Abstract
Cancer cells are characterized by the aberrant activation of signaling pathways governing proliferation, survival, angiogenesis, migration and immune evasion. These processes are partially regulated by the transcription factor STAT3. This factor is inappropriately activated in diverse tumor types. Since tumor cells can become dependent on its persistent activation, STAT3 is a favorable drug target. Here, we describe the functional characterization of the recombinant STAT3 inhibitor, rS3-PA. This inhibitor is based on a 20 amino acid peptide which specifically interacts with the dimerization domain of STAT3. It is integrated into a thioredoxin scaffold and fused to a protein transduction domain. Protein gel blot and immunofluorescence analyses showed that rS3-PA is efficiently taken up by cells via an endocytosis independent mechanism. Intracellularly, it reduces the phosphorylation of STAT3 and enhances its degradation. This leads to the downregulation of STAT3 target gene expression on the mRNA and protein levels. Subsequently, tumor cell proliferation, survival and migration and the induction of angiogenesis are inhibited. In contrast, normal cells remain unaffected. Systemic administration of rS3-PA at doses of 7.5 mg/kg reduced P-STAT3 levels and significantly inhibited tumor growth up to 35% in a glioblastoma xenograft mouse model.
Introduction
The comparison of normal and tumor cells with respect to their genomic sequences,Citation1 their gene expression patternsCitation2 and activated signaling pathwaysCitation3 has led to the identification of a large number of promising drug targets. A class of signaling molecules that confer an “addiction” phenotype to tumor cells have received particular attention.Citation4 These molecules are temporarily dispensable in normal cells, whereas tumor cells react to their inhibition by growth arrest and induction of apoptosis, and appear particularly suited as targets for innovative drugs.Citation5 Downregulation of the expression of “addiction-conferring” genes by RNA interference served as a proof-of-principle and encouraged the development of targeted compounds.
STAT3 is present in cells as a latent transcription factor. It is activated by receptor associated tyrosine kinases and mediates the action of many cytokines and growth factors. In normal cells, the extent and duration of STAT3 signaling is strictly controlled and downregulated by the action of e.g., phosphatases, Socs and Pias proteins.Citation6 In tumor cells, STAT3 is persistently activated and functions as an oncogene. The sphingosine-1-phosphate receptor-1, S1PR1, and its enhancing effects on tyrosine kinases is a major contributor to this process.Citation7 In addition, somatic mutations activating STAT3 have been discovered.Citation8 Persistently activated STAT3 has been detected in solid tumors of the breast, brain, colon, prostate, lung, pancreas, pituitary, gastrointestines, ovary, cervix and skin and has also been found in lymphomas and leukemias.Citation9 It activates target genes contributing to cell proliferation, cell survival, angiogenesis and to the suppression of immune surveillance.Citation10-Citation12 STAT3 also fulfills the definition of an “addiction factor.”Citation13
Transcription factors usually do not exhibit enzyme activities, nor do they comprise binding pockets for small molecular weight molecules. For this reason they are difficult to target with synthetic, small molecular weight compounds. However, biological macromolecules, especially peptides, can be used as competitive inhibitors to prevent protein-protein interactions required for particular protein functions.Citation14,Citation15 The delimitation of the binding domain of known target-interaction partners and the selection of specific sequences from random peptide libraries can be exploited to derive peptides which exert inhibitory functions.Citation16,Citation17
The peptide aptamer technology has been extensively used to identify peptides with binding specificities for particular functional domains of target proteins. Peptide aptamers are short peptides, usually 12 to 20 amino acids in length, which can be selected from random, high-complexity peptide libraries in yeast-two-hybrid screens.Citation18,Citation19 Our experiments have shown that a variant of the human thioredoxin (hTrx), devoid of cysteine residues, provides a favorable scaffold for the display of such target interacting peptides. Appropriately appended scaffolds allow the presentation of the peptide in a constrained conformation, the production as recombinant proteins and the cellular delivery of specific peptide aptamers.Citation20,Citation21
Here, we further modified the STAT3-specific peptide aptamer hTrxΔcys-DD3.8ΔcysCitation20 to optimize its functional properties. A tagged version, rS3-PA, was derived and detailed analyses of its cellular uptake into cultured cells and the molecular mode of STAT3 inhibition were conducted. We found that rS3-PA efficiently enters cells, causes the reduction of STAT3 phosphorylation and enhances the proteasomal degradation of P-STAT3. This results in STAT3 target gene inhibition and impaired tumor cell proliferation, migration and survival. Finally, i.v. administration of rS3-PA into mice showed that the systemic application of rS3-PA inhibits STAT3 activation in vivo and retarded the growth kinetics of transplanted glioma cells.
Materials and Methods
Reagents
Recombinant IL-6, INFγ chloroquine, heparan sulfates, mitomycin C, anti-Actin, anti-Flag and -Tubulin were obtained from Sigma-Aldrich. STAT3 and STAT5 antibodies were from Santa Cruz. P-STAT1, P-STAT5, P-STAT3Y705, His-tag and c-Myc antibodies were purchased from CellSignaling. Anti-Survivin was obtained from Acris Antibodies and anti-Lamin B1 was obtained from Abcam. MG-132 was purchased from Calbiochem.
Cell lines
Human (MZ-54, MZ-18 and U-373) and murine high-grade glioma cell lines (Tu-9648, Tu-2449), human breast cancer cell lines (SK-BR-3, MDA-MB-468), the A431 epithelial cancer cell line, the murine breast cancer cell line 4T1, the K562 leukemia cell line and immortalized fibroblasts NIH-3T3, were cultivated in DMEM with 10% FCS. Primary human umbilical vein endothelial cells (HUVEC) were kindly provided at passage 2 by Carmen Döbele (Institute of Cardiovascular Regeneration, Frankfurt, Germany) and grown in EGM (endothelial basal medium with supplements and 10% FCS). The immortalized breast cell line MCF-10A cells was grown in DMEM/F12 medium supplied with 5% FCS, 100 U/ml penicillin, 100 U/ml streptomycin, 2 mM L-glutamine, 20 ng/ml human EGF, 100 ng/ml cholera toxin, 10 ng/ml insulin and 500 ng/ml hydrocortisone.
Plasmid construction
For yeast-two-hybrid (YTH) analyses, the different members of the STAT transcription factor family, without their C-terminal transactivation domains, were fused to the Gal4-DNA-binding domain (pGBK-T7, Clontech-Takara Bio Europe). The different peptide aptamer variants were fused to the Gal4-activation domain (pGAD-T7, Clontech-Takara Bio Europe) to create the prey constructs. To improve the recombinant expression of the peptide aptamer, the pFlag vector (Sigma-Aldrich) was used. A NLS, generated by annealing two complementary oligonucleotides (5′- aaa aag cct cca aag aag aag agg aag gaa ttc ttt -3′ and 5′- aaa gaa ttc ctt cct ctt ctt ctt tgg aag ctt ttt -3′) was inserted between the Hind III/EcoR I restriction sites of pFlag. The sequence encoding the peptide aptamer inserted into the hTrx scaffold, was amplified from plasmid pET-hTrxDD3–3.8ΔcysCitation20 using primers with EcoR I restriction sites (5′- aaa gaa ttc atg ggt aag cag atc gag -3′ and 5′- aaa gaa ttc gac taa ttc att aat ggt -3′). Insertion of the product in pFlag-NLS vector resulted in construct pFlag-hTrxΔcys-DD3.8Δcys encoding rS3-PA (see ).
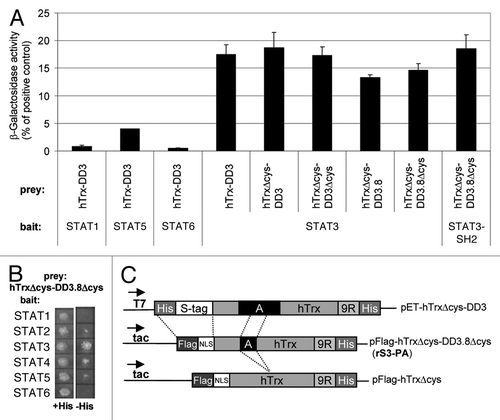
Figure 1. Structure and binding specificity of recombinant peptide aptamer rS3-PA. (A) YTH interaction analysis of the peptide aptamer rS3-PA with members of the STAT family. Yeast cells (Y187) were co-transformed with bait and prey constructs. Interaction of these proteins was quantified in standard β galactosidase assays (displayed in Miller Units). (B) Interaction of bait and prey fusion proteins described in (A) also results in the growth of colonies on plates lacking histidine. (C) Schematic presentation of peptide aptamer DD3 40 amino acids (A) inserted into the hTrx scaffold (upper construct). Modifications performed to generate pFlag-hTrxΔcys-DD3.8Δcys encoding rS3-PA are shown (middle construct). rS3-PA encodes a 20mer peptide fragment of DD3 devoid of cysteines. The peptide aptamer present in rS3-PA corresponds to the sequence N-VRH SAL HMA VGP LSW PAR VS-C. The bait used to select this peptide aptamer comprises the STAT3 sequence 655 to 755.Citation21 Secondary structure prediction programs from the Expasy website (e.g., Porter, NtSurfP, JPred) were used and a comparison of the results suggests that the 20 amino acid peptide aptamer sequence consists largely of a α-helical structure in its N-terminal region and a coiled-coil structure in its C-terminal region. The prediction for the 655–755 amino acid sequence of STAT3 suggests that it is largely composed of a coiled-coil domain with a possible α-helical insert at position 76–84. The pFlag-hTrxΔcys control construct was generated by Rsr II digestion removing the aptamer sequences (lower line). NLS, nuclear localization signal; 9R, nine arginine PTD.
Protein purification
BL21 codonPlus (DE3)-RP competent cells (Stratagene) were transformed with the recombinant expression plasmids (). A 5 ml culture of the cells in standard TB medium was grown for 8 h at 37°C. This culture was used to inoculate a 2 L TB culture with ampicillin and chloramphenicol which was grown for 16 h at 37°C. Cells were harvested at an OD600 of 3.5–5 and lysed under denaturing conditions using urea buffer (8 M urea, 500 mM NaCl in PBS, pH 7.5). Proteins were purified using affinity chromatography on an FPLC system (GE Healthcare) as described earlier.Citation21
Immunofluorescence microscopy
Cells were grown on coverslips and treated for 30 min with peptide aptamers. Slides were prepared for microscopy as described earlier.Citation20 Anti-Flag M2 (mouse) and anti-EEA1 (rabbit) antibodies were diluted 1:100 in blocking buffer (0.5% gelatin from cold water fish skin, 0.1% ovalbumin in PBS) and incubated overnight. Anti-mouse Alexa-Fluor-568 and anti-rabbit Alexa-Fluor-488 coupled secondary antibodies were used in a 1:100 dilution for detection. Nuclei were stained with 1 µM ToPRO-3 iodide (Molecular Probes). To visualize the ER, cells were washed with 0.2 M acetic acid and stained with 100 µg/ml Concanavalin A-Alexa-Fluor-488. Confocal laser scanning microscopy was used to visualize the cells.
mRNA analyses
Total RNA was isolated from cell lysates using the RNeasy Mini Kit (Qiagen) and the SuperScript II Reverse Transcriptase kit (Invitrogen) was used for synthesis of cDNA. To amplify transcripts of STAT3 target genes in Tu-9648 cells, the following primers were used: CyclinD1 5′- tgg aac ctg gcc gcc atg -3′ and 5′- gtg gcc ttg ggg tcg acg -3′, BclXL 5′- agt ttg gat gcg cgg gag -3′ and 5′- gcc aca gtc atg ccc gtc -3′, Survivin 5′- tgg cag ctg tac ctc aag -3′ and 5′- tca aga att cac tga cgg -3′, Actin 5′- atg gcc act gcc gca tcc -3′ and 5′- tcc aca tct gct gga agg -3′. PCR products were generated at 58°C annealing temperature and 20 cycles for semi-quantitative analysis.
Cell viability assays
Two thousand cells were seeded in 96-well plates and the next day, the medium was removed. One micromolar of the peptide aptamers (or PBS as control) were diluted in 100 µl medium and added to the cells every 24 h. Proliferation of the cells was measured on day 3 by adding 50 µl XTT solution (Roche). After 4 h, substrate turnover was determined in a plate reader at 490 nm.
Mouse xenograft transplantations
Female NMRI nu/nu mice (Charles River) were kept in individual ventilated cages. Experiments were performed according to German government guidelines (Regierungspräsidium Darmstadt). Tu-9648 cells (3 × 106) were injected into the right flanks of 6-week-old nude mice. Mice were treated daily by tail vein injection with 150 µl of recombinant proteins (7.5 mg/kg rS3-PA, 7.5 mg/kg hTrx) or temozolomide (60 mg/kg) or with 150 µl PBS. Tumor volumes and body weights were measured every second day and volumes were calculated using the formula: (length × π × width2)/6.
Preparation of tissue lysates
Mouse organs or tumors were dissected and dissociated in 5 ml standard RIPA buffer/g tissue using an Ultra-Turrax (IKA, Staufen, Germany). Samples were incubated on ice for 20 min and sonified. After centrifugation (15,000 rpm, 20 min, 4°C) supernatants were stored at −20°C. For protein gel blot analysis, 100 µg of protein of each sample was loaded onto a polyacrylamide gel. Protein gel blots were performed according to standard procedures.
Results
Development of rS3-PA, a recombinant peptide aptamer with binding specificity for STAT3
We previously identified the 40 amino acid peptide aptamer bTrx-DD3. This molecule was selected to specifically bind to the SH2 domain of STAT3 as shown in YTH assays and co-immunoprecipitation experiments.Citation21 In a subsequent study, the original bacterial bTrx scaffold was replaced by the human hTrx scaffold.Citation20 We verified the binding specificity of this molecule with respect to its interaction with the other members of the STAT family of transcription factors in YTH assays. Aptamer hTrx-DD3 was cloned in a GAL4 prey construct and the genes encoding the STAT family members were cloned in GAL4 bait constructs. The transactivation domains of the STAT proteins were omitted. The interaction of the Stat proteins with the aptamer was investigated using β-galactosidase assays () or determined by growth of the yeast cells on plates lacking histidine (). We found that the DD3 aptamer binds efficiently to STAT3, but not to STAT1, 2, 4, 5A and 6.
Subsequently, the three cysteines present in hTrx as well as one cysteine present in the peptide aptamer sequence were replaced by serines and the 40 amino acid aptamer was delimited to 20 amino acids, resulting in the hTrxΔcys-DD3.8Δcys construct.Citation20 This modified peptide aptamer exhibits very similar binding affinity and specificity for the SH2 domain of STAT3 when compared with the original DD3 peptide aptamer (). Finally, the T7 promoter was replaced by a tac promoter and the N-terminal His- and S-tags of hTrxΔcys-DD3.8Δcys were replaced by a nuclear localization signal and a Flag-tag. The Flag-tag facilitates the intracellular detection of the molecule. This construct has been named rS3-PA (). A construct lacking the aptamer sequence, pFlag-hTrxΔcys, was prepared for control experiments. Upon bacterial expression and affinity purification via its His-tag, we obtained yields of about 25 µg rS3-PA per ml of culture and concentrations of 1.5–2.0 µg/µl. Both rS3-PA and hTrxΔcys-DD3.8Δcys were taken up from the medium and retained by the cultured cells very similarly (data not shown). Intracellular uptake is mediated by the nine arginine protein transduction domain (PTD).
Peptide aptamer rS3-PA is taken up by cultured cells in an endocytosis independent fashion
Protein transduction depends upon interactions between the positively charged PTD and negatively charged heparan sulfate proteoglycans (HS) present on the cell surface. Internalization can involve a form of fluid phase endocytosis, called marcropinocytosis, or occur through the direct penetration of the cellular membrane. We used inhibitors affecting different steps of the endocytosis process to investigate the mechanism of internalization of rS3-PA (). The compounds were added to the medium of the cells 30 min before rS3-PA. After 4 h the cells were washed with acetic acid to remove the proteins associated with the cell surface. We found that none of the endocytosis inhibitors affected rS3-PA uptake when compared with the non-treated cells (). Only heparan sulfates (HS) exerted an effect on the intracellular accumulation of rS3-PA in NIH-3T3, 4T1 and Tu-9648 cells ( and ). The addition of HS, however, is known to sequester PTD fusion proteins in the medium without directly affecting uptake processes.Citation22
Table 1. Effects of different inhibitors on internalization of proteins via endocytosis-like mechanisms
Figure 2. Cellular uptake of rS3-PA. (A) Cells were transduced for 4 h with 1 µM rS3-PA in the presence (+) or absence (-) of 50 µg/ml heparan sulfate (HS). Intracellular rS3-PA was visualized by protein gel blotting of cell lysates with a His-tag specific antibody. (B) Detection of intracellular rS3-PA by confocal laser scanning microscopy. SK-BR-3 cells were transduced with 2 µM rS3-PA for 30 min. The peptide aptamer was visualized with an anti-Flag antibody (green) (panel 1), the endoplasmic reticulum (ER) is stained with concanavalin A (red) (panel 2). Uptake of Flag-hTrxΔcys is shown in panel 3, visualized with an anti-Flag antibody (red), and the endosomes were detected using an anti-EEA1 antibody (green) (panel 4). The nuclei were stained with 1 µM TO-PRO-3 (blue). Panels 5 and 10 show higher magnifications of transduced MZ-54 and SK-BR-3 cells to visualize the separate locations of the endosomes (green) and rS3-PA (red).
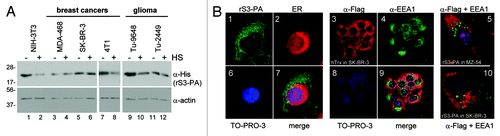
Uptake of rS3-PA into cells was also visualized by immunofluorescence confocal laser scanning microscopy (CLSM). Thirty min after exposure of the cells to rS3-PA, the molecule can be found evenly distributed in the cytoplasm as well as in the nucleus (, panel 1). rS3-PA does not accumulate in dotted structures which would be characteristic for endosomal uptake. To confirm this conclusion, endosomes were visualized with an early endosome-specific antibody (anti-EEA1), and Flag-hTrxΔcys or rS3-PA proteins were detected with an anti-Flag antibody. We observed that the signals for rS3-PA and Flag-hTrx did not co-localize with the signals for the endosomes (, panels 5, 9 and 10). This indicates that the uptake of rS3-PA is independent of the endosomal pathway and favors the accumulation of functional rS3-PA in the cytoplasm.
Addition of rS3-PA to the medium specifically reduces P-STAT3 levels in cancer cells
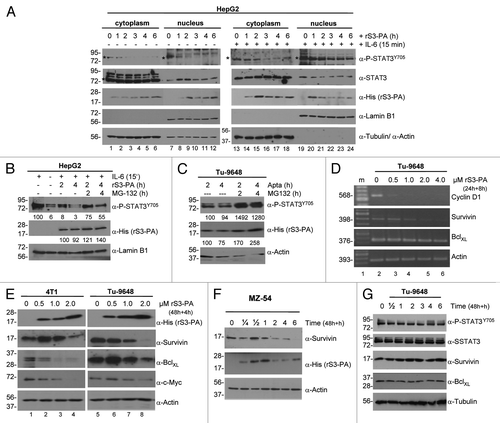
We analyzed the molecular mechanisms by which rS3-PA interferes with the function of STAT3. In serum deprived HepG2 cells, STAT3 is not activated and addition of IL-6 for 15 min caused the accumulation of P-STAT3 in the nucleus (). However, exposure of the cells to rS3-PA for up to 6 h caused a time dependent decrease in P-STAT3 levels. In IL-6 induced cells, rS3-PA treatment reduced P-STAT3 to 32% in the cytoplasm and 44% in the nucleus (). Total STAT3 levels were reduced to 45% when compared with control cells. P-STAT3 levels were also reduced by rS3-PA in Tu-9648 glioma cells (not shown). These cells exhibit a high level of persistently activated STAT3.Citation16 Our results show that the presence of rS3-PA interferes with the phosphorylation of STAT3.
Figure 3. rS3-PA suppresses STAT3 phosphorylation, enhances P-STAT3 degradation and inhibits STAT3 target gene expression. (A) HepG2 cells were transduced with rS3-PA for 0 to 6 h. IL-6 (10 ng/ml) was added to the cells for 15 min (lanes 13 to 24) before the cells were harvested. Cytoplasmic and nuclear fractions were prepared and analyzed in protein gel blots using specific antibodies as indicated. Non-induced cells (lanes 1 to 12) served as controls. (B) Protein gel blot analysis of HepG2 cells treated with rS3-PA for 0, 2 or 4 h and induced with IL-6 for 15 min at the end of these periods. To detect proteasomal degradation of P-STAT3, 50 µM MG-132 was added to the cells in lanes 5 and 6. Nuclear extracts were prepared and P-STAT3, the uptake of rS3-PA and the nuclear marker Lamin B1 were visualized on the blot. Numbers below the blots represent relative band intensities. (C) Same experiment as shown in (B) showing proteasomal degradation of P-STAT3 in Tu-9648 cells after rS3-PA treatment. (D) Tu-9648 cells were treated for 24 h with increasing concentrations (0.5–4 µM) of rS3-PA. After 24 h medium was replaced with fresh rS3-PA and after 8 h total RNA was prepared. Semi-quantitative RT-PCR (20 cycles) products were loaded onto a 2% agarose gel. M: 100 bp size marker (lane 1). (E) Tu-9648 cells and 4T1 cells were treated in 24 h intervals with increasing amounts of rS3-PA. After 48 h fresh rS3-PA was added again for 4 h and cellular protein lysates were prepared for protein gel blot analysis. A His-tag antibody was used to show the uptake of rS3-PA into the cells. Detection of Actin was used as a loading control. (F) MZ-54 cells were treated with 2 µM rS3-PA for 48 h and on the third day for the indicated time points lysates for protein gel blots were prepared. Anti-Actin was used as a control. (G) Tu-9648 cells were treated with 2 µM of the control protein Flag-hTrxΔcys (hTrx) for 48 h and on the third day for the indicated time points. Expression of STAT3, P-STAT3 and of the target genes Survivin and BclXL was analyzed, as well as the uptake of the Flag-hTrxΔcys with a His antibody.
To confirm the specificity of rS3-PA action, we analyzed the effects of rS3-PA on K562 leukemia cells, expressing high levels of P-STAT5. We found that rS3-PA treatment did not affect P-STAT5 and STAT5 levels in these cells (not shown). In addition, the effect of rS3-PA on STAT1 was investigated by inducing MDA-MB-468 cells treated with INFγ. The results showed that P-STAT1 levels, which increase upon INFγ stimulation, are not affected by rS3-PA. A strong and specific effect on P-STAT3 signaling was observed in these cells (not shown).
We observed also that rS3-PA accumulated in the cytoplasm and the nucleus of non-induced HepG2 cells as a function of time. In IL-6 treated cells, rS3-PA accumulated to a maximal level within 1 to 2 h, but then decreased steadily until the end of the observation period at 6 h (). This suggests that the stability of rS3-PA is a function of the presence of P-STAT3 and the presence of P-STAT3 is related to the degradation of rS3-PA. We transduced HepG2 cells and Tu-9648 cells with rS3-PA in the presence of a proteasome inhibitor. MG-132 prevented the downregulation of rS3-PA as well as the degradation of P-STAT3 (). These results suggest that at least three molecular mechanisms are involved in the inhibition of STAT3 function by rS3-PA: it interferes with dimer formation, it inhibits STAT3 phosphorylation and it causes the proteasomal degradation of the P-STAT3-rS3-PA complex.
rS3-PA suppresses STAT3 target gene expression
We analyzed the consequences of STAT3 inhibition by rS3-PA in tumor cell lines and measured the immediate effects on the expression of known STAT3-dependent target genes. Exposure of Tu-9648 glioma cells and 4T1 mammary tumor cells to rS3-PA resulted in a dose-dependent downregulation of cyclin D1 and survivin mRNA within 32 h (). A moderate downregulation was found for BclxL. Exposure of 4T1 and Tu-9648 cells to rS3-PA for 48 h also resulted in decreased levels of the Survivin, BclxL and c-Myc proteins (). Time-dependent treatment of e.g., MZ-54 cells with rS3-PA caused the continuous decrease in Survivin protein (). Treatment of the cells with the control vector protein Flag-hTrxΔcys did not influence the expression of STAT3 target genes ().
rS3-PA inhibits the migration and the angiogenic induction potential of cultured tumor cells
We analyzed the effects of rS3-PA treatment on additional STAT3 regulated target genes which play crucial roles in cell motility.Citation23,Citation24 MZ-54 cells were grown for 2 d in the presence of 1 µM rS3-PA. Then, the cell layer was scratched with a pipette tip and photographed (time point 0) and again 16 h later. We found that control treated MZ-54 cells had largely filled the gap, whereas rS3-PA treated cells had lost their motility (). Similar observations were made with Tu-2449 cells and the results were confirmed in transwell migration assays (not shown).
Figure 4. Suppression of cellular migration and secretion of angiogenic factors by rS3-PA. (A) To study migration of MZ-54 cells, cells were seeded in 6-well plates and grown for two days in the presence of PBS (control), 1 µM Flag-hTrxΔcys (hTrx) or 1 µM rS3-PA. The cell monolayers were scratched with a pipette tip and photographed (0 h, not shown). Mitomycin C (10 µg/ml) was added to prevent further proliferation of the cells. Shown are scratches 16 h after injury of the cell monolayer. (B) By comparing the photographs taken at 0 h and 16 h the cells that had moved into the scratch area were counted. The number of cells present in the scratch after PBS treatment was considered 100%. (C) By measuring the width of the open scratch before and after treatment, the reduction in scratch width was calculated. Scratch width at 0 h was set to 100%. (D) 4T1 cells were grown in DMEM with 2% FCS for 3 d in the presence of 1 µM rS3-PA or Flag-hTrxΔcys (hTrx). On day four, the conditioned DMEM medium was mixed 1:1 with EBM. To assay the presence of angiogenic factors, HUVEC cells at passage 3 were seeded in 12-well plates containing matrigel and 500 µL of the conditioned EBM was added. In positive control experiments, EBM was mixed 1:1 with DMEM/5%FCS and tube formation was considered 100%. Cells were grown overnight at 37°C and the next day, seven representative microscopic fields of all wells were photographed. The total length of the tubes per field (E) and the number of multicentric junctions (F, arrows in D) are displayed in bar graphs. The relative length or amount compared with control treated HUVEC cells was calculated. Error bars in (B, C, E and F) represent SD, *p < 0.05 and **p < 0.01.
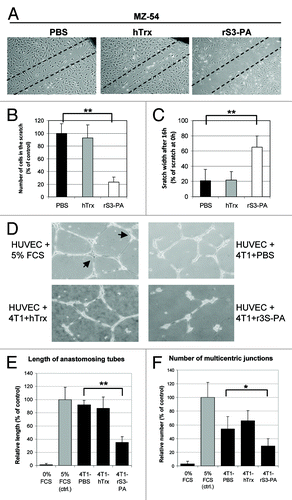
The formation of tubular structures can be induced in cultures of endothelial cells (HUVEC) by exposure to angiogenic factors, present in supernatants collected from tumor cell lines. We found that factors present in the conditioned medium from 4T1, SK-BR-3 and Tu-9648 cells all induced tube formation of HUVEC cells (not shown). Since VEGF transcription is regulated by STAT3,Citation25 we studied the effects of rS3-PA on the secretion of angiogenic factors by 4T1 cells. The cells were grown with or without rS3-PA and after 4 d the growth medium was collected. This conditioned medium was mixed with EBM (1:1) without serum and added to HUVEC cells seeded on matrigel. Tube formation was analyzed after 16 h by measuring tube length and the number of multicentric junctions (). Conditioned medium from rS3-PA treated 4T1 cells had a strongly reduced capacity to induce tube formation (). We conclude that rS3-PA inhibits the secretion of angiogenic factors by these cancer cells.
Effects of rS3-PA on the proliferation and induction of apoptosis of cultured tumor cells
Previous studies have shown that downregulation of STAT3 expression by siRNA can impede the proliferation of tumor cells.Citation13,Citation26-Citation28 We expect that the treatment of tumor cells with 1 µM rS3-PA should result in similar phenotypes. The cellular growth of cells exposed to rS3-PA was analyzed microscopically () and by XTT assays (). Compared with control cells, MZ-54, Tu-9648, Tu-2449, MDA-MB-468 and 4T1 cells are highly sensitive to the inhibition by rS3-PA. SK-BR-3 cells were only modestly affected by 1 µM of rS3-PA, but growth inhibition was more pronounced at higher concentrations of 2–4 µM (not shown). The proliferation of non-tumorigenic NIH-3T3 fibroblasts and MCF-10A cells remained unaffected. In addition, no adverse effects were observed after treatment of primary endothelial and primary mammary epithelial cells (not shown). This excludes a general cytotoxic effect of rS3-PA.
Figure 5. The rS3-PA induced downregulation of P-STAT3, which affects the proliferation and survival of cancer cells, but not of normal cells. (A) Different tumors and normal cells (NIH-3T3 and MCF-10A) were cultured in the presence of 1 µM Flag-hTrxΔcys (hTrx), rS3-PA or an equal volume of PBS. The medium was exchanged daily and the cells were photographed after 96 h of treatment. (B) Cancer cells and normal cells were transduced for 72 h with 1 µM hTrx or rS3-PA. The viability of PBS-treated control cells was considered 100% at 72 h for each cell line. (C) Cells were treated with rS3-PA, hTrx or PBS, for 24 h or 48 h. Cells were lysed and the enrichment of nucleosomes in the cytoplasm was determined in an ELISA-based assay (Cell Death Detection ELISA, Roche). The nucleosome enrichment factor was calculated according to the formula: Treated cells (A405nm – A490nm)/PBS control cells (A405nm – A490nm). The absorbance (A405) of control treated cells (PBS) was set to 1. Error bars in (B and C) represent SD. NS, not significant, *p < 0.05, **p < 0.01.
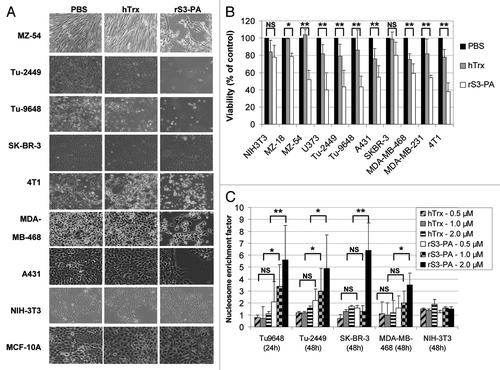
The expression of anti-apoptotic STAT3 target genes is suppressed by rS3-PA treatment (). We analyzed if rS3-PA causes the induction of apoptosis by determining the amount of DNA fragmentation in treated and non-treated cells. The enrichment in cytoplamic nucleosomes is measured in an ELISA based assay using histone antibodies. The data obtained showed that treatment of cells with 2 µM rS3-PA for 24–48 h caused a clear increase in DNA fragmentation in Tu-9648, Tu-2449 and SK-BR-3 cells (5–6-fold) and a moderate increase in MDA-MB-468 cells (3-fold). NIH-3T3 cells were not affected by this treatment (). We conclude, that rS3-PA inhibits proliferation and also induces apoptosis in cancer cells, but not in normal cells.
Systemic application of rS3-PA inhibits STAT3 phosphorylation and reduces the growth of transplanted tumor cells
To investigate the tumor suppressive effects of rS3-PA in animals, 3 × 106 Tu-9648 cells were transplanted into NMRI-Nu/Nu mice and tumor growth was monitored for 15 d. The experiment was performed with seven animals per group in the first experiment and eight animals per group in the second experiment. Mice were treated once a day with PBS, Temozolomide, Flag-hTrxΔcys (7.5 mg/kg) or rS3-PA (7.5 mg/kg). Temozolomide (Temodal, Essex Pharma) is a DNA alkylating agent used in the therapy of glioblastoma patients. The compounds were administered intravenously each day for 15 d. The tumor volumes reached about 2000 mm3 in the PBS treated control animals (). In two independent experiments we observed a reproducible therapeutic effect of rS3-PA causing a reduction in the average tumor volume of about 35% and slightly exceeding the therapeutic effect of temozolomide ().
Figure 6. The rS3-PA induced downregulation of P-STAT3 inhibits tumor cell growth in vivo. (A) Tu-9648 cells (3 × 106/50 µl) were subcutaneously injected into the flanks of nude mice. In experiment 1, each treatment group consisted of seven mice (n = 7), in experiment 2 of eight mice (n = 8). The mice were i.v. injected daily with 150 µl hTrx (7.5 mg/kg), with 150 µl rS3-PA (7.5 mg/kg) or with 150 µl of PBS. Representative tumors and the levels of P-STAT3 and STAT3 in the tumors are shown. Tumors shown in lanes 1, 2, 5, 6, 9 and 10, were obtained 4 h, tumors in lanes 3, 4, 7, 8, 11 and 12 were obtained 15 min after the last treatment. (B) Diagram showing growth of tumors monitored for 15 d. An additional group of animals were treated with 150 µl of temozolomide (60 mg/kg). Error bars represent SEM with *p < 0.05. (C) Kidney and liver extracts were prepared 15 min (PBS mouse #1, hTrx, #2 and rS3PA-#6), or 4 h (rS3-PA mouse #1) after the last injection. Levels of P-STAT3, STAT3 and Tubulin expression, as well as the presence of administered rS3-PA in the tissue extracts are shown. (D) HE stained liver sections of PBS and rS3-PA treated mice.
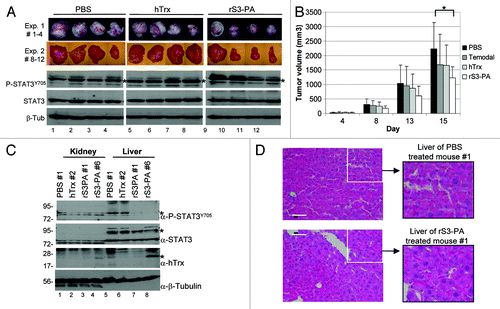
Proteins were extracted from the tumors and the levels of STAT3 and P-STAT3 were determined. Similar to control treated animals, no changes in P-STAT3 levels was observed if tumors were prepared 4 h after the last rS3-PA administration (, lanes 9 and 10). In contrast, a reduction in P-Stat3 is clearly detectable, if tumors were prepared within 15 min (, lanes 11 and 12). This indicates that the suppressive effect of rS3-PA is limited in time and can be observed only relatively shortly after rS3-PA injection. Nevertheless, this transient inhibition of STAT3 seems sufficient to cause an appreciable reduction of tumor growth.
We investigated, if rS3-PA is also taken up in other tissues and exerts an inhibitory effect on activated STAT3. We found that rS3-PA can be only detected in extracts from kidney and liver cells 15 min after the last injection (, lanes 4 and 8). Although we detected very low P-STAT3 levels in kidney cells (, lanes 1 to 4), these cells were not affected by the peptide aptamer. However, STAT3 was found slightly activated in normal liver cells of control animals (, lanes 5 and 6) and treatment of the animals with rS3-PA resulted in a complete suppression of STAT3 activation (, lanes 7 and 8). No peptide aptamer was detected in liver cells when the tissue extracts were prepared 4 h after injection (, lanes 3 and 7). These results again indicate a relatively fast clearance.
We examined sections of liver tissue from control mice and rS3-PA treated mice and did not detect pathological changes (). Also the body weight and the behavior of the mice treated for 15 d with rS3-PA was not different from that of control mice (not shown). We conclude that peptide aptamer rS3-PA is able to inhibit STAT3 activation in tumors as well as in mouse organs upon systemic administration without causing obvious side effects.
Discussion
Persistently activated STAT3 plays a central role in cellular transformation.Citation29,Citation30 It induces target genes which promote tumor cell proliferation, survival and invasion, regulates the communication between tumor cells and normal cells and thus contributes to the immune evasion of tumors.Citation31-Citation33 These functional properties make STAT3 a very promising drug target.Citation11 In particular cases, the therapeutic value of STAT3 inhibition might be restricted by additional genetic lesions in tumor cells. The expression of the p19/p14ARF gene, e.g., determines the tumor enhancing and the tumor suppressing effects of STAT3 expression and activation in Ras transformed liver tumor cells.Citation34 Nevertheless, different classes of compounds are being investigated as STAT3 inhibitors and anti-cancer drugs. Small molecular weight compounds, metal complexes, oligonucleotides, cell permeable peptides and peptidomimetics have been taken into consideration.Citation16,Citation35-Citation40 However, crucial parameters, e.g., cellular permeability, potency, target specificity and systemic stability still have to be optimized to make these molecules clinically applicable.
Here, we describe the characterization and functional analysis of a specific STAT3 inhibitor, rS3-PA. This peptide aptamer is fused to a PTD, a small cationic peptide sequence which interacts with the anionic cell surface through electrostatic forces to mediate intracellular uptake. Mechanistic studies showed that the strength of the peptide-lipid interactions induces either an endocytotic pathway or a mechanism involving membrane disorganization causing a direct translocation.Citation41-Citation44 Our results indicate that rS3-PA enters cells via direct translocation (). This is an important advantage, since rS3-PA is not trapped in the endosomes or lysosomes and is able to directly exert its function in the cytoplasm. It has been suggested that the cargo contributes to the choice between direct translocation and an endocytotic process and the nature of the hTrx scaffold protein could play a role in the direct penetration ability of rS3-PA.Citation45
The sequence of the bait construct used for the selection of the rS3-PA peptide aptamer was initially designed to obtain a molecule able to prevent the formation of functional STAT3 dimers. The peptide aptamer binds and masks the STAT3 dimerization domain. However, additional effects of rS3-PA on STAT3 can be observed. Upon entry into cells, rS3-PA clearly interfered with the IL-6 induced phosphorylation of STAT3 (). It is possible that the rS3-PA-STAT3 complex cannot be recruited to the IL-6 receptor; alternatively the bulky rS3-PA molecule could mask the tyrosine phosphorylation site in such a way that it is not accessible for the kinase. rS3-PA also downregulates STAT3 function through effects on its stability. The exposure of HepG2 cells to rS3-PA causes a decrease in P-STAT3 when compared with untreated cells. This decrease can be compensated by the simultaneous exposure of the cells to the proteasome inhibitor MG-132 () and suggests that rS3-PA-STAT3 complexes are being recognized and destined for degradation.
The downregulation of P-STAT3 by rS3-PA has immediate consequences for transcriptional programs regulated via the STAT3 signaling pathway, which in turn result in phenotypic alterations.Citation46-Citation48 Exposure of the highly motile MZ-54 and Tu-2449 human glioma cells to rS3-PA for 48 h almost entirely inhibited their motility (). STAT3 has been found to bind to β-PIX, a Rac1 activator, and it has been proposed that this is a mechanism by which cytoplasmic STAT3 regulates Rac1 activity and modulates the organization of the actin cytoskeleton and directional migration.Citation24 This process could possibly be affected by the complex formation with rS3-PA.
In addition to a loss of migration potential, we observed a downregulation of various STAT3 target genes (). rS3-PA reduced the proliferation of all STAT3-dependent cancer cell lines and enhanced apoptosis processes () implying that this aptamer triggers the expected biological consequences. These effects were not observed in normal cells, which do not express P-STAT3 and are not dependent on P-STAT3 signaling events, indicating that rS3-PA is not generally cytotoxic. This is in contrast to several other STAT3 inhibitors which have been investigated, e.g., the natural compound cucurbitacin, which also have severe effects on normal cells.Citation49 Our analysis confirmed that rS3-PA acts specifically and affects only the phosphorylation of STAT3, but not that of the closely related family members STAT1 and STAT5.
Interacting peptide domains and peptide aptamers have been established as effective inhibitors of intracellular signaling molecules.Citation16,Citation40,Citation50 However, there are only very few examples in which the direct inhibition of a transcription factor complex by an externally supplied interacting peptide have been studied in vivo. A recent report describes the delimitation of a binding domain from the Notch1 interaction partner Maml1. The intracellular delivery of this Maml1 peptide, constrained in a stable helical conformation, inhibits the Notch transcription factor complex in T-cell acute lymphoblastic leukemia cells and represses Notch-mediated gene expression. The treatment of mice with this peptide retarded leukemic tumor cell growth.Citation17 The molecule depends on a “peptide stapling system” that stabilizes two turns in an α-helical peptide region and provides a new class of inhibitory compounds. The small stapled peptide can be produced in cell permeable forms, have a high binding affinities for their target structures and exhibit favorable stabilities and bioactivities.Citation51
We report that i.v. administration of rS3-PA slows the growth of transplanted tumor cells in mice up to 35%, a value exceeding the therapeutic effect of temozolomide in this system. Despite the therapeutic effects observed, our experiments suggest that rS3-PA is limited in its systemic stability and that the effects on STAT3 inhibition are temporary. The inhibition of STAT3 declines a few hours after rS3-PA administration. This conclusion corroborates earlier studies in which investigators administered TAT-β-galactosidase protein to mice. A quantitative analysis showed that the enzyme activity was rapidly detectable in all tissues examined, but that the activity peaked within the first hour after administration.Citation52 For this reason we performed in vivo experiments in which more frequent injections and higher doses were applied. When rS3-PA was injected 3 times per day at a dose of 20 mg/kg, the therapeutic efficacy further improved and tumor growth was reduced by 65% when compared with PBS treated controls (not shown).
The short time of action and the resulting limited therapeutic efficacy of rS3-PA are probably caused by its uptake into the large majority of normal cells, since PTD mediated uptake is not accompanied by a preference for a particular cell type. This is most likely the reason that the levels of activated P-STAT3 in the liver were strongly reduced shortly after the application of rS3-PA. The reduction is more pronounced in the liver than in the tumor cells, most likely due to differences in vascularization and cellular accessibility. The inhibition of STAT3 activation also seems to last longer in the liver tissue when compared with the tumors. The properties of rS3-PA can possibly be further improved, e.g., by the provision with a tumor cell targeting domain to selectively direct the peptide aptamer to tumor cells. In addition, measures to enhance protein stability, e.g., by PEGylation and by further manipulations of the scaffold structure could become favorable. Also, the choice of suitable indications, e.g., the topical application of rS3-PA in the treatment of chronic inflammatory skin diseases and skin cancer appears promising.Citation53,Citation54 These applications enhance the concentration of the peptides at the tumor or diseased site and lower random tissue distribution and rapid clearance. The therapeutic effects of rS3-PA can probably be enhanced by combination treatment with cytotoxic agents. Promising results have been obtained, e.g., for the growth inhibition of lung cancer cells,Citation55 but further technology will have to be developed to improve bioavailability, stability and specific targeting. Finally, specifically interacting peptide sequences can be used as valuable tools for the derivation of small molecular weight compounds with similar functional properties.Citation35,Citation56
Acknowledgments
We like to thank Nancy Hynes (Friedrich Miescher Insitute, Basel, Switzerland), Cord Hartman (Georg-Speyer-Haus, Institute for Biomedical Research, Frankfurt, Germany), Donat Kögel and Jakob Weissenberger (Neuroscience Center, Goethe University, Frankfurt, Germany) for providing tumor cell lines and helpful discussions. The help of Ralf Brandes (Institute for Cardiovascular Physiology, Goethe University, Frankfurt, Germany) with confocal laser scanning microscopy is gratefully acknowledged. This work was supported by the Deutsche Forschungsgemeinschaft (GR 536/11–1 and GR 536/12–1) and the LOEWE program “Oncogenic signal transduction” (OSF, Frankfurt) of the state of Hessen.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Salk JJ, Fox EJ, Loeb LA. Mutational heterogeneity in human cancers: origin and consequences. Annu Rev Pathol 2010; 5:51 - 75; http://dx.doi.org/10.1146/annurev-pathol-121808-102113; PMID: 19743960
- van't Veer LJ, Bernards R. Enabling personalized cancer medicine through analysis of gene-expression patterns. Nature 2008; 452:564 - 70; http://dx.doi.org/10.1038/nature06915; PMID: 18385730
- Cerami E, Demir E, Schultz N, Taylor BS, Sander C. Automated network analysis identifies core pathways in glioblastoma. PLoS ONE 2010; 5:e8918; http://dx.doi.org/10.1371/journal.pone.0008918; PMID: 20169195
- Sharma SV, Settleman J. Exploiting the balance between life and death: Targeted cancer therapy and “oncogenic shock”. Biochem Pharmacol 2010; http://dx.doi.org/10.1016/j.bcp.2010.03.001; PMID: 20211150
- Felsher DW. Oncogene addiction versus oncogene amnesia: perhaps more than just a bad habit?. Cancer Res 2008; 68:3081 - 6, discussion 6; http://dx.doi.org/10.1158/0008-5472.CAN-07-5832; PMID: 18451131
- Groner B, Lucks P, Borghouts C. The function of Stat3 in tumor cells and their microenvironment. Semin Cell Dev Biol 2008; 19:341 - 50; http://dx.doi.org/10.1016/j.semcdb.2008.06.005; PMID: 18621135
- Lee H, Deng J, Kujawski M, Yang C, Liu Y, Herrmann A, et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med 2010; 16:1421 - 8; http://dx.doi.org/10.1038/nm.2250; PMID: 21102457
- Pilati C, Amessou M, Bihl MP, Balabaud C, Van Nhieu JT, Paradis V, et al. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J Exp Med 2011; 208:1359 - 66; http://dx.doi.org/10.1084/jem.20110283; PMID: 21690253
- Al Zaid Siddiquee K, Turkson J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res 2008; 18:254 - 67; http://dx.doi.org/10.1038/cr.2008.18; PMID: 18227858
- Oh YM, Kim JK, Choi Y, Choi S, Yoo JY. Prediction and experimental validation of novel STAT3 target genes in human cancer cells. PLoS ONE 2009; 4:e6911; http://dx.doi.org/10.1371/journal.pone.0006911; PMID: 19730699
- Aggarwal BB, Vijayalekshmi RV, Sung B. Targeting inflammatory pathways for prevention and therapy of cancer: short-term friend, long-term foe. Clin Cancer Res 2009; 15:425 - 30; http://dx.doi.org/10.1158/1078-0432.CCR-08-0149; PMID: 19147746
- Ecker A, Simma O, Hoelbl A, Kenner L, Beug H, Moriggl R, et al. The dark and the bright side of Stat3: proto-oncogene and tumor-suppressor. Front Biosci 2009; 14:2944 - 58; http://dx.doi.org/10.2741/3425; PMID: 19273247
- Sherry MM, Reeves A, Wu JK, Cochran BH. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 2009; 27:2383 - 92; http://dx.doi.org/10.1002/stem.185; PMID: 19658181
- Borghouts C, Kunz C, Groner B. Current strategies for the development of peptide-based anti-cancer therapeutics. J Pept Sci 2005; 11:713 - 26; http://dx.doi.org/10.1002/psc.717; PMID: 16138387
- Borghouts C, Kunz C, Groner B. Peptide aptamers: recent developments for cancer therapy. Expert Opin Biol Ther 2005; 5:783 - 97; http://dx.doi.org/10.1517/14712598.5.6.783; PMID: 15952909
- Borghouts C, Tittmann H, Delis N, Kirchenbauer M, Brill B, Groner B. The intracellular delivery of a recombinant peptide derived from the acidic domain of PIAS3 inhibits STAT3 transactivation and induces tumor cell death. Mol Cancer Res 2010; 8:539 - 53; http://dx.doi.org/10.1158/1541-7786.MCR-09-0417; PMID: 20371673
- Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC, et al. Direct inhibition of the NOTCH transcription factor complex. Nature 2009; 462:182 - 8; http://dx.doi.org/10.1038/nature08543; PMID: 19907488
- Borghouts C, Kunz C, Groner B. Peptide aptamer libraries. Comb Chem High Throughput Screen 2008; 11:135 - 45; http://dx.doi.org/10.2174/138620708783744462; PMID: 18336207
- Bickle MB, Dusserre E, Moncorge O, Bottin H, Colas P. Selection and characterization of large collections of peptide aptamers through optimized yeast two-hybrid procedures. Nat Protoc 2006; 1:1066 - 91; http://dx.doi.org/10.1038/nprot.2006.32; PMID: 17406388
- Borghouts C, Kunz C, Delis N, Groner B. Monomeric recombinant peptide aptamers are required for efficient intracellular uptake and target inhibition. Mol Cancer Res 2008; 6:267 - 81; http://dx.doi.org/10.1158/1541-7786.MCR-07-0245; PMID: 18314488
- Nagel-Wolfrum K, Buerger C, Wittig I, Butz K, Hoppe-Seyler F, Groner B. The interaction of specific peptide aptamers with the DNA binding domain and the dimerization domain of the transcription factor Stat3 inhibits transactivation and induces apoptosis in tumor cells. Mol Cancer Res 2004; 2:170 - 82; PMID: 15037656
- Gump JM, June RK, Dowdy SF. Revised role of glycosaminoglycans in TAT protein transduction domain-mediated cellular transduction. J Biol Chem 2010; 285:1500 - 7; http://dx.doi.org/10.1074/jbc.M109.021964; PMID: 19858185
- Arulanandam R, Geletu M, Feracci H, Raptis L. Activated Rac1 requires gp130 for Stat3 activation, cell proliferation and migration. Exp Cell Res 2010; 316:875 - 86; http://dx.doi.org/10.1016/j.yexcr.2009.10.017; PMID: 19852956
- Teng TS, Lin B, Manser E, Ng DC, Cao X. Stat3 promotes directional cell migration by regulating Rac1 activity via its activator betaPIX. J Cell Sci 2009; 122:4150 - 9; http://dx.doi.org/10.1242/jcs.057109; PMID: 19861492
- Loeffler S, Fayard B, Weis J, Weissenberger J. Interleukin-6 induces transcriptional activation of vascular endothelial growth factor (VEGF) in astrocytes in vivo and regulates VEGF promoter activity in glioblastoma cells via direct interaction between STAT3 and Sp1. Int J Cancer 2005; 115:202 - 13; http://dx.doi.org/10.1002/ijc.20871; PMID: 15688401
- Gao L, Zhang L, Hu J, Li F, Shao Y, Zhao D, et al. Down-regulation of signal transducer and activator of transcription 3 expression using vector-based small interfering RNAs suppresses growth of human prostate tumor in vivo. Clin Cancer Res 2005; 11:6333 - 41; http://dx.doi.org/10.1158/1078-0432.CCR-05-0148; PMID: 16144938
- Gritsko T, Williams A, Turkson J, Kaneko S, Bowman T, Huang M, et al. Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin Cancer Res 2006; 12:11 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-04-1752; PMID: 16397018
- Mora LB, Buettner R, Seigne J, Diaz J, Ahmad N, Garcia R, et al. Constitutive activation of Stat3 in human prostate tumors and cell lines: direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res 2002; 62:6659 - 66; PMID: 12438264
- Schindler C, Plumlee C. Inteferons pen the JAK-STAT pathway. Semin Cell Dev Biol 2008; 19:311 - 8; http://dx.doi.org/10.1016/j.semcdb.2008.08.010; PMID: 18765289
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 2009; 9:798 - 809; http://dx.doi.org/10.1038/nrc2734; PMID: 19851315
- Kortylewski M, Xin H, Kujawski M, Lee H, Liu Y, Harris T, et al. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 2009; 15:114 - 23; http://dx.doi.org/10.1016/j.ccr.2008.12.018; PMID: 19185846
- Stewart CA, Trinchieri G. Reinforcing suppression using regulators: a new link between STAT3, IL-23, and Tregs in tumor immunosuppression. Cancer Cell 2009; 15:81 - 3; http://dx.doi.org/10.1016/j.ccr.2009.01.008; PMID: 19185840
- Aggarwal BB, Kunnumakkara AB, Harikumar KB, Gupta SR, Tharakan ST, Koca C, et al. Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship?. Ann N Y Acad Sci 2009; 1171:59 - 76; http://dx.doi.org/10.1111/j.1749-6632.2009.04911.x; PMID: 19723038
- Schneller D, Machat G, Sousek A, Proell V, van Zijl F, Zulehner G, et al. p19(ARF) /p14(ARF) controls oncogenic functions of signal transducer and activator of transcription 3 in hepatocellular carcinoma. Hepatology 2011; 54:164 - 72; http://dx.doi.org/10.1002/hep.24329; PMID: 21452288
- Uehara Y, Mochizuki M, Matsuno K, Haino T, Asai A. Novel high-throughput screening system for identifying STAT3-SH2 antagonists. Biochem Biophys Res Commun 2009; 380:627 - 31; http://dx.doi.org/10.1016/j.bbrc.2009.01.137; PMID: 19285012
- Nelson EA, Sharma SV, Settleman J, Frank DA. A chemical biology approach to developing STAT inhibitors: molecular strategies for accelerating clinical translation. Oncotarget 2011; 2:518 - 24; PMID: 21680956
- Costantino L, Barlocco D. STAT 3 as a target for cancer drug discovery. Curr Med Chem 2008; 15:834 - 43; http://dx.doi.org/10.2174/092986708783955464; PMID: 18473793
- Fletcher S, Turkson J, Gunning PT. Molecular approaches towards the inhibition of the signal transducer and activator of transcription 3 (Stat3) protein. ChemMedChem 2008; 3:1159 - 68; http://dx.doi.org/10.1002/cmdc.200800123; PMID: 18683176
- Yue P, Turkson J. Targeting STAT3 in cancer: how successful are we?. Expert Opin Investig Drugs 2009; 18:45 - 56; http://dx.doi.org/10.1517/13543780802565791; PMID: 19053881
- Zhao W, Jaganathan S, Turkson J. A cell-permeable Stat3 SH2 domain mimetic inhibits Stat3 activation and induces antitumor cell effects in vitro. J Biol Chem 2010; 285:35855 - 65; http://dx.doi.org/10.1074/jbc.M110.154088; PMID: 20807764
- Deshayes S, Morris M, Heitz F, Divita G. Delivery of proteins and nucleic acids using a non-covalent peptide-based strategy. Adv Drug Deliv Rev 2008; 60:537 - 47; http://dx.doi.org/10.1016/j.addr.2007.09.005; PMID: 18037526
- Morris MC, Deshayes S, Heitz F, Divita G. Cell-penetrating peptides: from molecular mechanisms to therapeutics. Biol Cell 2008; 100:201 - 17; http://dx.doi.org/10.1042/BC20070116; PMID: 18341479
- Eiri´ksdo´ttir E, Konate K, Langel U, Divita G, Deshayes S. Secondary structure of cell-penetrating peptides controls membrane interaction and insertion. Biochim Biophys Acta 2010; 1798:1119 - 28; http://dx.doi.org/10.1016/j.bbamem.2010.03.005; PMID: 20214875
- Meade BR, Dowdy SF. Enhancing the cellular uptake of siRNA duplexes following noncovalent packaging with protein transduction domain peptides. Adv Drug Deliv Rev 2008; 60:530 - 6; http://dx.doi.org/10.1016/j.addr.2007.10.004; PMID: 18155315
- Go YM, Jones DP. Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta 2008; 1780:1273 - 90; http://dx.doi.org/10.1016/j.bbagen.2008.01.011; PMID: 18267127
- Aoki Y, Feldman GM, Tosato G. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood 2003; 101:1535 - 42; http://dx.doi.org/10.1182/blood-2002-07-2130; PMID: 12393476
- Masuda M, Suzui M, Yasumatu R, Nakashima T, Kuratomi Y, Azuma K, et al. Constitutive activation of signal transducers and activators of transcription 3 correlates with cyclin D1 overexpression and may provide a novel prognostic marker in head and neck squamous cell carcinoma. Cancer Res 2002; 62:3351 - 5; PMID: 12067972
- Zushi S, Shinomura Y, Kiyohara T, Miyazaki Y, Kondo S, Sugimachi M, et al. STAT3 mediates the survival signal in oncogenic ras-transfected intestinal epithelial cells. Int J Cancer 1998; 78:326 - 30; http://dx.doi.org/10.1002/(SICI)1097-0215(19981029)78:3<326::AID-IJC12>3.0.CO;2-4; PMID: 9766567
- Dantas IN, Gadelha GC, Chaves DC, Monte FJ, Pessoa C, de Moraes MO, et al. Studies on the cytotoxicity of cucurbitacins isolated from Cayaponia racemosa (Cucurbitaceae). Z Naturforsch C 2006; 61:643 - 6; PMID: 17137107
- Bouquier N, Fromont S, Zeeh JC, Auziol C, Larrousse P, Robert B, et al. Aptamer-derived peptides as potent inhibitors of the oncogenic RhoGEF Tgat. Chem Biol 2009; 16:391 - 400; http://dx.doi.org/10.1016/j.chembiol.2009.02.006; PMID: 19389625
- Verdine GL, Walensky LD. The challenge of drugging undruggable targets in cancer: lessons learned from targeting BCL-2 family members. Clin Cancer Res 2007; 13:7264 - 70; http://dx.doi.org/10.1158/1078-0432.CCR-07-2184; PMID: 18094406
- Cai SR, Xu G, Becker-Hapak M, Ma M, Dowdy SF, McLeod HL. The kinetics and tissue distribution of protein transduction in mice. Eur J Pharm Sci 2006; 27:311 - 9; http://dx.doi.org/10.1016/j.ejps.2005.10.011; PMID: 16376528
- Kong LY, Gelbard A, Wei J, Reina-Ortiz C, Wang Y, Yang EC, et al. Inhibition of p-STAT3 enhances IFN-alpha efficacy against metastatic melanoma in a murine model. Clin Cancer Res 2010; 16:2550 - 61; http://dx.doi.org/10.1158/1078-0432.CCR-10-0279; PMID: 20388845
- R´cz E, Prens EP. Molecular pathophysiology of psoriasis and molecular targets of antipsoriatic therapy. Expert Rev Mol Med 2009; 11:e38; http://dx.doi.org/10.1017/S146239940900129X; PMID: 20003607
- Kluge A, Dabir S, Kern J, Nethery D, Halmos B, Ma P, et al. Cooperative interaction between protein inhibitor of activated signal transducer and activator of transcription-3 with epidermal growth factor receptor blockade in lung cancer. Int J Cancer 2009; 125:1728 - 34; http://dx.doi.org/10.1002/ijc.24553; PMID: 19569236
- Oikawa T, Unno Y, Matsuno K, Sawada J, Ogo N, Tanaka K, et al. Identification of a small-molecule inhibitor of the interaction between Survivin and Smac/DIABLO. Biochem Biophys Res Commun 2010; 393:253 - 8; http://dx.doi.org/10.1016/j.bbrc.2010.01.113; PMID: 20117083