

Abstract
Aim: An insufficient functional β-cell mass is a prerequisite to develop diabetes. Thus, means to protect or restore β-cell mass are important goals in diabetes research. Inflammation and proinflammatory cytokines play important roles in β-cell dysfunction and death, and recent data show that 2 miRNAs, miR-21 and miR-34a, may be involved in mediating cytokine-induced β-cell dysfunction. Therefore, manipulation of miR-21 and miR-34a levels may potentially be beneficial to β cells. To study the effect of long-term alterations of miR-21 or miR-34a levels upon net β-cell number, we stably overexpressed miR-21 and knocked down miR-34a, and investigated essential cellular processes.
Materials and Methods: miRNA expression was manipulated using Lentiviral transduction of the β-cell line INS-1. Stable cell lines were generated, and cell death, NO synthesis, proliferation, and total cell number were monitored in the absence or presence of cytokines.
Results: Overexpression of miR-21 decreased net β-cell number in the absence of cytokines, and increased apoptosis and NO synthesis in the absence and presence of cytokines. Proliferation was increased upon miR-21 overexpression. Knockdown of miR-34a increased net β-cell number in the absence of cytokines, and reduced apoptosis and NO synthesis in the absence and presence of cytokines. Proliferation was decreased upon miR-34a knockdown.
Conclusion: As overexpression of miR-21 increased proliferation, but also apoptosis and NO synthesis, the potential of miR-21 as a therapeutic agent to increase β-cell survival is doubtful. Knockdown of miR-34a slightly decreased proliferation, but as apoptosis and NO synthesis were highly reduced, miR-34a may be further investigated as a therapeutic target to reduce β-cell death and dysfunction.
Introduction
Diabetes mellitus develops as the functional β-cell mass fails to meet the demand for insulin. In Type 1 diabetes mellitus (T1D), loss of β-cell mass is caused by an autoimmune inflammatory destruction leading to absolute insulin deficiency. Type 2 diabetes mellitus (T2D) arises when the functional β-cell mass cannot compensate for insulin resistance. There is increasing agreement that both T1D and T2D are diseases in which inflammatory β-cell destruction is essential.Citation1,Citation2 Proinflammatory cytokines are produced by infiltrating immune cellsCitation1 and induce β-cell apoptosisCitation2 through activation of the transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB).Citation3,Citation4 Cytokine antagonists are under study as novel anti-diabetic treatments.Citation5,Citation6
MicroRNAs (miRNAs) are small noncoding ~22 nucleotide transcripts regulating protein expression by binding to complementary mRNA binding regions thereby targeting them for either degradation or translational inhibition.Citation7 Since their discovery in 2001, miRNAs have received great attention, owing to predictions indicating that up to 50% of all genes are regulated through the miRNA pathway.Citation8 Screening approaches have revealed that β cells express numerous miRNAs,Citation9 and several functional roles of these miRNAs have been described: β-cell differentiation,Citation9,Citation10 regulation of expression of essential β-cellular transcription factors as well as insulin,Citation9 and regulation of insulin secretion.Citation10,Citation11 Recently, miRNAs were shown to regulate β-cell apoptosis following exposure to fatty acids and cytokines.Citation12,Citation13 Our understanding of the functional effects of miRNAs in the β cell is however incomplete.
miR-21, a miRNA generally considered to be anti-apoptotic, has mostly been investigated as a target for knockdown in the treatment of cancers, since it inhibits the proinflammatory tumor suppressor programmed cell death protein 4 (PDCD4) translation.Citation14 In non-obese diabetic (NOD) mice, deficiency of PDCD4 was found to protect β cells from streptozotocin-induced cell death, and conferred resistance to spontaneous autoimmune diabetes.Citation15 β-cell miR-21 overexpression might therefore exert a protective effect through reducing levels of PDCD4. Interestingly, the important mediator of proinflammatory cytokine signaling, NFκB, induces expression of miR-21.Citation15,Citation16 In several cell types, miR-21 is an inhibitor of NFκB activity through its target PDCD4, as reviewed in,Citation17 and thus provide negative feedback on NFκB activity. Proinflammatory cytokines increase miR-21 expression in human islets and the mouse insulinoma (MIN6) cell line,Citation13 possibly as a protective response, and correspondingly, transient knockdown of miR-21 in MIN6 cells increased apoptosis.
miR-34a is a pro-apoptotic miRNA by activating the p53 apoptosis pathway, and increased expression of miR-34a leads to increased cell death.Citation18 In the β-cell line MIN6, transient knockdown of miR-34a reduced apoptosis.Citation13 These results suggest that miR-21 and miR-34a are potential targets to manipulate β-cell apoptotic pathways.
Islet transplantation is a promising experimental therapy for T1D, but the procedure is hampered by immediate and late graft failure resulting in recurrence of T1D over the course of a few years. As there are insufficient β cells available for the current transplantation demand, any increase of β-cell survival in the transplants would be of great benefit. The transplantation procedure is an obvious opportunity to improve β-cell survival through gene therapy as unwanted side effects of altering gene expression in non-islet cells are reduced. However, as β cells are slow-dividing cells, episomal plasmid-based gene therapy is difficult, since it necessitates plasmid entry into the nucleus during cell division. Lentiviral vectors are therefore required to manipulate gene expression in non-dividing cells, and furthermore to achieve long-term alterations. Published data designating miR-34a and miR-21 as promising targets for manipulation are produced using transient transfection methods, and differences in response to short-term vs. long-term miRNA alterations have been observed.Citation19 Therefore, it is crucial to address potential long-term effects of stable miRNA alterations in regard of gene therapy application.
We have created rat insulinoma (INS-1) cell lines stably overexpressing miR-21 or with stable knockdown of miR-34a with the aim of comprehensively analyzing the effects of these miRNA manipulations on net β-cell number in the absence of cytokines, and on β-cell apoptosis, NO synthesis and cellular proliferation in the absence or presence of cytokines.
Results
Overexpression of miR-21 and knockdown of miR-34a in INS-1 cells
Verification of altered expression of the 2 miRNAs was performed by quantitative PCR (qPCR). Expression was investigated compared with 2 different types of control cells: wild-type cells (“wild type”), and cells transduced with an empty vector (“miR-neg”). Altered expression was verified for miR-21 and miR-34a. In the stably transduced INS-1 cells, miR-21 expression was increased over 9-fold, while miR-34a expression was reduced to under half of the control (Fig. S1).
Total cell number is reduced by miR-21 overexpression, but increased by miR-34a knockdown
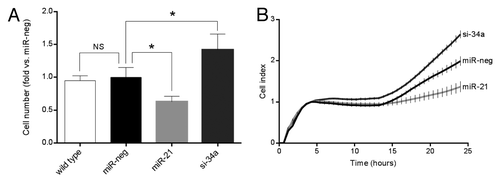
The transduced cell lines revealed different total cell numbers upon passaging the cells under standard culture conditions. To investigate this further, we seeded 50000 cells and counted the total cell number after 3 d of culture. There were no significant differences between wild-type and miR-neg cells. In the absence of cytokines overexpression of miR-21 resulted in 33% fewer cells compared with miR-neg cells, while knockdown of miR-34a lead to 57% more cells compared with miR-neg cells (). We further investigated this difference in total cell number using the xCELLigence system (Roche Diagnostics) to measure cell-to-plate surface contact (cell index) real-time for 24 h (). Data obtained with the xCELLigence system confirmed results obtained through manual counting, with increased number of miR-34a-deficient cells compared with miR-neg, while the number of miR-21-overexpressing cells was decreased in the absence of cytokines.
Figure 1. Overexpression of miR-21 reduces cell number, while knockdown of miR-34a increases cell number in the absence of cytokines. (A) Cells (50 000 cells) were cultured for 72 h prior to trypsinization and manual counting in a counting chamber. Results of 5 independent experiments are shown as means + SEM. Statistical significance was determined using a 2-way paired t test. * P < 0.05 vs. miR-neg. (B) 20 000 cells were seeded pr well in the xCELLigence E-plate and cell index measured in real-time for 24 h. The graph depicts the average cell index ± SEM determined from triplicates from each time point from a single experiment.
Apoptosis is potentiated by miR-21 overexpression, but decreased by miR-34a knockdown in absence or presence of cytokines
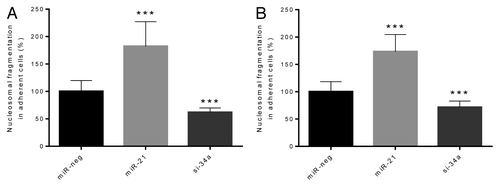
To gain insight into the mechanisms leading to altered cell numbers, we investigated cell death at different stages. Early apoptosis was measured by detecting nucleosomal fragments in adherent cell lysates in the absence or presence of cytokines (). Overexpression of miR-21 significantly increased apoptosis compared with miR-neg cells in the absence (P < 0.05) or presence (P < 0.001) of cytokines, whereas miR-34a knockdown significantly reduced apoptosis compared with miR-neg cells in the absence (P < 0.05) or presence (P < 0.001) of cytokines. To measure late apoptosis/necrosis we investigated nucleosomal fragments in lysates of non-adherent cells and in supernatants (Fig. S2). Overexpression of miR-21 significantly increased late apoptosis in the absence (P < 0.01) and presence of cytokines (P < 0.05), whereas knockdown of miR-34a reduced late apoptosis in both the absence (P < 0.05) and presence (P < 0.05) of cytokines.
Figure 2. Early β-cell apoptosis is potentiated by overexpression of miR-21, but reduced by knockdown of miR-34a both in the absence and the presence of cytokines. Cells (50 000 cells) were left untreated (A) or were exposed to IL-1β and IFN-γ for 24 h (B). Nucleosomal fragments in adherent cell lysates were detected by ELISA (Cell Death Detection ELISA). Results of 10 independent experiments are shown as means + SEM. Statistical significance was determined using a 2-way paired t test. *** P < 0.001 vs. miR-neg.
Cytokine-induced NO formation is potentiated by miR-21 overexpression and decreased by miR-34a knockdown
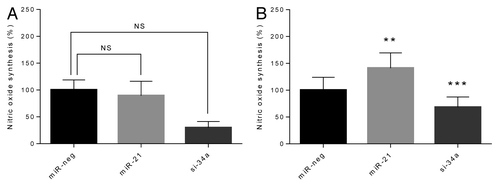
When exposed to cytokines, the production of NO, an indicator of cytokine-induced β-cell stress, was significantly increased in both cell lines as expected (). In the presence of cytokines, overexpression of miR-21 significantly increased cytokine-induced NO synthesis (P < 0.01), while miR-34a knockdown significantly reduced cytokine-induced NO synthesis (P > 0.001) (). No statistically significant changes were detected in the absence of cytokines.
Figure 3. Synthesis of NO is potentiated by overexpression of miR-21, and reduced by knockdown of miR-34a in the presence of cytokines. Cells (50 000 cells) were left untreated (A) or were exposed to IL-1β and IFN-γ for 24 h (B). Culture medium was retrieved and accumulated nitric oxide was determined using the Griess reaction. Results of 9 independent experiments are shown as means + SEM. Statistical significance was determined using a 2-way paired t test. ** P < 0.01 vs. miR-neg, *** P < 0.001 vs. miR-neg.
Proliferation is affected by alteration of miR-21 and miR-34a levels
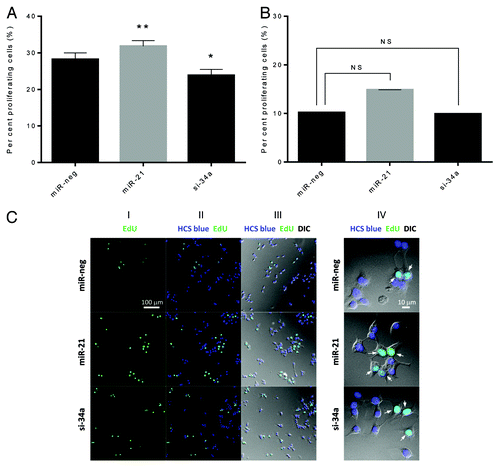
Finally, we investigated whether manipulation of miR-21 and miR-34a levels altered cell proliferation, determined by incorporation of EdU (5-ethynyl-2’-deoxyuridine) into DNA during active DNA synthesis and HCS Blue staining of all cell nuclei. Overexpression of miR-21 increased proliferation, while knockdown of miR-34a reduced proliferation in the absence of cytokines () (absolute numbers of proliferating and total cells are shown in Fig. S3). Upon exposure to proinflammatory cytokines no significant differences were found (). Representative photomicrographs are shown ().
Figure 4. miR-21 overexpression and miR-34a knockdown affect β-cell proliferation. (A and B) Cells (25 000 cells) were left untreated (A) or were exposed to IL-1β and IFN-γ for 24 h (B). Nuclei were stained using EdU and HCS Blue, and fixed with mounting reagent. Nuclei were counted by 2 blinded observers as total number of cells (blue nuclei) and proliferating cells (green nuclei). The ratio of proliferating cells was calculated. Results of 4 independent experiments are shown as means + SEM. Statistical significance was determined using a 2-way paired t test. * P < 0.05 vs. miR-neg, ** P < 0.01 vs. miR-neg. (C) Representative photomicrographs of β-cell proliferation. Panel I shows proliferating cells identified by EdU staining, panel II shows a merge of total number of cells (blue nuclei) and proliferating cells (green nuclei), and panel III shows a merge between panel I and II and an image taken using digital interference contrast microscopy (DIC). Panel IV shows a close-up of panel III (arrows indicate proliferating cells).
Discussion
Here we show that miRNAs miR-21 and miR-34a are involved in key cellular processes regulating β-cell number. Although slightly increasing proliferation, we found that overexpression of miR-21 potentiated cell death of the β-cell line INS-1 both under control conditions as well as when exposed to proinflammatory cytokines, ultimately leading to a reduced net β-cell number. We further show that knockdown of miR-34a protected β cells from cytokine-induced cell death and decreased cell death under control conditions, thereby increasing the net number of β cells.
miR-21 is considered as a pro-proliferative miRNA,Citation15,Citation20-Citation30 and we therefore anticipated that overexpression of miR-21 increased β-cell proliferation. Under control conditions, increased levels of miR-21 did indeed promote β-cell proliferation as determined by EdU staining. However, β-cell death was augmented to such a degree that the net effect of miR-21 overexpression was a decreased net number of β cells, possibly due to forcing the cells through cell-cycle checkpoints and thereby triggering checkpoint-induced apoptosis pathways. In support of this, overexpression of miR-21 tended to alter the expression of 2 cell-cycle genes involved in the G1 phase preceding the first apoptosis-checkpoint (Fig. S4). Despite targeting various tumor suppressors and the classification as an anti-apoptotic miRNA, the effect of miR-21 overexpression is ambiguous. Several studies demonstrate harmful effects of miR-21, casting doubt on the categorization of miR-21 as an anti-apoptotic miRNA.Citation13,Citation31 In human islets and MIN6 cells, proinflammatory cytokines induce expression of miR-21 through activation of NFκB,Citation13,Citation16 and NFκB leads to expression of iNOS and NO production.Citation3 One possible mechanism whereby miR-21 overexpression harms the cells is thereby through an increased cytokine-induced NO synthesis, ultimately leading to increased apoptosis. Depending on cell type, miR-21 can either activate or inhibit NFκB,Citation32,Citation33 and although miR-21 may inhibit NFκB activity through reduced PDCD4 levels, long-term overexpression of miR-21 in our experiments appears to be detrimental. Although reduced levels of PDCD4 in NOD mice protected β cells from cytokine-induced apoptosis and delays diabetes development, the level of diabetes incidence reached that of wild-type mice after 25 wk.Citation15 An explanation for the lack of long-term protection of PDCD4 knockdown may be due to elevated apoptosis through decreased translational repression of procaspase 3 mRNA as suggested in.Citation34 We were not able to detect a reduction in PDCD4 mRNA levels (Fig. S5), presumably due to miR-21 targeting PDCD4 mRNA with only partial complementarity which does not degrade the mRNA, but prevents it from being translated into protein. However, tight control of PDCD4 levels is required for maintaining a functional β-cell mass, and long-term alterations of miR-21 expression may perturb this balance. Other anti-apoptotic miRNAs such as the miR-17–92 cluster control proteins with a similar tight regulation, e.g., the eukaryote transcription factor E2F1 where both too high and too low levels result in depletion of testicular cells.Citation35
Apart from the importance of miR-21 expression levels, the temporal expression pattern seems decisive for cellular outcomes. The net action of a miRNA overexpression can shift from anti-apoptotic to pro-apoptotic, depending on whether a miRNA alteration is transient or stable, respectively.Citation19 Since most studies finding anti-apoptotic/pro-proliferative effects of miR-21 are based upon transient alterations of miR-21 expression, this offers further explanation of the detrimental long-term effect seen in our study of stable cell lines.
We observed that knockdown of miR-34a slightly but significantly decreased proliferation of INS-1 cells under control conditions. However, as cells were protected from cell death both under control conditions and when exposed to proinflammatory cytokines, the net effect of miR-34a knockdown was an increase of β-cell mass. Our findings are in agreement with other studies showing that miR-34a is a pro-apoptotic miRNA which exerts its effects through promoting p53 mediated apoptosis.Citation13,Citation18,Citation36-Citation41 Among the known targets of miR-34a is the protein silent information regulator (SIRT1), and downregulation of SIRT1 leads to apoptosis through an increase in p53.Citation41 SIRT1 is additionally involved in glucose homeostasisCitation42 and in β-cells SIRT1 increases glucose-stimulated insulin secretion.Citation43 Recently, a monogenic form of T1D has been identified, which is caused by a mutation in the SIRT1 gene.Citation44 Knockdown of miR-34a might therefore not only reduce apoptosis in β-cells, but also improve insulin secretion through regulating SIRT1 levels. We show that knockdown of miR-34a tended to increase the expression of SIRT1 (P = 0.10), which therefore might substantiate the beneficial effects of knocking down miR-34a (Fig. S5).
A limitation of this study was the INS-1 cell line model to investigate the effects of stable alteration of 2 miRNAs in β cells. Although immortalized cell lines are generally not the most biologically relevant model systems for the primary β cell, they are well described, thereby facilitating interpretation of data. Additionally, when investigating altered proliferation and growth rates INS-1 cells provide an advantage, as detecting such alterations in primary β cells can be difficult due to their very low basal proliferation.Citation45,Citation46 The current data however warrants the design of future experiments investigating the roles of miR-21 and miR-34a in primary β cells and animal models.
In conclusion, we have found that through stable miRNA alterations in the β-cell line INS-1, miR-21 overexpression potentiated, and miR-34a knockdown attenuated apoptosis both in the absence or presence of proinflammatory cytokines. The observed effect on apoptosis in the absence of cytokines suggests that miR-21 and miR-34a not only regulate translation of proteins involved in cytokine-induced apoptosis, but also proteins implicated in general mechanisms regulating apoptosis under unstimulated conditions. While the pro-apoptotic effects of miR-21 overexpression in β cells require further investigations, we conclude that stable knockdown of miR-34a exerts protective effects both in the absence or presence of cytokines, suggesting Lentiviral knockdown of miR-34a as a therapeutic method to prevent β-cell death in islets transplantation.
Materials and methods
Cytokines
Recombinant mouse interleukin (IL)-1β was from BD PharMingen (554577) and was used at a concentration of 150 pg/ml. Recombinant rat interferon (IFN)-γ was from R&D Systems (585–1F) and was used at a concentration of 5 ng/ml.
Cells
INS-1 cells were a gift from C Wollheim, Department of Cell Physiology and Metabolism, University Medical Center, Geneva, Switzerland. Cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2 in RPMI 1640 medium with glutamax (GIBCO, 153732), supplemented with 10% (heat-inactivated) fetal calf serum (GIBCO, 26140), 100 U/ml penicillin, 100 µg/ml streptomycin (GIBCO, 15140), and 50 µmol/l β-mercaptoethanol (Sigma, M7522) (complete medium). Cells were passaged weekly, and precultured for 2 d prior to cytokine exposure.
Lentivirus production and transduction
Lentiviral expression vectors and viral particles were generated using the BLOCK-iT™ Lentiviral Pol II miR RNAi Expression System (Invitrogen/Gibco, K4938–00), following the manufacturer’s protocols. Sequences for miR-21 and miR-34a were found at http://www.mirbase.org, and primers for expression of miR-21 and a siRNA targeting a unique loop sequence of the pre-miR-34a transcript, thereby selectively knocking down miR-34a, were ordered from TAG Copenhagen A/S. Sequences cloned into the vectors were:
5′-TGCTGTAGCT TATCAGACTG ATGTTGAGTT TTGGCCACTG ACTGACTCAA CATCTCTGAT AAGCTA-3′ for miR-21 overexpression, and
5′-TGCTGTTAGC TAATACTCAC AACAACGTTT TGGCCACTGA CTGACGTTGT TGTGTATTAG CTAA-3′ for knockdown of miR-34a. As a negative control, we transfected cells with a virus containing an artificial sequence not expressed in the rat genome (miR-neg). Virus was generated by cotransfection of HEK293FT cells with plasmids from the ViraPower packaging mix (Invitrogen) using Lipofectamine™ 2000 (Invitrogen, 11668). After 72 h, virus was harvested by ultracentrifugation of the supernatant fraction at 20 000 g for 2 h, and the pellet was resuspended in medium. Titers were determined in HT1080 cells. Stable INS-1 cells were generated by seeding 1 million cells in 2 ml medium. After 2 d, medium was removed and virus-containing medium added, at a multiplicity of infection (MOI) of 10. Medium was replaced with 2 ml complete medium 6h after transduction, and transduced cells were selected for using 6 µg/ml blasticidin for 2–3 wk, during which approximately half the INS-1 cells died.
qPCR
Overexpression of miR-21 or knockdown of miR-34a was verified using qPCR. INS-1 cells (1.0 × 106 in 5 ml per condition) were seeded in 6-well plates 48 h prior to experiments. Total RNA was harvested and extracted using TRIzol (Invitrogen). Quantity of the extracted RNA was assessed using a NanoDrop 1000 (Thermo Scientific, ND-1000). qPCR was performed using Applied Biosystems 7900HT PCR System using TaqMan® Universal PCR Master Mix (Applied Biosystems), according to the manufacturer’s guidelines. TaqMan miRNA detection kits were: miR-21 (000397), miR-34a (000426), U6 (001973). To investigate the expression of CDK4, cyclin D2, SIRT1, and PDCD4, we used SYBR® green PCR Master Mix (Applied Biosystems), according to the manufacturer’s guidelines. Primers were provided by TAG Copenhagen as the following sequences: CDK4 forward (5′-ACCTACGGAC ATACCTGGAC A-3′); CDK4 reverse (5′-TTGCATGAAG GAAATCTAGG C-3′); SIRT1 forward (5′-TCCTGTGGGA TACCTGACTT C-3′); SIRT1 reverse (5′-CAAGGCGAGC ATAAATACCA T-3′); PDCD4 forward (5′-ATGATGTGGA GGAGGTGGAT GTG-3′); PDCD4 reverse (5′- CCAATGCTAA GGATACTGCC AAC-3′); HPRT forward (5′-GCAGACTTTG CTTTCCTTGG-3′); HPRT reverse (5′-GCCTGTATCC AACACTTCGA G-3′). Probes were provided by Applied Biosystems: cyclin D2 (Rn0142401_m1), HPRT (cat. no. Rn01428093_m1). Data was analyzed using SDS 2.3 (Applied Biosystems), and was quantified using the 2-ΔΔct methodCitation47 with the house-keeping genes U6 or HPRT as endogenous controls and mRNA from the control INS-1 cells as calibrator.
Cell counting
Cells were plated in 48-well dishes (50 000 cells/well), cultured for 72 h, and trypsinized using 200 µl Trypsin-EDTA (Invitrogen, 15090–046). Cells were counted manually in a counting chamber. The means from 5 independent experiments were subsequently used to correct for varying growth rates of the cell lines.
Cell death detection assay
As a measure of cell death, histone/DNA complexes released from the nucleus to the cytosol were measured according to the manufacturer’s instructions (Cell Death Detection ELISAPLUS, Roche). Cells were plated in 48-well dishes (50 000 cells/well), exposed to cytokines after 48 h, and evaluated after 72 h using the ELISA kit. Early β-cell apoptosis was determined through analysis of lysates of the adherent cells, while late apoptosis/necrosis was determined through analysis of detached cells floating in the media and harvested by centrifugation of the culture supernatants at 1200 x rpm for 5 min.
NO measurement
NO, an indicator of β-cell stress, was determined in cell supernatants from the above experiments. NO was measured as accumulated nitrite in the medium by mixing equal volumes of cell culture medium and Griess reagent (0.1% [wt/vol.] naphthylethene diamine hydrochloride; Sigma) in H2O2, and 1% (wt/vol.) sulphanilamide (Bie and Berntsen) in 5% (vol./vol.) H3PO4 (Merck # 1005631000). Absorbance was measured at 550 nm and accumulated nitrite calculated from a NaNO2 standard curve.
Proliferation assay
Cells were grown according to protocol given by the Click-iT® EdU Cell Proliferation Assay (Invitrogen, C10351) and fixed for differential interference contrast (DIC) and fluorescence microscopy analysis (FM) as described.Citation48 Nuclei were stained with blue-fluorescent HCS NuclearMask™ Blue Stain (Invitrogen), and EdU (5-ethynyl-2’-deoxyuridine) (Invitrogen) was applied for staining cycling cells having entered the S-phase. Proliferating cells were found as the ratio between the total number of cells (with blue nuclei) and cells during S-phase (with green nuclei). Nuclei were counted by 2 blinded observers and calculated as means. Images were obtained with an Olympus DP72 digital camera on an Olympus BX63 motorized reflected epifluorescence microscope using Olympus cellSense Dimension imaging software. Images were digitally processed with Adobe Photoshop CS4 for overlays.
Cell viability
Cell viability was assessed using the xCELLigence platform (RTCA SP, Roche Diagnostics), which through a cell index reflects real-time attachment, morphology, and proliferation. E-plates (96-wells) were precoated with poly-d-lysine (Sigma, CAS no. 27964–99). Twenty-thousand INS-1 cells were added per well in 20 µl medium followed by 30 min incubation at room temperature. The E-plate was inserted into the xCELLigence platform and placed at 37 °C with 5% CO2. Cell index was monitored over a time period of 24 h.
Statistical analysis
Results are presented as means + SEM. Statistical significance (P < 0.05) between single conditions was determined using a 2-way paired or unpaired t test with the Tukey correction using SAS® 9.1.3 for normally distributed data. For Fig. S5, a 1-way unpaired t test was used.
| Abbreviations: | ||
| CDK4 | = | cyclin-dependent kinase 4 |
| CO2 | = | carbon dioxide |
| DIC | = | differential interference contrast |
| EDTA | = | ethylenediaminetetra acetic acid |
| EdU | = | 5-ethynyl-2’-deoxyuridine |
| FM | = | fluorescence microscopy |
| HCS | = | high-content screening |
| HPRT | = | hypoxanthine-guanine phosphoribosyltransferase |
| H2O2 | = | hydrogen peroxide |
| H3PO4 | = | phosphoric acid |
| IFN | = | interferon |
| IL | = | interleukin |
| INS-1 | = | insulinoma 1 |
| LPS | = | lipopolysaccharide |
| miRNA | = | micro RNA |
| MOI | = | multiplicity of infection |
| NaNO2 | = | sodium nitrite |
| NFκB | = | nuclear factor kappa-light-chain-enhancer of activated B-cells |
| NO | = | nitric oxide |
| NOD | = | non-obese diabetic |
| PDCD4 | = | programmed cell death protein 4 |
| PTEN | = | phosphatase and tensin homolog |
| qPCR | = | quantitative polymerase chain reaction |
| SEM | = | standard error of the mean |
| SIRT | = | silent information regulator |
| T1D | = | type 1 diabetes mellitus |
| T2D | = | type 2 diabetes mellitus |
Additional material
Download Zip (531.7 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Funding for the support of this work was as follows: Backe MB was supported by a SHARE (Synergy in Human and Animal Research, LIFE) PhD fellowship. Novotny GW was supported by the European Community’s Seventh Framework Program FP7/2007–2013-FP7-NMP-In vivo Imaging of Beta-cell by Applied Nano Technology (VIBRANT). Christensen DP was supported by JDRF (Juvenile Diabetes Research Foundation) (#26–2008–893) and the EFSD/JDRF/NovoNordisk European Program in Type 1 Diabetes Research. Grunnet LG and Mandrup-Poulsen T were supported by Novo Nordisk and the Novo Nordisk Foundation. We thank Helle Fjordvang, Søren Tvorup Christensen and Thorkil Ploug for technical assistance.
References
- Donath MY, Størling J, Berchtold LA, Billestrup N, Mandrup-Poulsen T. Cytokines and beta-cell biology: from concept to clinical translation. Endocr Rev 2008; 29:334 - 50; http://dx.doi.org/10.1210/er.2007-0033; PMID: 18048762
- Donath MY, Størling J, Maedler K, Mandrup-Poulsen T. Inflammatory mediators and islet beta-cell failure: a link between type 1 and type 2 diabetes. J Mol Med (Berl) 2003; 81:455 - 70; http://dx.doi.org/10.1007/s00109-003-0450-y; PMID: 12879149
- Giannoukakis N, Rudert WA, Trucco M, Robbins PD. Protection of human islets from the effects of interleukin-1beta by adenoviral gene transfer of an Ikappa B repressor. J Biol Chem 2000; 275:36509 - 13; http://dx.doi.org/10.1074/jbc.M005943200; PMID: 10967112
- Heimberg H, Heremans Y, Jobin C, Leemans R, Cardozo AK, Darville M, Eizirik DL. Inhibition of cytokine-induced NF-kappaB activation by adenovirus-mediated expression of a NF-kappaB super-repressor prevents beta-cell apoptosis. Diabetes 2001; 50:2219 - 24; http://dx.doi.org/10.2337/diabetes.50.10.2219; PMID: 11574401
- Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, Seifert B, Mandrup-Poulsen T, Donath MY. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 2007; 356:1517 - 26; http://dx.doi.org/10.1056/NEJMoa065213; PMID: 17429083
- Moran A, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, et al. Interleukin-1 antagonism in type one diabetes of recent onset: two multicentre, randomized, double-blind, placebo-controlled trials. Lancer 2013; 38:1905 - 15; http://dx.doi.org/10.1016/S0140-6736(13)60023-9
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116:281 - 97; http://dx.doi.org/10.1016/S0092-8674(04)00045-5; PMID: 14744438
- Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 2009; 19:92 - 105; http://dx.doi.org/10.1101/gr.082701.108; PMID: 18955434
- Lynn FC, Skewes-Cox P, Kosaka Y, McManus MT, Harfe BD, German MS. MicroRNA expression is required for pancreatic islet cell genesis in the mouse. Diabetes 2007; 56:2938 - 45; http://dx.doi.org/10.2337/db07-0175; PMID: 17804764
- Tang X, Tang G, Ozcan S. Role of microRNAs in diabetes. Biochim Biophys Acta 2008; 1779:697 - 701; http://dx.doi.org/10.1016/j.bbagrm.2008.06.010; PMID: 18655850
- Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, Pfeffer S, Tuschl T, Rajewsky N, Rorsman P, et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature 2004; 432:226 - 30; http://dx.doi.org/10.1038/nature03076; PMID: 15538371
- Lovis P, Roggli E, Laybutt DR, Gattesco S, Yang JY, Widmann C, Abderrahmani A, Regazzi R. Alterations in microRNA expression contribute to fatty acid-induced pancreatic beta-cell dysfunction. Diabetes 2008; 57:2728 - 36; http://dx.doi.org/10.2337/db07-1252; PMID: 18633110
- Roggli E, Britan A, Gattesco S, Lin-Marq N, Abderrahmani A, Meda P, Regazzi R. Involvement of microRNAs in the cytotoxic effects exerted by proinflammatory cytokines on pancreatic beta-cells. Diabetes 2010; 59:978 - 86; http://dx.doi.org/10.2337/db09-0881; PMID: 20086228
- Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O’Leary JJ, Ruan Q, Johnson DS, Chen Y, O’Neill LA. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol 2010; 11:141 - 7; http://dx.doi.org/10.1038/ni.1828; PMID: 19946272
- Ruan Q, Wang T, Kameswaran V, Wei Q, Johnson DS, Matschinsky F, Shi W, Chen YH. The microRNA-21-PDCD4 axis prevents type 1 diabetes by blocking pancreatic beta cell death. Proc Natl Acad Sci U S A 2011; 108:12030 - 5; http://dx.doi.org/10.1073/pnas.1101450108; PMID: 21730150
- Zhou R, Hu G, Liu J, Gong AY, Drescher KM, Chen XM. NF-kappaB p65-dependent transactivation of miRNA genes following Cryptosporidium parvum infection stimulates epithelial cell immune responses. PLoS Pathog 2009; 5:e1000681; http://dx.doi.org/10.1371/journal.ppat.1000681; PMID: 19997496
- Ma X, Buscaglia LEB, Barker JR, Li Y. MicroRNAs in NF-κB signaling. J Mol Cell Biol 2011; 0:1 - 8
- Ji Q, Hao X, Zhang M, Tang W, Yang M, Li L, Xiang D, Desano JT, Bommer GT, Fan D, et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One 2009; 4:e6816; http://dx.doi.org/10.1371/journal.pone.0006816; PMID: 19714243
- Kasar S, Salerno E, Yuan Y, Underbayev C, Vollenweider D, Laurindo MF, Fernandes H, Bonci D, Addario A, Mazzella F, et al. Systemic in vivo lentiviral delivery of miR-15a/16 reduces malignancy in the NZB de novo mouse model of chronic lymphocytic leukemia. Genes Immun 2012; 13:109 - 19; http://dx.doi.org/10.1038/gene.2011.58; PMID: 21881595
- Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res 2005; 65:6029 - 33; http://dx.doi.org/10.1158/0008-5472.CAN-05-0137; PMID: 16024602
- Krichevsky AM, Gabriely G. miR-21: a small multi-faceted RNA. J Cell Mol Med 2009; 13:39 - 53; http://dx.doi.org/10.1111/j.1582-4934.2008.00556.x; PMID: 19175699
- Gaur AB, Holbeck SL, Colburn NH, Israel MA. Downregulation of Pdcd4 by mir-21 facilitates glioblastoma proliferation in vivo. Neuro Oncol 2011; 13:580 - 90; http://dx.doi.org/10.1093/neuonc/nor033; PMID: 21636706
- Hu H, Li Y, Gu J, Zhu X, Dong D, Yao J, Lin C, Fei J. Antisense oligonucleotide against miR-21 inhibits migration and induces apoptosis in leukemic K562 cells. Leuk Lymphoma 2010; 51:694 - 701; http://dx.doi.org/10.3109/10428191003596835; PMID: 20141427
- Park JK, Lee EJ, Esau C, Schmittgen TD. Antisense inhibition of microRNA-21 or -221 arrests cell cycle, induces apoptosis, and sensitizes the effects of gemcitabine in pancreatic adenocarcinoma. Pancreas 2009; 38:e190 - 9; http://dx.doi.org/10.1097/MPA.0b013e3181ba82e1; PMID: 19730150
- Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007; 133:647 - 58; http://dx.doi.org/10.1053/j.gastro.2007.05.022; PMID: 17681183
- Zhu S, Si ML, Wu H, Mo YY. MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1). J Biol Chem 2007; 282:14328 - 36; http://dx.doi.org/10.1074/jbc.M611393200; PMID: 17363372
- Selcuklu SD, Donoghue MT, Spillane C. miR-21 as a key regulator of oncogenic processes. Biochem Soc Trans 2009; 37:918 - 25; http://dx.doi.org/10.1042/BST0370918; PMID: 19614619
- Lu TX, Lim EJ, Itskovich S, Besse JA, Plassard AJ, Mingler MK, Rothenberg JA, Fulkerson PC, Aronow BJ, Rothenberg ME. Targeted ablation of miR-21 decreases murine eosinophil progenitor cell growth. PLoS One 2013; 8:e59397; http://dx.doi.org/10.1371/journal.pone.0059397; PMID: 23533623
- Li J, Huang H, Sun L, Yang M, Pan C, Chen W, Wu D, Lin Z, Zeng C, Yao Y, et al. MiR-21 indicates poor prognosis in tongue squamous cell carcinomas as an apoptosis inhibitor. Clin Cancer Res 2009; 15:3998 - 4008; http://dx.doi.org/10.1158/1078-0432.CCR-08-3053; PMID: 19509158
- Papagiannakopoulos T, Shapiro A, Kosik KS. MicroRNA-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Res 2008; 68:8164 - 72; http://dx.doi.org/10.1158/0008-5472.CAN-08-1305; PMID: 18829576
- Cheng AM, Byrom MW, Shelton J, Ford LP. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res 2005; 33:1290 - 7; http://dx.doi.org/10.1093/nar/gki200; PMID: 15741182
- Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell 2010; 39:493 - 506; http://dx.doi.org/10.1016/j.molcel.2010.07.023; PMID: 20797623
- Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O’Leary JJ, Ruan Q, Johnson DS, Chen Y, O’Neill LA. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol 2010; 11:141 - 7; http://dx.doi.org/10.1038/ni.1828; PMID: 19946272
- Eto K, Goto S, Nakashima W, Ura Y, Abe SI. Loss of programmed cell death 4 induces apoptosis by promoting the translation of procaspase-3 mRNA. Cell Death Differ 2012; 19:573 - 81; http://dx.doi.org/10.1038/cdd.2011.126; PMID: 21959934
- Novotny GW, Sonne SB, Nielsen JE, Jonstrup SP, Hansen MA, Skakkebaek NE, Rajpert-De Meyts E, Kjems J, Leffers H. Translational repression of E2F1 mRNA in carcinoma in situ and normal testis correlates with expression of the miR-17-92 cluster. Cell Death Differ 2007; 14:879 - 82; http://dx.doi.org/10.1038/sj.cdd.4402090; PMID: 17218954
- Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, Bentwich Z, Oren M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell 2007; 26:731 - 43; http://dx.doi.org/10.1016/j.molcel.2007.05.017; PMID: 17540598
- Guo Y, Li S, Qu J, Wang S, Dang Y, Fan J, Yu S, Zhang J. MiR-34a inhibits lymphatic metastasis potential of mouse hepatoma cells. Mol Cell Biochem 2011; 354:275 - 82; http://dx.doi.org/10.1007/s11010-011-0827-0; PMID: 21553024
- Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 2007; 26:745 - 52; http://dx.doi.org/10.1016/j.molcel.2007.05.010; PMID: 17540599
- Li WB, Ma MW, Dong LJ, Wang F, Chen LX, Li XR. MicroRNA-34a targets notch1 and inhibits cell proliferation in glioblastoma multiforme. Cancer Biol Ther 2011; 12:477 - 83; http://dx.doi.org/10.4161/cbt.12.6.16300; PMID: 21743299
- Welch C, Chen Y, Stallings RL. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene 2007; 26:5017 - 22; http://dx.doi.org/10.1038/sj.onc.1210293; PMID: 17297439
- Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A 2008; 105:13421 - 6; http://dx.doi.org/10.1073/pnas.0801613105; PMID: 18755897
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005; 434:113 - 8; http://dx.doi.org/10.1038/nature03354; PMID: 15744310
- Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Méneur C, Permutt MA, Imai S. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab 2005; 2:105 - 17; http://dx.doi.org/10.1016/j.cmet.2005.07.001; PMID: 16098828
- Biason-Lauber A, Böni-Schnetzler M, Hubbard BP, Bouzakri K, Brunner A, Cavelti-Weder C, Keller C, Meyer-Böni M, Meier DT, Brorsson C, et al. Identification of a SIRT1 mutation in a family with type 1 diabetes. Cell Metab 2013; 17:448 - 55; http://dx.doi.org/10.1016/j.cmet.2013.02.001; PMID: 23473037
- Parnaud G, Bosco D, Berney T, Pattou F, Kerr-Conte J, Donath MY, Bruun C, Mandrup-Poulsen T, Billestrup N, Halban PA. Proliferation of sorted human and rat beta cells. Diabetologia 2008; 51:91 - 100; http://dx.doi.org/10.1007/s00125-007-0855-1; PMID: 17994216
- Russ HA, Bar Y, Ravassard P, Efrat S. In vitro proliferation of cells derived from adult human beta-cells revealed by cell-lineage tracing. Diabetes 2008; 57:1575 - 83; http://dx.doi.org/10.2337/db07-1283; PMID: 18316362
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 2008; 3:1101 - 8; http://dx.doi.org/10.1038/nprot.2008.73; PMID: 18546601
- Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, Christensen ST. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr Biol 2005; 15:1861 - 6; http://dx.doi.org/10.1016/j.cub.2005.09.012; PMID: 16243034

