical methods. We report that mTORC1 plays a central role in lipid accumulation as well as rat islet cell expansion, which, in turn, is intricately associated with β-cell defects. Human and rat islets showed similar ectopic lipid accumulation and insulin release in response to nutrient overload. Unlike rat islets, however, human islets did not increase in size under nutrient overload, suggesting that nutrient-mediated mTORC1 activation alone is not sufficient to stimulate human islet cell growth. The lack of islet cell expansion under nutrient overload appears to have a significant impact in insulin biosynthesis and storage in human islets.
Results
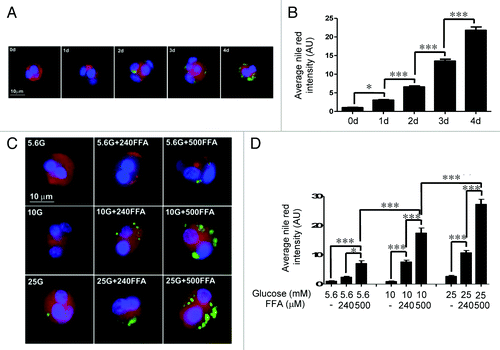
Lipid droplet formation in primary rat β-cells
Lipid droplet formation in dispersed rat islet cells treated with 25 mM glucose and 500 μM FFAs (oleate: palmitate = 1:1 v/v) was monitored by capturing phase contrast digital images at 30 min intervals for 40 hours using a 20× objective. shows that lipid droplets appeared after ~15 h and increased in size and number over 40 hours. Over time, the initially round islet cells became more flattened and appeared to form aggregates. Next, we quantitated lipid droplets in clusters (< 5 cells) of single rat β-cells by co-localizing insulin and Nile red, an indicator of TG content. shows a time course of lipid droplet formation in β-cells treated with 25 mM glucose and 500 μM FFAs. Lipid droplets were detected after 1 day and increased steadily over 4 days. displays quantitation of the average Nile red intensity for ~80 clusters of β-cells per condition. illustrate glucose and FFAs dose-response of lipid droplet formation in β-cells after 4 days. Basal (5.6 mM) or moderately elevated (10 mM) glucose alone did not result in lipid droplet formation, whereas high (25 mM) glucose alone resulted in a small increase in lipid droplet formation. Increasing concentrations of FFAs (240, 500 μM) in combination with basal glucose (5.6 mM) showed increases in lipid droplet formation but at a low level. A combination of elevated glucose and FFAs markedly increased lipid droplet formation in a dose-dependent manner with the maximal effect at 25 mM glucose + 500 μM FFAs.
Figure 1. Time lapse studies monitoring lipid droplet formation in dispersed islet cells. Phase contrast digital images of dispersed islet cells treated with 25 mM glucose and 500 μM FFAs (oleate:palmitate = 1:1v/v) were acquired at 30 min intervals for 40 hours using a 20× objective. Representative images at the indicated time (h) are shown. An enlarged image indicated by the white arrow (far right panel) demonstrates that lipid droplets were uniform in size. Images were rescaled using Adobe Photoshop software. Results are representative of three independent experiments.

Figure 2. (A) Time course of lipid droplet formation in dispersed rat β-cells. Dispersed rat islet cells (105) were treated for the indicated time periods with 25 mM glucose and 500 μM FFAs. After treatment, cells were processed for immunostaining. Insulin (red), nuclei (blue), and Nile red staining (green) are shown. (B) The bar graph shows the average Nile red intensity for a total of ~80 clusters of insulin-positive cells per time point from three independent experiments is shown. (C) Dose-dependent effects of glucose and FFAs on lipid droplet formation. Dispersed rat islet cells (105) were treated for 4 days as indicated. After treatment, cells were processed for immunostaining. Insulin (red), nuclei (blue), and Nile red staining (green) are shown. 5.6G and 240FFA denote 5.6 mM glucose and 240 μM FFAs, respectively. (D) The bar graph shows the average Nile red intensity for a total of ~80 clusters of insulin-positive cells per condition from three independent experiments.
The role of mTORC1 in the formation of lipid droplets and ADRP expression in rat islets
The role of mTORC1 in lipid droplet formation was studied by determining the effect of nutrients and rapamycin (a specific and potent inhibitor of mTORC1) on the phosphorylation of S6 (an indicator of mTORC1 activation). We determined the ratio of p-S6 to β-actin levels to normalize for alterations in protein levels caused by different nutrient treatment conditions. For these experiments, we used moderately elevated concentrations of nutrients to study the synergistic actions of glucose and FFAs. indicates that rat islets treated with 10 mM glucose (lane 1) showed low levels of mTORC1 activation, while islets treated with 240 μM FFAs in the presence of 5.6 mM glucose (lane 5) showed no detectable mTORC1 activation. Islets treated with a combination of 10 mM glucose and 240 μM FFAs (lane 3) exhibited ~3-fold increase in S6 phosphorylation, indicating a synergistic effect of glucose and FFAs on mTORC1 activation. Rapamycin completely blocked S6 phosphorylation under all conditions. shows that treatment of intact islets with 25 mM glucose + 500 μM FFAs for 4 days caused a significant increase in islet TG content compared with those treated with 5.6 mM glucose alone, and rapamycin significantly blocked TG accumulation by 30%. The effects of rapamycin on lipid droplet formation in β- and α-cells exposed to excess nutrients for 4 days were also determined. shows that insulin-positive β-cells treated with 25 mM glucose + 500 μM FFAs (panel b) contained numerous lipid droplets (green), whereas control (5.6 mM glucose alone, panel a) or rapamycin treated condition (panel c) is devoid of lipid droplets. Panels d, e and f show that glucagon-positive α-cells contained no lipid droplets regardless of experimental conditions. shows the quantitation of the average Nile red intensity of ~80 β- or α-cells/condition illustrated in the above panels. Next, we studied nutrient-mediated lipid droplet formation in intact rat islets by immunohistochemistry of frozen islet sections. An islet treated with 25 mM glucose + 500 μM FFAs for 4 days (, panel g) resulted in elevated lipid droplet formation compared with control islet (panel c). Rapamycin significantly blocked nutrient-mediated lipid droplet formation (panel k). A comparison of the nuclear staining (DAPI) of two islets (panels b and f) indicate that nutrient overload caused islet cell expansion as shown by greater distance between the nuclei in panel f compared with panel b. Rapamycin, as anticipated, completely blocked nutrient-mediated islet cell expansion, shown by more compact clusters of nuclei (panel j). Insulin content was greatly reduced under the condition of excess nutrients and rapamycin did not prevent this reduction (compare panels a, e and i). shows quantitation of insulin content and lipid accumulation determined by the average intensity of insulin and Nile red of the three conditions.
Figure 3. Nutrient-mediated activation of mTORC1. Rat islets (100) were treated for 4 days as indicated. Samples were processed for Western blotting for phosphorylated S6, stripped, and reblotted for β-actin. Band intensities were quantitated by Biorad ChemiDoc XRS Image Lab Software. The ratio of p-S6/β-actin was obtained as described in the Methods section. Data are the means ± SEM of n = 3 experiments.
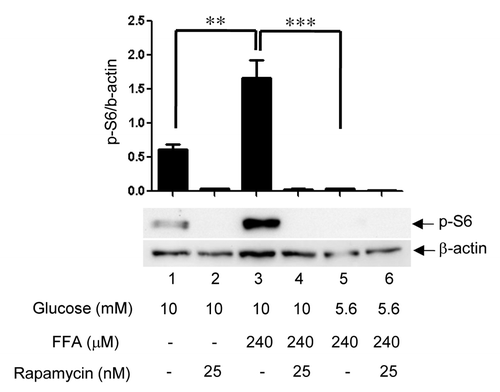
Figure 4.(A) mTORC1-dependent TG accumulation. Rat islets (150) were cultured for 4 days as indicated. After treatment, lipid extraction by chloroform:methanol was performed and TG quantitated. Data are the means ± SEM of n = 3 experiments with duplicate samples in each experiment. (B) mTORC1 mediates lipid droplet formation exclusively in β-cells, sparing α-cells. Dispersed rat islet cells (105) were cultured for 4 days in cCMRL-1066 containing 5.6 mM glucose, 25 mM glucose + 500 μM FFAs, or 25 mM glucose + 500μM FFAs+25 nM rapamcyin. After treatment, cells were processed for immunostaining. Representative images of β-cells (panels a, b and c) and α-cells (panels d, e and f) under the three conditions [insulin and glucagon (red), nuclei (blue), and Nile red staining (green)]. (C) The bar graph shows the average Nile red intensity of a total of ~50 β- or α-cells per condition from three independent experiments is shown.
![Figure 4.(A) mTORC1-dependent TG accumulation. Rat islets (150) were cultured for 4 days as indicated. After treatment, lipid extraction by chloroform:methanol was performed and TG quantitated. Data are the means ± SEM of n = 3 experiments with duplicate samples in each experiment. (B) mTORC1 mediates lipid droplet formation exclusively in β-cells, sparing α-cells. Dispersed rat islet cells (105) were cultured for 4 days in cCMRL-1066 containing 5.6 mM glucose, 25 mM glucose + 500 μM FFAs, or 25 mM glucose + 500μM FFAs+25 nM rapamcyin. After treatment, cells were processed for immunostaining. Representative images of β-cells (panels a, b and c) and α-cells (panels d, e and f) under the three conditions [insulin and glucagon (red), nuclei (blue), and Nile red staining (green)]. (C) The bar graph shows the average Nile red intensity of a total of ~50 β- or α-cells per condition from three independent experiments is shown.](/cms/asset/77343559-911f-4275-966f-c29b8fd00d0e/kisl_a_10922720_f0004.gif)
Figure 5.(A) Rat islets (50) were cultured for 4 days in cCMRL containing 5.6 mM glucose, 25 mM glucose + 500 μM FFAs, or 25 mM glucose + 500 μM FFAs + 25 nM rapamycin. Frozen sections (5 μm) of islets were processed for immunostaining [insulin (green), nuclei (blue), and Nile red (red)] as described in the Methods section. Images were rescaled using Adobe Photoshop software. (B) The bar graph shows the average intensities of Nile red for lipid accumulation and DyLight 647 for insulin, respectively, of 15–20 islet sections per condition from three different experiments.
![Figure 5.(A) Rat islets (50) were cultured for 4 days in cCMRL containing 5.6 mM glucose, 25 mM glucose + 500 μM FFAs, or 25 mM glucose + 500 μM FFAs + 25 nM rapamycin. Frozen sections (5 μm) of islets were processed for immunostaining [insulin (green), nuclei (blue), and Nile red (red)] as described in the Methods section. Images were rescaled using Adobe Photoshop software. (B) The bar graph shows the average intensities of Nile red for lipid accumulation and DyLight 647 for insulin, respectively, of 15–20 islet sections per condition from three different experiments.](/cms/asset/06bf8ca6-c7da-47d4-9766-ae1100ec0a94/kisl_a_10922720_f0005.gif)
Next, we determined nutrient-mediated ADRP expression in islets (100 islets per condition) by determining the ratio of ADRP to the internal control β-actin to account for the alteration in protein levels under different experimental conditions. demonstrates that ADRP expression increased in a dose-dependent manner with nutrient overload. shows that nutrient activation of mTORC1 upregulated ADRP expression in rat islets. Treatment of islets with 25 mM glucose alone (lane 2) resulted in an ~2-fold increase in ADRP expression compared with 5.6 mM glucose (lane 1). The combination of 25 mM glucose and 500 μM FFAs (lane 4) resulted in an ~3-fold increase in ADRP expression compared with 5.6 mM glucose. Rapamycin treatment (lanes 3 and 5) prevented the nutrient mediated increase in ADRP expression. Immunohistochemical studies indicated that ADRP (red) was co-localized with lipid droplets as shown by the appearance of ADRP around small distinct spheres in the cytosol (, panel b). also demonstrates that rapamycin inhibited nutrient-mediated lipid droplet formation as well as β-cell expansion (compare panel c vs. panel b). Moreover, insulin content (intensity of green) was significantly reduced by treatment with 25 mM glucose and 500 μM FFAs (compare panel a vs. panel b) and rapamycin did not alter this effect. shows the quantitation of the average ADRP and insulin intensity of ~90 β-cells/condition from 3 independent experiments.
Figure 6.(A) Dose-dependent effects of nutrients on ADRP expression. Rat islets (100) were cultured for 4 days in cCMRL-1066 containing 5.6 mM glucose, 10 mM glucose + 240μM FFAs, or 25 mM glucose + 500 μM FFAs. Samples were processed for Western blotting and quantitated by densitometry. β-actin was used as a protein loading control. Data are the means ± SEM of n = 3 experiments. (B) Glucose and FFAs synergize in increasing mTORC1-mediated ADRP expression. Rat islets (100) were cultured for 4 days in cCMRL-1066 under various conditions as indicated. Samples were processed for western blotting and quantitated by densitometry. β-actin was used as a protein loading control. Data are the means ± SEM of n = 3 experiments. (C) ADRP expression and insulin content in single β-cells determined by immunohistochemistry. Dispersed rat islet cells (105) were cultured for 4 days in cCMRL-1066 containing 5.6 mM glucose, 25 mM glucose + 500 μM FFAs, or 25 mM glucose + 500μM FFAs + 25 nM rapamcyin. After treatment, cells were processed for immunostaining. Representative images of β-cells under the three conditions are shown [insulin (green), nuclei (blue), and ADRP (red)]. (D) The bar graph shows the average intensities of DyLight 647 for insulin and Dylight 488 for ADRP, respectively, of a total of ~80 β-cells per condition from three independent experiments.
![Figure 6.(A) Dose-dependent effects of nutrients on ADRP expression. Rat islets (100) were cultured for 4 days in cCMRL-1066 containing 5.6 mM glucose, 10 mM glucose + 240μM FFAs, or 25 mM glucose + 500 μM FFAs. Samples were processed for Western blotting and quantitated by densitometry. β-actin was used as a protein loading control. Data are the means ± SEM of n = 3 experiments. (B) Glucose and FFAs synergize in increasing mTORC1-mediated ADRP expression. Rat islets (100) were cultured for 4 days in cCMRL-1066 under various conditions as indicated. Samples were processed for western blotting and quantitated by densitometry. β-actin was used as a protein loading control. Data are the means ± SEM of n = 3 experiments. (C) ADRP expression and insulin content in single β-cells determined by immunohistochemistry. Dispersed rat islet cells (105) were cultured for 4 days in cCMRL-1066 containing 5.6 mM glucose, 25 mM glucose + 500 μM FFAs, or 25 mM glucose + 500μM FFAs + 25 nM rapamcyin. After treatment, cells were processed for immunostaining. Representative images of β-cells under the three conditions are shown [insulin (green), nuclei (blue), and ADRP (red)]. (D) The bar graph shows the average intensities of DyLight 647 for insulin and Dylight 488 for ADRP, respectively, of a total of ~80 β-cells per condition from three independent experiments.](/cms/asset/abc18697-3651-4530-b34d-8a93ce7093fa/kisl_a_10922720_f0006.gif)
Rat islet cell growth and new DNA synthesis under nutrient overload
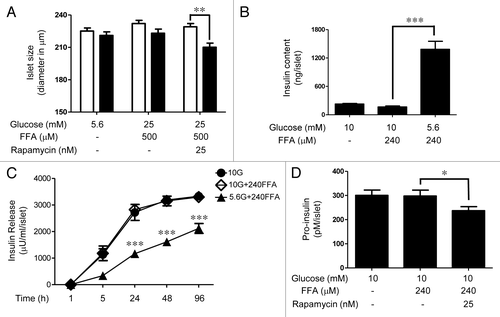
demonstrates that islets exposed for 4 days to 25 mM glucose and 500 μM FFAs (2nd black bar) showed a significant increase in islet size as compared with those under 5.6 mM glucose alone (1st black bar) or 25 mM glucose + 500 μM FFAs + 25 nM rapamcyin (3rd black bar). Of note, islets incubated with 5.6 mM glucose showed a significant decrease in size after 4 days of incubation. Nutrient-mediated islet cell expansion was also studied by immunohistochemistry of frozen islet sections. We observed that actin filaments were highly enriched along the plasma membrane of primary islet cells, which allowed visual assessment of intra-islet cell size by staining with Alexa 488 phalloidin, a high affinity actin binding toxin tagged with the fluorescent probe Alexa 488. An islet treated with 25 mM glucose + 500 μM FFAs for 4 days (, panel g) resulted in overall increases in intra-islet cell size compared with control islet (panel c), shown by the enlarged black areas surrounded by the green actin staining. A comparison of the nuclear staining (DAPI) of two islet sections (panels b and f) suggested that the increase in islet size caused by excess nutrients was primarily due to hypertrophy rather than hyperplasia since the number of nuclei did not significantly increase, and since the nuclei were much farther apart in panel f compared with panel b. Rapamycin blocked the increase in intra-islet cell size caused by excess nutrients as shown in panels j and k. Quantitation of the intra-islet cell sizes under different conditions is shown in . Insulin content was greatly reduced under the condition of excess nutrients and rapamycin did not prevent this reduction (compare panels a, e and i), consistent with and . Next, we studied if excess nutrients also stimulate islet cell proliferation by measuring new DNA synthesis as indicated by [3H]thymidine incorporation. For these experiments, we used moderately elevated concentrations of nutrients to study the synergistic actions of glucose and FFAs and to correlate with mTORC1 activity () and insulin release studies (). Treatment of islets with FFAs (240 μM) in the presence of 5.6 mM glucose (, 7th bar) resulted in a ~2-fold increase of [3H]thymidine incorporation compared with 5.6 mM glucose alone (, 1st bar), whereas 10 mM glucose alone showed no significant increase. A combination of 10 mM glucose and 240 μM FFAs produced a synergic increase in [3H]thymidine incorporation (, 5th bar) in a rapamycin sensitive manner.
Figure 7.(A) mTORC1-dependent increase in rat islet size under nutrient overload. Phase contrast images of rat islets (40) before and after the 4 days incubation period under various conditions as indicated were acquired using a 20× objective. The diameter of each islet was computed by converting pixels to μm using the Metamorph image analysis software. The white and the black bars indicate the sizes of the islets before and after the treatment, respectively. Data are the means ± SEM of n = 3 experiments. (B) mTORC1-dependent new DNA synthesis in rat islets under nutrient overload. Rat islets (100) were cultured for 4 days under various conditions as indicated. [3H]thymidine was added to each dish 24 h prior to the end or the 4-day period. Data are the means ± SEM of n = 3 experiments. dpm, disintegrations per minute. (C) Islet cell expansion under nutrient overload determined by immunohistochemistry. Rat islets (50) were cultured for 4 days in cCMRL containing 5.6 mM glucose, 25 mM glucose + 500 μM FFAs, or 25 mM glucose + 500 μM FFAs+25 nM rapamycin. Frozen sections (10 μm) of islets were processed for immunostaining (insulin [red], nuclei [blue], and phalloidin [green]). Images were rescaled using Adobe Photoshop software. (D) The bar graph shows the average area of intra-islet cells determined using the Metamorph image analysis software. Data are the means ± SEM of ~250 cells obtained from 10–15 islet sections per condition.
![Figure 7.(A) mTORC1-dependent increase in rat islet size under nutrient overload. Phase contrast images of rat islets (40) before and after the 4 days incubation period under various conditions as indicated were acquired using a 20× objective. The diameter of each islet was computed by converting pixels to μm using the Metamorph image analysis software. The white and the black bars indicate the sizes of the islets before and after the treatment, respectively. Data are the means ± SEM of n = 3 experiments. (B) mTORC1-dependent new DNA synthesis in rat islets under nutrient overload. Rat islets (100) were cultured for 4 days under various conditions as indicated. [3H]thymidine was added to each dish 24 h prior to the end or the 4-day period. Data are the means ± SEM of n = 3 experiments. dpm, disintegrations per minute. (C) Islet cell expansion under nutrient overload determined by immunohistochemistry. Rat islets (50) were cultured for 4 days in cCMRL containing 5.6 mM glucose, 25 mM glucose + 500 μM FFAs, or 25 mM glucose + 500 μM FFAs+25 nM rapamycin. Frozen sections (10 μm) of islets were processed for immunostaining (insulin [red], nuclei [blue], and phalloidin [green]). Images were rescaled using Adobe Photoshop software. (D) The bar graph shows the average area of intra-islet cells determined using the Metamorph image analysis software. Data are the means ± SEM of ~250 cells obtained from 10–15 islet sections per condition.](/cms/asset/7bf6926c-90b8-4f3b-910a-688f06ab8c11/kisl_a_10922720_f0007.gif)
Figure 8.(A) Insulin release into the culture medium under nutrient overload. Islets (35) were cultured in 2 ml cCMRL containing 10 mM glucose or 240 μM FFAs alone or in combination in the absence and presence of 25 nM rapamycin. Cultured medium (50 μl) was removed at the indicated times and the amount of insulin released into the culture medium was determined by radioimmunoassay. Data are the means ± SEM of n = 3 experiments. (B) Insulin content in islets after 4 days treatment under nutrient overload. After treatment, islets were washed and resuspended in 500 μl of 0.1% BSA in PBS, followed by sonication using a microtip probe. Insulin content was determined by radioimmunoassay. Data are the means ± SEM of n = 3 experiments with triplicate samples in each experiment. (C) Pro-insulin levels in islets after 4 days treatment under nutrient overload. After treatment, islets were washed and resuspended in 200 μl of 0.1% BSA in PBS, followed by sonication using a microtip probe. Pro-insulin levels were determined as described in the Methods section. Data are the means ± SEM of n = 3 experiments.
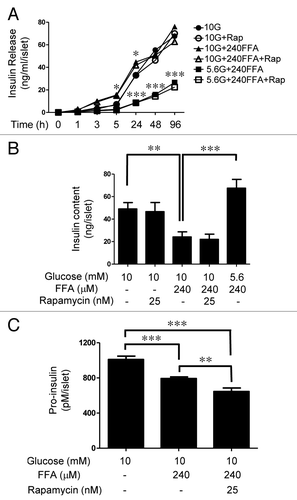
Rat islet insulin release and content under nutrient overload
Next, we determined insulin release into the medium under moderately elevated nutrients over a 4-day period. shows that insulin release into the medium steadily increased under all conditions. Islets exposed to the combination of 10 mM glucose and 240 μM FFAs showed significantly higher levels of insulin release than those to 10 mM glucose alone at 5 and 24 h time points but this difference was negligible after 4-day incubation. A significantly lower insulin release under 5.6 mM glucose and 240 μM FFAs was observed. Rapamycin had no significant effect on insulin release into the medium under any conditions. shows insulin content after a 4-d incubation period. Although there was no significant difference in insulin release into the medium under 10 mM glucose alone and 10 mM glucose + 240μM FFAs, insulin content was significantly reduced under the condition of the combination of glucose and FFAs (, 3rd bar) compared with 10 mM glucose alone (, 1st bar). A high level of insulin content under 5.6 mM glucose and 240 μM FFAs was consistent with the low level of insulin release during the 4-day incubation period. To test if reduction in insulin biosynthesis accounts for the reduced levels of insulin content in islets treated with 10 mM glucose + 240μM FFAs, we determined pro-insulin levels in islets as an indicator of insulin biosynthesis after 4-d incubation. shows that pro-insulin levels in islets treated with 10 mM glucose + 240 μM FFAs were significantly lower than those from islets treated with 10 mM glucose alone. Rapamycin treatment reduced pro-insulin levels below those of islets treated with the combination of the two nutrients.
Human β-cell metabolic alterations under nutrient overload
We examined metabolic and morphological changes of human islets under nutrient overload using similar approaches to the rat islet studies. shows a time-dependent lipid accumulation during a 4-day incubation period in dispersed human β-cells under nutrient overload. demonstrates that 25 mM glucose + 500 μM FFAs (lane 2) stimulated mTORC1 activation indicated by S6 phosphorylation compared with 5.6 mM glucose alone (lane 1). Rapamycin completely blocked this effect. Next, we studied nutrient-mediated lipid droplet formation in human islets by immunohistochemistry of frozen islet sections. Islets treated with 25 mM glucose + 500 μM FFAs for 4 days (, panel g) resulted in elevated lipid droplet formation compared with control islets (panel c). Rapamycin significantly blocked nutrient-mediated lipid droplet formation (panel k). A comparison of the nuclear staining (DAPI) of islets under the two conditions (panels b and f) indicate that nutrient overload did not cause islet cell expansion. Insulin content was greatly reduced under the conditions of excess nutrients and rapamycin did not prevent this reduction (compare panels a, e and i). shows quantitation of insulin content and lipid accumulation determined by the average intensity of insulin and Nile red of the three conditions. shows that treatment of human islets with 25 mM glucose and 500 μM FFAs for 4 days did not increase the size of islets, consistent with the results shown in . While it may not be obvious from , rapamycin slightly reduced the size of human islets after 4-day treatment. shows that insulin release into the medium under 10 mM glucose or 10 mM glucose + 240 μM FFAs increased during the first 24 h exposure, then plateaued. Insulin release under 5.6 mM glucose + 240 μM FFAs, on the other hand, increased steadily throughout the 4-day incubation period but at significantly lower levels. shows insulin content in human islets after 4-day incubation. Insulin content in islets treated with 10 mM glucose or 10 mM glucose + 240 μM FFAs was approximately 5 fold lower than that in islets treated with 5.6 mM glucose + 240 μM FFAs. Unlike in rat islets, there was no significant difference in insulin content in human islets treated with 10 mM glucose and 10 mM glucose + 240 μM FFAs. shows pro-insulin levels under indicated conditions. There was no difference in pro-insulin levels in islets treated with 10 mM glucose and 10 mM glucose + 240 μM FFAs. These results are consistent with no difference in insulin content under the two conditions. As in rat islets, rapamycin reduced pro-insulin levels below those of islets treated with elevated levels of nutrients.
Figure 9.(A) Time course of lipid droplet formation in dispersed human β-cells. Dispersed human islet cells (105) were treated for the indicated time periods with 25 mM glucose and 500 μM FFAs. After treatment, cells were processed for immunostaining. The average Nile red intensity for a total of ~80 clusters of insulin-positive cells per time point from three independent experiments is shown. (B) Nutrient-mediated activation of mTORC1. Human islets (100) were treated for 4 days as indicated. Samples were processed for Western blotting and quantitated by densitometry. Data are the means ± SEM of n = 3 experiments. (C) Human islets (50) were cultured for 4 days in cCMRL containing 5.6 mM glucose, 25 mM glucose + 500 μM FFAs, or 25 mM glucose + 500 μM FFAs + 25 nM rapamycin. Frozen sections (10 μm) of islets were processed for immunostaining [insulin (green), nuclei (blue), and Nile red (red)]. Images were rescaled using Adobe Photoshop software. (D) The bar graph shows the average intensities of Nile red for lipid accumulation and DyLight 647 for insulin, respectively, of 15–20 islet sections per condition from three different experiments is shown.
![Figure 9.(A) Time course of lipid droplet formation in dispersed human β-cells. Dispersed human islet cells (105) were treated for the indicated time periods with 25 mM glucose and 500 μM FFAs. After treatment, cells were processed for immunostaining. The average Nile red intensity for a total of ~80 clusters of insulin-positive cells per time point from three independent experiments is shown. (B) Nutrient-mediated activation of mTORC1. Human islets (100) were treated for 4 days as indicated. Samples were processed for Western blotting and quantitated by densitometry. Data are the means ± SEM of n = 3 experiments. (C) Human islets (50) were cultured for 4 days in cCMRL containing 5.6 mM glucose, 25 mM glucose + 500 μM FFAs, or 25 mM glucose + 500 μM FFAs + 25 nM rapamycin. Frozen sections (10 μm) of islets were processed for immunostaining [insulin (green), nuclei (blue), and Nile red (red)]. Images were rescaled using Adobe Photoshop software. (D) The bar graph shows the average intensities of Nile red for lipid accumulation and DyLight 647 for insulin, respectively, of 15–20 islet sections per condition from three different experiments is shown.](/cms/asset/b074b968-007a-4b20-9fdf-d4a97e2dcb16/kisl_a_10922720_f0009.gif)
Figure 10.(A) Absence of human islet cell growth under nutrient overload. Phase contrast images of human islets (40) before and after the 4-day incubation period under various conditions as indicated were acquired using a 20× objective. The diameter of each islet was computed by converting pixels to μm using the Metamorph image analysis software. The white and the black bars indicate the sizes of the islets before and after the treatment, respectively. Data are the means ± SEM of n = 3 experiments. (B) Insulin release into the culture medium under nutrient overload. Human islets (30) were cultured in 2 ml cCMRL containing 10 mM glucose or 240 μM FFAs alone or in combination of the two nutrients. Cultured medium (50 μl) was removed at the indicated times and the amount of insulin released into the culture medium was determined by radioimmunoassay. Data are the means ± SEM of n = 3 experiments. (C) Insulin content in human islets after 4-day treatment under nutrient overload. After treatment, islets were washed and resuspended in 500 μl of 0.1% BSA in PBS, followed by sonication using a microtip probe. Insulin content was determined by radioimmunoassay. Data are the means ± SEM of n = 3 experiments with triplicate samples in each experiment. (D) Pro-insulin levels in islets after 4-day treatment under nutrient overload. After treatment, islets were washed and resuspended in 200 μl of 0.1% BSA in PBS, followed by sonication using a microtip probe. Pro-insulin levels were determined as described in the Methods section. Data are the means ± SEM of n = 3 experiments.
Discussion
Using a combination of microscopy and biochemical methods, we studied β-cell metabolic and morphological changes under nutrient overload in rat and human islets. A significant finding of this study is that the nutrient sensor mTORC1 plays a central role in lipid droplet formation and islet cell expansion in rat islets. Nutrient activation of mTORC1 played a pivotal role in lipid droplet formation in rat β-cells by upregulating the expression of ADRP, which is known to stabilize lipid droplets. Importantly, lipid droplet formation was observed only in β-cells but not in α-cells. Islets treated with a combination of glucose and FFAs showed a significant reduction in insulin biosynthesis and content compared with islets treated with glucose alone despite the similar levels of insulin release into the culture media. Unlike rat islets, human islets did not demonstrate islet cell expansion under nutrient overload despite their similar metabolic responses to nutrient overload in lipid droplet formation and insulin release. Overall, this study indicates that adaptive responses of β-cells to metabolic perturbation, lipid accumulation and islet cell expansion (in the case of rodent islets), are associated with β-cell dysfunction rather than compensatory enhancement of β-cell function.
In this study we provide quantitative evidence of time- and dose-dependent lipid droplet formation in primary rat β-cells in response to chronic nutrient overload. To our knowledge, it is the first in-depth study of time-dependent lipid droplet formation in primary β-cells using photomicroscopy and an immunohistochemical approach. Intriguingly, lipid droplet formation occurred exclusively in β-cells in an mTORC1-dependent manner. Selective β-cell loss but α-cell expansion have been associated with T2DM,Citation31,Citation32 suggesting that α- and β-cells respond differently to local environments such as excess nutrients. The mechanisms responsible for sparing α-cells from the effects of excess nutrients are unknown. In comparison to β-cells, α-cells may express reduced FFA transporters, undergo β-oxidation at a higher rate, or lack enzymes that are required for the biogenesis of lipid droplets. (panel g) shows lipid droplets in the periphery of an islet. However, these are not localized in the α-cells because the islet architecture is perturbed under these conditions, resulting in the translocation of α-cells to the center of the islet (data not shown).
Specific molecular mechanisms involved in nutrient-mediated ectopic lipid accumulation in β-cells have not been clearly understood. Our study demonstrates that mTORC1 exerts a pivotal role in lipid droplet formation in rat and human pancreatic β-cells. In support of a major role for mTORC1 in this process, β-cell specific accumulation of lipid droplets was significantly blocked by rapamycin. In addition, elevated glucose in combination with FFAs potentiated mTORC1 activation, based on S6 phosphorylation, in a manner that mirrored the degree of increase in ADRP expression and lipid droplet accumulation in β-cells. Thus, it appears that mTORC1, a nutrient sensor, plays a role in storing excess nutrients in the form of neutral lipid TG inside lipid droplets in the β-cell by upregulating the expression of ADRP which stabilizes lipid droplets. De novo synthesis of TG must be also elevated under nutrient overload through the stimulation of the esterification pathway. Interestingly, suppression of ADRP expression by rapamycin was sufficient to reduce the level of TG in the β-cell. Whether destabilizing lipid droplets by insufficient amounts of ADRP facilitates lipolysis of TG into intermediate lipid species such as diacylglycerols, lysophosphatidic acid, etc. is under investigation. If this hypothesis is true, rapamycin is anticipated to cause toxic effects rather than preserving β-cell function although it blocks lipid droplet formation. Our data showing that rapamycin did not preserve insulin content under nutrient overload supports this hypothesis.
The causal relation between lipid accumulation and β-cell function is complex. Numerous in vitro studiesCitation33-Citation36 have shown that the saturated FFA palmitate is highly toxic, whereas the monounsaturated FFA oleate is not. Unsaturated fatty acids are thought to rescue palmitate-induced apoptosis by channeling palmitate into triglyceride pool and away from pathways leading to apoptosis.Citation34,Citation35 Recent evidence indicates that oleate attenuate palmitate-induced endoplasmic reticulum stress and β-cell apoptosis.Citation36 Our present study, however, shows that an increase in triglyceride pool is associated with β-cell dysfunction. It is possible that oleate rescues β-cells from palmitate-induced apoptosis but is unable to prevent β-cell dysfunction caused by metabolic perturbation associated with chronic nutrient overload.
Rat islet cell expansion under nutrient overload appears to be due primarily to hypertrophy () rather than hyperplasia. Although a combination of nutrients caused a significant increase in [3H]thymidine incorporation, the extent to which β-cell proliferation occurs under these conditions is unknown. Islet size did not increase significantly under 5.6 mM glucose and 240 μM (data not shown), yet [3H]thymidine incorporation was substantially elevated. This suggests that the elevated DNA synthesis activity represents a small percentage of the islet cell population or islet cells in S phase that did not complete the cell cycle to produce two new daughter cells. Our previous studies have also shown that the enhanced [3H]thymidine incorporation observed under conditions of prolonged elevated glucose was not translated to an increase in cell proliferation.Citation37 Recent evidence indicates that activation of mTORC1 increased mitochondrial DNA synthesis,Citation38 suggesting that the increase in new DNA synthesis observed in may reflect mitochondrial DNA synthesis. The regulation of human islet cell growth seems to be different from rodents since nutrient activation of mTORC1 failed to promote hypertrophy in human islets ( and ). Although experimental data are limited, increased β-cell mass in obesity has been observed in morphometric analyses of human pancreatic tissues obtained at autopsy.Citation39,Citation40 The extent of this adaptive increase (~0.5-fold), however, was modest compared with that in obese rodents (~10-fold). Moreover, the increased β-cell mass in humans is thought to occur through new islet formation rather than hypertrophy or replication of existing β-cells.Citation39 The results of our in vitro studies are consistent with this hypothesis. The impact of nutrient overload associated with obesity in β-cell defects in humans is yet to be elucidated.
It is noteworthy that islet cell expansion was correlated with a significant reduction of insulin content as shown in and under both high and moderate concentrations of nutrients. Other evidence indicated that an increase in β-cell mass was not always correlated with an enhanced β-cell function.Citation12,Citation15-Citation17,Citation41 These findings are important in that they challenge a well-accepted view that β-cell mass expansion and enhanced β-cell function under insulin resistance and nutrient overload provide compensatory responses that lead to hyperinsulinemia. Perhaps, hypersecretion of insulin under nutrient overload is not the consequence of increased β-cell mass and/or enhanced β-cell function but an unsustainable response leading to depletion of insulin stores. The findings of the present study support rat islet cell expansion under nutrient overload as indicative of metabolic distress rather than compensatory enhancement of β-cell function.
Continuous prolonged exposure of both rat and human islets to elevated nutrients caused over- secretion of insulin into the culture media, as expected. Interestingly, there was no significant difference in the amount of insulin released into the media between a combination of nutrients (10 mM glucose and 240 μM FFAs) and glucose (10 mM) alone over a 4-day incubation period. Moreover, rapamycin had no effect on nutrient-mediated insulin release by rat islets (). Insulin content in rat islets after the 4-day treatment, however, differed strikingly depending on the experimental conditions. The reduction in insulin content in islets treated with the combination of nutrients as compared with glucose alone was apparently due to a decrease in insulin biosynthesis (). Unlike rat islets, human islets did not show islet cell expansion under nutrient overload. Furthermore, insulin content in human islets treated with the combination of nutrients was not significantly different from islets treated with glucose alone (). Therefore, the 50% reduction of insulin content in rat islets may be due to marked islet cell expansion. The idea that dedifferentiation or immaturity of newly expanded β-cellsCitation16,Citation17 under nutrient overload causes the reduction of insulin content supports these findings.
In summary, we studied multifactorial β-cell responses to chronic nutrient overload in rat and human islets using biochemical and microscopic methodologies. We provide clear evidence that excess nutrients cause lipid droplet formation in both rat and human islets in an mTORC1-dependent manner. We have observed a striking difference in the metabolic responses of rat and human islets to nutrient overload. Unlike rat islets, human islets did not exhibit islet cell expansion. We believe that islet cell expansion under nutrient overload represents a facet of metabolic perturbation that leads to impaired β-cell function rather than a compensatory response that enhances β-cell function. Lipid droplet formation in both rat and human β-cells in response to high nutrients also appears to be correlated with β-cell dysfunction or perturbed metabolic state. As future directions, we are planning to study the effects of other pharmacological agents (i.e., acyl CoA:diacylglycerol acyltransferase 1 inhibitor) to further study the causal relationship between β-cell adaptive responses and β-cell defects under chronic nutrient overload.
Materials and Methods
Male Sprague-Dawley rats were purchased from Harlan Sprague-Dawley. Collagenase type XI (C9263), Hanks' balanced salt solution (H1387), palmitic acid (# P0500), oleic acid (O3880), Nile red (N3013), and fibronectin (F1141) were obtained from Sigma. Infinity Triglyceride Reagent (TR-22421), Triglyceride Standard (TR22923), fetal bovine serum (SH3007003), Penicillin-streptomycin (ICN1670049), and West Pico Chemiluminescent Substrate (34080) were from Fisher Scientific. Hybond ECl (RPN3032D) was obtained from GE Healthcare Life Sciences. Tissue culture medium, CMRL-1066 (11530037), and trypsin-EDTA (25200072) were from Invitrogen. BSA (fraction V, fatty acid-free, 08823234) was from ICN Biomedical. Rapamycin (BML-A275) was obtained from Biomol. Monoclonal rabbit insulin antibody (3014S) and monoclonal rabbit p-S6 antibody (4856S) were purchased from Cell Signaling. Polyclonal rabbit ADRP antibody was generated in Dr. Andrew Greenberg’s laboratory. Monoclonal mouse actin antibody (A3853) and monoclonal mouse anti-glucagon antibody (G2654) were from Sigma. The secondary antibodies, peroxidase-conjugated donkey anti-rabbit IgG (711–035–152), peroxidase-conjugated donkey anti-mouse IgG (715–035–150), DyLight 649-conjugated donkey anti-rabbit IgG (711–495–152), Alexa 488-conjugated donkey anti-mouse antibody (715–545–150) were obtained from Jackson ImmunoResearch Laboratories. Alexa 488 donkey anti-rabbit IgG (A21206), Alexa 647 donkey anti-mouse IgG (A31571), Alexa 488 phalloidin (A12379), and 4'-6-diamidino-2-phenylindole (DAPI) (D3571) were obtained from Invitrogen. [3H]thymidine (ART 0178H) was obtained from American Radiolabeled Chemicals, Inc. Rat insulin RIA kit (RI-13K) and human insulin RIA kit (HI-11K) were from Millipore. Rat (80-PINRT-E01) and human (80-PINHU-E01.1) pro-insulin ELISA kits were obtained from ALPCO. All other chemicals were from commercially available sources.
Rat islet isolation and culture
Islets were isolated from male Sprague-Dawley rats (250–270 g) by collagenase digestion. Pancreases were inflated with 15 ml of Hank's balanced salt solution (HBSS) containing 0.5 mg/ml collagenase, isolated, minced, and incubated for 5 min at 37°C while shaking. The tissue was washed 2 times with HBSS and resuspended in 20 ml of HBSS. The cell suspension was filtered through a 70 μm cell strainer and washed with 20 ml of HBSS to remove acinar cells. Islets and remaining tissue were transferred to a Petri dish by inverting the cell strainer and washing it with 20 ml of CMRL-1066. Islets were hand-picked under a stereomicroscope and cultured in an atmosphere of 95% air, 5% CO2 in “complete” CMRL-1066 tissue culture medium (cCMRL) containing 5.6 mM glucose, 2 mM L-glutamine, 10% (v/v) heat-inactivated fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. These studies were approved by the Southern Illinois University Edwardsville Institutional Animal Care and Use Committee.
Human islets
Human islets (13 shipments) were obtained from 6 different islet centers through Integrated Islet Distribution Program (IIDP) from January 2011 to January 2012. We restricted the donor selection criteria as BMI < 32 and no type 2 diabetes to obtain intact and healthy islets. The range of donor age, gender, and BMI are as follows: age (19–58), gender (6 males, 4 females, 3 unknown), BMI (19–31.3).
Fatty acid preparation
Fatty acid supplemented medium was prepared as described previously.Citation42 Briefly, a 20 mM solution of fatty acid dissolved in 0.01M NaOH was incubated at 70°C for 30 min with intermittent mixing. The fatty acid soaps were complexed with 5% fatty acid-free BSA in PBS. The BSA complexed fatty acid was added to the 10% serum-containing cell culture medium to achieve the desired total fatty acid concentrations at a 3:1 fatty acid to BSA molar ratio. The pH of the medium containing complexed fatty acids was adjusted to 7.4. For all experiments, oleate and palmitate were prepared separately and combined to achieve the desired concentration with a 1:1 ratio.
Time lapse studies
Rat islets (50) were dispersed into single cells by incubation in 0.05% Trypsin/0.53 mM EDTA in HBSS for 4 min at 37°C while shaking. Dispersed islet cells were washed once with cCMRL and resuspended in 2 ml of cCMRL supplemented with 25 mM glucose and 500 μM FFAs (oleate:palmitate = 1:1v/v). The cell suspension was placed in a Petri dish (35 × 10 mm) with glass bottom coated with fibronectin. The dish was placed on the stage of a Leica DMI inverted microscope. Using the Delta T system (Bioptechs, Inc.) chamber temperature was held at 37°C and infused with humidified 5% CO2 to maintain a suitable environment for a long-term, live cell imaging. Phase contrast digital images were acquired at 30 min intervals for 40h using a 20X objective. Images were rescaled using Adobe Photoshop software.
Quantitation of lipid droplets by immunohistochemistry
Dispersed rat islet cells (105 cells/dish) were cultured on fibronectin coated coverslips in Petri dishes (35 × 10 mm) under various experimental conditions as indicated in the figure legends. After incubation, cells were fixed with 4% paraformaldehyde in PBS for 20 min, followed by incubation in permeabilization solution (0.1% Triton X-100) for 20 min. Cells were then incubated in blocking solution (0.2% BSA in PBS) for 20 min, followed by treatment with primary rabbit anti-insulin antibody (1:300 dilution in blocking solution) for 20 min at 37°C. Cells were washed and incubated for 20 min at 37°C with secondary DyLight 647-conjugated donkey anti-rabbit IgG (1:300), DAPI (1:5000) for nuclear staining, and Nile red (5 μM) for neutral lipid staining. Cells were washed twice in PBS and mounted on slides. Using a 63× objective in a Leica DMI inverted fluorescent microscope, insulin staining cells were selected and images of Nile red staining were captured. Average Nile red intensity was determined for ~80 clusters of insulin-positive cells per condition. Background, taken as an average intensity of cells not stained with Nile red, was subtracted from the total intensity. For quantitation of lipid droplets in α-cells, monoclonal donkey anti-mouse antibody (1:300) and Alexa 488-conjugated donkey anti-mouse IgG (1:300) were used as a primary and secondary antibody, respectively.
Western blot analysis
Proteins were separated by polyacrylamide gels and transferred to Hybond ECL. For p-S6 and ADRP detection, monoclonal rabbit p-S6 antibody (1:1000) and anti-ADRP (1:1000) antibodies, respectively were used as primary antibodies. HRP-conjugated donkey anti-rabbit IgG was used as a secondary antibody. Detection was performed using West Pico Chemiluminescent Substrate and p-S6 and ADRP were quantitated by Biorad ChemiDoc XRS Image Lab Software. The Hybond membranes were stripped and reblotted for β-actin using monoclonal mouse anti-β-actin antibody and HRP-conjugated donkey anti-mouse IgG as a primary and a secondary antibody, respectively. The ratio of p-S6/β-actin or ADRP/β-actin was obtained by first determining the relative increase over the control condition (lane 1) for p-S6, ADRP, or β-actin, then calculating the ratio of the respective pair.
Quantitation of TG content in islets
Rat islets (150/condition) were incubated in cCMRL supplemented with 25 mM glucose and 500 μM FFAs in the absence and presence of 25 nM rapamycin as indicated in the figure. After 48 h, islets were rinsed once with PBS, resuspended in 200 μl of 2:1 (v/v) chloroform:methanol, and sonicated until no intact islet structure was visible by light microscopy. Solvents were evaporated to dryness in an Eppendorf Vacufuge at 45°C. The amount of TG was quantitated by an enzymatic colorimetric assay based on lipase-mediated glycerol production.
Quantitation of ADRP expression and insulin content by immunohistochemistry
After treatment, dispersed islet cells were fixed, permeabilized, and treated with monoclonal rabbit anti-insulin antibody (1:300 dilution in blocking solution) and mouse anti-ADRP monoclonal antibody (Progen) as primary antibodies. Cells were washed and treated with secondary antibodies Cy5-conjugated donkey anti-rabbit IgG (1:300) and Alexa 488-conjugated anti-mouse IgG (1:300), and DAPI (1:5000) for nuclear staining. Using a 63× objective in a Leica DMI inverted fluorescent microscope, insulin staining cells were selected and images of ADRP staining were captured. Average intensity of Dylight 649 for insulin and Alexa 488 for ADRP was determined for a total of ~90 cells per condition from 3 independent experiments.
Insulin secretion and content
Islets were treated for 4 days under various conditions as indicated in the figures. Triplicates of 5 islets for each treatment group were preincubated in CMRL-1066/0.1% BSA containing 3 mM glucose for 30 min. Culture medium was replaced with CMRL-1066 containing either 3 mM or 20 mM glucose and further incubated for 1 h. Supernatants were assayed for insulin secretion by radioimmunoassay (RI-13K for rat islets and HI-14K for human islets, Millipore). Islets were washed and resuspended in 500 μl of 0.1% BSA in PBS, followed by sonication using a microtip probe. Insulin content in rat or human islets was determined by radioimmunoassay using the kits from Millipore.
Determination of pro-insulin level
Islets were treated for 4 days under various conditions as indicated in the figures. After treatment, triplicates of 5 islets were washed and resuspended in 200 μl of 0.1% BSA in PBS, followed by sonication using a microtip probe. Pro-insulin levels in rat and human islets were determined using pro-insulin ELISA kits following the manufacturer’s instructions. Data are the means ± SEM of n = 3 experiments.
Determination of islet size
Rat or human islets (30–50 islets/condition) with similar sizes were placed in Falcon Petri dishes (35 × 10 mm). Phase contrast images of islets using a 20× objective were acquired before and after treatment as indicated in the figure. Using MetaMorph image analysis software (Molecular Devices), the diameter of each islet was determined.
Frozen sectioning and immunohistochemistry
After treatment, islets were washed and resuspended in 100 μl of Cryo-Gel embedding medium, snap-frozen in liquid nitrogen, and stored at -70°C until use. Frozen sections of 10 μm thickness were prepared using a Vibratome and transferred to coverslips. Tissues were fixed and permeabilized in 4% paraformaldehyde and 1% Triton-X 100 in PBS. Tissues were washed in PBS to remove the residual glutaraldehyde, blocked in 2% BSA in PBS, and treated with appropriate primary and secondary antibodies, DAPI for nuclear staining, and Alexa 488 phalloidin for actin staining as indicated in the figure legend. Fluorescent images were obtained using a 63× objective in a Leica DMI inverted fluorescent microscope. Images were rescaled using Adobe Photoshop software.
[3H]thymidine incorporation
Rat islets (100/condition) were counted into Falcon Petri dishes (35 × 10 mm) and were cultured for 4 days as indicated in the figure legends. During the final 24 h of the 96 h incubation, 10 μCi of [3H]thymidine was added to each dish. [3H]thymidine incorporation was determined by trichloroacetic acid extraction and scintillation counting.Citation37
Statistics
Results are expressed as mean ± SEM. Differences between means were evaluated using ANOVA or the Student t-test as appropriate. Significant differences are indicated by *p < 0.05, **p < 0.01, ***p < 0.001.
| Abbreviations: | ||
| ADRP | = | adipose differentiation-related protein |
| FFAs, free fatty acids (oleate | = | palmitate=1:1) |
| mTORC1 | = | mammalian target of rapamycin complex 1 |
| TG | = | triglyceride |
Acknowledgments
This work was supported by NIH Grant 1R15DK094142 (G.K.), SIUE Internal Grants (G.K.), American Association of Colleges of Pharmacy (G.K.), NIH Grant DK06181 (M.L.M.), DK00618146S1 (M.L.M.),Washington University Diabetes Research and Training Center Morphology and BioImaging Core DK20579 (M.L.M.), ADA 7-08-RA-57 (A.S.G.), USDA, Agricultural Research Service 58-1950-7-707 (A.S.G.), NIH RO1-DK0822574 (A.S.G.),1RC2ES018781 (A.S.G.) and R24DK087669 (A.S.G.). All procedures performed on animals were approved by the SIUE Institutional Animal Care and Use Committee.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Unger RH, Zhou YT. Lipotoxicity of beta-cells in obesity and in other causes of fatty acid spillover. Diabetes 2001; 50:Suppl 1 S118 - 21; http://dx.doi.org/10.2337/diabetes.50.2007.S118; PMID: 11272168
- Unger RH. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology 2003; 144:5159 - 65; http://dx.doi.org/10.1210/en.2003-0870; PMID: 12960011
- Raz I, Eldor R, Cernea S, Shafrir E. Diabetes: insulin resistance and derangements in lipid metabolism. Cure through intervention in fat transport and storage. Diabetes Metab Res Rev 2005; 21:3 - 14; http://dx.doi.org/10.1002/dmrr.493; PMID: 15386813
- Milburn JL Jr., Hirose H, Lee YH, Nagasawa Y, Ogawa A, Ohneda M, et al. Pancreatic beta-cells in obesity. Evidence for induction of functional, morphologic, and metabolic abnormalities by increased long chain fatty acids. J Biol Chem 1995; 270:1295 - 9; PMID: 7836394
- Sone H, Kagawa Y. Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia 2005; 48:58 - 67; http://dx.doi.org/10.1007/s00125-004-1605-2; PMID: 15624098
- Londos C, Brasaemle DL, Schultz CJ, Segrest JP, Kimmel AR. Perilipins, ADRP, and other proteins that associate with intracellular neutral lipid droplets in animal cells. Semin Cell Dev Biol 1999; 10:51 - 8; http://dx.doi.org/10.1006/scdb.1998.0275; PMID: 10355028
- Hickenbottom SJ, Kimmel AR, Londos C, Hurley JH. Structure of a lipid droplet protein; the PAT family member TIP47. Structure 2004; 12:1199 - 207; http://dx.doi.org/10.1016/j.str.2004.04.021; PMID: 15242596
- Londos C, Sztalryd C, Tansey JT, Kimmel AR. Role of PAT proteins in lipid metabolism. Biochimie 2005; 87:45 - 9; http://dx.doi.org/10.1016/j.biochi.2004.12.010; PMID: 15733736
- Borg J, Klint C, Wierup N, Ström K, Larsson S, Sundler F, et al. Perilipin is present in islets of Langerhans and protects against lipotoxicity when overexpressed in the beta-cell line INS-1. Endocrinology 2009; 150:3049 - 57; http://dx.doi.org/10.1210/en.2008-0913; PMID: 19299455
- Faleck DM, Ali K, Roat R, Graham MJ, Crooke RM, Battisti R, et al. Adipose differentiation-related protein regulates lipids and insulin in pancreatic islets. Am J Physiol Endocrinol Metab 2010; 299:E249 - 57; PMID: 20484013
- Coleman DL. Diabetes-obesity syndromes in mice. Diabetes 1982; 31:Suppl 1 Pt 2 1 - 6; PMID: 7160533
- Hull RL, Kodama K, Utzschneider KM, Carr DB, Prigeon RL, Kahn SE. Dietary-fat-induced obesity in mice results in beta cell hyperplasia but not increased insulin release: evidence for specificity of impaired beta cell adaptation. Diabetologia 2005; 48:1350 - 8; http://dx.doi.org/10.1007/s00125-005-1772-9; PMID: 15937671
- Bonner-Weir S, Deery D, Leahy JL, Weir GC. Compensatory growth of pancreatic beta-cells in adult rats after short-term glucose infusion. Diabetes 1989; 38:49 - 53; http://dx.doi.org/10.2337/diabetes.38.1.49; PMID: 2642434
- Steil GM, Trivedi N, Jonas JC, Hasenkamp WM, Sharma A, Bonner-Weir S, et al. Adaptation of beta-cell mass to substrate oversupply: enhanced function with normal gene expression. Am J Physiol Endocrinol Metab 2001; 280:E788 - 96; PMID: 11287362
- Fontés G, Zarrouki B, Hagman DK, Latour MG, Semache M, Roskens V, et al. Glucolipotoxicity age-dependently impairs beta cell function in rats despite a marked increase in beta cell mass. Diabetologia 2010; 53:2369 - 79; http://dx.doi.org/10.1007/s00125-010-1850-5; PMID: 20628728
- Delghingaro-Augusto V, Nolan CJ, Gupta D, Jetton TL, Latour MG, Peshavaria M, et al. Islet beta cell failure in the 60% pancreatectomised obese hyperlipidaemic Zucker fatty rat: severe dysfunction with altered glycerolipid metabolism without steatosis or a falling beta cell mass. Diabetologia 2009; 52:1122 - 32; http://dx.doi.org/10.1007/s00125-009-1317-8; PMID: 19294363
- Kargar C, Ktorza A. Anatomical versus functional beta-cell mass in experimental diabetes. Diabetes Obes Metab 2008; 10:Suppl 4 43 - 53; http://dx.doi.org/10.1111/j.1463-1326.2008.00940.x; PMID: 18834432
- McDaniel ML, Marshall CA, Pappan KL, Kwon G. Metabolic and autocrine regulation of the mammalian target of rapamycin by pancreatic beta-cells. Diabetes 2002; 51:2877 - 85; http://dx.doi.org/10.2337/diabetes.51.10.2877; PMID: 12351422
- Harris TE, Lawrence JC, Jr. TOR signaling. Sci STKE 2003; 2003:re15.
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev 2004; 18:1926 - 45; http://dx.doi.org/10.1101/gad.1212704; PMID: 15314020
- Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 2004; 23:3151 - 71; http://dx.doi.org/10.1038/sj.onc.1207542; PMID: 15094765
- Pende M, Kozma SC, Jaquet M, Oorschot V, Burcelin R, Le Marchand-Brustel Y, et al. Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature 2000; 408:994 - 7; http://dx.doi.org/10.1038/35050135; PMID: 11140689
- Lee CH, Inoki K, Guan KL. mTOR pathway as a target in tissue hypertrophy. Annu Rev Pharmacol Toxicol 2007; 47:443 - 67; http://dx.doi.org/10.1146/annurev.pharmtox.47.120505.105359; PMID: 16968213
- Wang X, Beugnet A, Murakami M, Yamanaka S, Proud CG. Distinct signaling events downstream of mTOR cooperate to mediate the effects of amino acids and insulin on initiation factor 4E-binding proteins. Mol Cell Biol 2005; 25:2558 - 72; http://dx.doi.org/10.1128/MCB.25.7.2558-2572.2005; PMID: 15767663
- Um SH, D’Alessio D, Thomas G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab 2006; 3:393 - 402; http://dx.doi.org/10.1016/j.cmet.2006.05.003; PMID: 16753575
- Patti ME, Kahn BB. Nutrient sensor links obesity with diabetes risk. Nat Med 2004; 10:1049 - 50; http://dx.doi.org/10.1038/nm1004-1049; PMID: 15459705
- Liu H, Remedi MS, Pappan KL, Kwon G, Rohatgi N, Marshall CA, et al. Glycogen synthase kinase-3 and mammalian target of rapamycin pathways contribute to DNA synthesis, cell cycle progression, and proliferation in human islets. Diabetes 2009; 58:663 - 72; http://dx.doi.org/10.2337/db07-1208; PMID: 19073772
- Balcazar N, Sathyamurthy A, Elghazi L, Gould A, Weiss A, Shiojima I, et al. mTORC1 activation regulates beta-cell mass and proliferation by modulation of cyclin D2 synthesis and stability. J Biol Chem 2009; 284:7832 - 42; http://dx.doi.org/10.1074/jbc.M807458200; PMID: 19144649
- Fu A, Ng AC, Depatie C, Wijesekara N, He Y, Wang GS, et al. Loss of Lkb1 in adult beta cells increases beta cell mass and enhances glucose tolerance in mice. Cell Metab 2009; 10:285 - 95; http://dx.doi.org/10.1016/j.cmet.2009.08.008; PMID: 19808021
- Rachdi L, Balcazar N, Osorio-Duque F, Elghazi L, Weiss A, Gould A, et al. Disruption of Tsc2 in pancreatic beta cells induces beta cell mass expansion and improved glucose tolerance in a TORC1-dependent manner. Proc Natl Acad Sci U S A 2008; 105:9250 - 5; http://dx.doi.org/10.1073/pnas.0803047105; PMID: 18587048
- Yoon KH, Ko SH, Cho JH, Lee JM, Ahn YB, Song KH, et al. Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab 2003; 88:2300 - 8; http://dx.doi.org/10.1210/jc.2002-020735; PMID: 12727989
- Gromada J, Franklin I, Wollheim CB. Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr Rev 2007; 28:84 - 116; http://dx.doi.org/10.1210/er.2006-0007; PMID: 17261637
- Cnop M, Hannaert JC, Hoorens A, Eizirik DL, Pipeleers DG. Inverse relationship between cytotoxicity of free fatty acids in pancreatic islet cells and cellular triglyceride accumulation. Diabetes 2001; 50:1771 - 7; http://dx.doi.org/10.2337/diabetes.50.8.1771; PMID: 11473037
- Maedler K, Spinas GA, Dyntar D, Moritz W, Kaiser N, Donath MY. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 2001; 50:69 - 76; http://dx.doi.org/10.2337/diabetes.50.1.69; PMID: 11147797
- Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr., Ory DS, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A 2003; 100:3077 - 82; http://dx.doi.org/10.1073/pnas.0630588100; PMID: 12629214
- Cunha DA, Hekerman P, Ladrière L, Bazarra-Castro A, Ortis F, Wakeham MC, et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J Cell Sci 2008; 121:2308 - 18; http://dx.doi.org/10.1242/jcs.026062; PMID: 18559892
- Kwon G, Marshall CA, Liu H, Pappan KL, Remedi MS, McDaniel ML. Glucose-stimulated DNA synthesis through mammalian target of rapamycin (mTOR) is regulated by KATP channels: effects on cell cycle progression in rodent islets. J Biol Chem 2006; 281:3261 - 7; http://dx.doi.org/10.1074/jbc.M508821200; PMID: 16344552
- Koyanagi M, Asahara S, Matsuda T, Hashimoto N, Shigeyama Y, Shibutani Y, et al. Ablation of TSC2 enhances insulin secretion by increasing the number of mitochondria through activation of mTORC1. PLoS One 2011; 6:e23238; http://dx.doi.org/10.1371/journal.pone.0023238; PMID: 21886784
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003; 52:102 - 10; http://dx.doi.org/10.2337/diabetes.52.1.102; PMID: 12502499
- Klöppel G, Löhr M, Habich K, Oberholzer M, Heitz PU. Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv Synth Pathol Res 1985; 4:110 - 25; PMID: 3901180
- Tomita T, Doull V, Pollock HG, Krizsan D. Pancreatic islets of obese hyperglycemic mice (ob/ob). Pancreas 1992; 7:367 - 75; http://dx.doi.org/10.1097/00006676-199205000-00015; PMID: 1594559
- Kwon G, Pappan KL, Marshall CA, Schaffer JE, McDaniel ML. cAMP Dose-dependently prevents palmitate-induced apoptosis by both protein kinase A- and cAMP-guanine nucleotide exchange factor-dependent pathways in beta-cells. J Biol Chem 2004; 279:8938 - 45; http://dx.doi.org/10.1074/jbc.M310330200; PMID: 14688288

