Abstract
Cancer growth is controlled by cancer cells (cell intrinsic phenomenon), but also by the immune cells in the tumor microenvironment (cell extrinsic phenomenon). Thus cancer progression is mediated by the activation of transcription programs responsible for cancer cell proliferation, but also induced proliferation/activation of immunosuppressive cells such as Th17, Treg or myeloid derived suppressor cells (MDSCs). One of the key transcription factors involved in these pathways is the signal transducer and activator of transcription 3 (STAT3). In this review we will focus on STAT3 activation in immune cells, and how it impacts on tumor progression.
Introduction
STAT3 belongs to the signal transducer and activator of transcription (STAT) family of signal responsive transcription factors which consists of seven members encoded by distinct genes. In non-stimulated cells, STAT3, like other STATs proteins, is kept in an inactive cytoplasmic form. Then, once activated, STAT3 translocates into the nucleus where it behaves as a transcription activator for a broad array of targeted genes. Typically, STAT3 activation is induced by phosphorylation on a critical tyrosine residue (Tyr 705) that triggers STAT3 dimerization thanks to reciprocal phosphotyrosine-SH2 domain interactions.Citation1 Even if multiple tyrosine kinases have been described as intracellular activators of STAT3 activity (such as EGFR, Src, ERK), the phosphorylation of STAT3 on tyrosine 705 is mainly regulated by members of Janus-activated kinases with JAK1 as key modulator.Citation2 In addition to tyrosine 705 phosphorylation, STAT3 is also activated through serine (Ser 727) phosphorylation. This phosphorylation is commonly regulated by protein kinase C, mitogen-activated protein kinases, and CDK5. Finally, the reversible acetylation of STAT3 by histone acetyltransferase on a single lysine residue (Lys 685) represents a third mechanism of STAT3 activation.Citation3 Acetylated STAT3 has been depicted as a way to enhance stability of STAT3 dimers which are required for DNA-binding and transcriptional activity.
Regulation of STATs protein activation is dictated by negative regulators such as PIAS (protein inhibitor of activated STAT) and SOCS (suppressors of cytokine signaling) proteins as well as protein tyrosine phosphatases. SOCS proteins prevent the JAK from activating STAT3 by direct and indirect interactions with tyrosine kinase SH2 domains.Citation4 They also drive the blockade of STAT3 to bind to receptor subunits. To date, SOCS protein family comprises eight members and SOCS3 seems to be the most specific inhibitor of STAT3 signaling pathway. Contrary to SOCS molecules, PIAS proteins are nuclear factors. They negatively regulate STAT transcriptional activity through several mechanisms, particularly by interacting and thus blocking DNA binding activity.Citation5 Protein tyrosine phosphatases remove phosphates from activated STATs which represent a third level of STAT modulation. Numerous phosphatases have been described to regulate STAT3, such as PTEN,Citation6 CD45,Citation7 SHP-1 and SHP-2.Citation8,Citation9 Moreover, kinases such as phosphatidylinositol 3-kinase (PI3K) could regulate STAT3 phosphorylation in a different way, depending on the model used. STAT3 pathway is inhibited by PI3K in melanoma cells, whereas it is activated in laryngeal papillomas or in breast cancer stem-like cells with an emphasis in PTEN deficient cells.Citation6,Citation10,Citation11 Lastly, STAT3 has also been shown to go through ubiquitination-dependent proteosomal degradation.Citation12
It is now well established that STAT3 signaling is a major intrinsic pathway driving apoptosis, inflammation, cellular transformation, survival, proliferation, invasion, angiogenesis and metastasis of cancer.Citation13,Citation14 However, compelling evidence has now showed that STAT3 is constitutively activated in many human cancers.Citation15 Indeed, many receptor signaling pathways are excessively stimulated in tumoral context and lead to persistent activation of STAT3. Many regulated genes induced by STAT3 in turn activate the same STAT3 pathways and keep a stable feedforward loop going between tumor cells and tumor-interacting immune cells. In addition, the own tumorigenic properties of STAT3 highlight its oncoprotein status by driving malignant properties related to chronic inflammation.Citation16,Citation17 Thereby, this explains that STAT3 has been characterized as a central actor for inflammation-induced cancer. STAT3 activation occurs in both cancer and stromal cells thereby allowing a crosstalk between these two cellular types. This activation is rapid and transient under normal biological conditions and mediated by a large number of extracellular stimuli including cytokines (IL-6, IL-10, IFNs, TNF-α…) and growth factors (i.e., EGF, G-CSF, GM-CSF, VEGF and Her2/Neu). Active oncogenic proteins, such as Src and Ras, as well as chemical carcinogens and other molecules also have the ability to activate STAT3 (see ref. Citation18 for extensive review).
It is now well established that carcinogenesis induces the appearance of danger signals which could drive tumor rejection by the immune system (a phenomenon called immunosurveillance). Some cancer cells could escape this rejection by limiting tumor antigen expression (a phenomenon called immunoediting) or by inducing active immune tolerance mechanisms.Citation19 These mechanisms include the proliferation and local accumulation of immunosuppressive cells, including regulatory T cells (Tregs), Th17 cells and myeloid-derived immunosuppressive cells (MDSCs). This tolerance (a phenomenon called immunoescape) prevents cancer rejection by the immune system and blunts the efficacy of immunotherapy as demonstrated in several mouse models.Citation20
This review is dedicated to the current knowledge of the impact of STAT3 activation in immune cells (summarized in ) on the balance between immunosurveillance and immunoescape.
Table 1. Main STAT3 modulators and their effects in immune cells
STAT3 and T Cells
T lymphocytes or T cells play a central role in the adaptative immune response of the host to cancer.Citation21 Tumor-infiltrating CD4 and CD8 T cells are associated with clinical outcome and survival in colorectal cancer,Citation22 breastCitation23 and lung cancers.Citation24 CD4 T cells called helper T cells are capital in the generation of T cell immune response by their capacity to drive and coordinate all components of the immune response. Current cancer immunotherapies are therefore designed to induce or enhance T cell reactivity against tumor antigens.Citation25 Among T cells, different subsets have been described (regulatory T cells, cytotoxic T cells and T helper) with distinct functions that could be regulated by STAT3.
Th1/Th2
T helper (Th) cells assist other hematopoietic cells in immunologic processes, including activation of CTL, natural killer (NK) cells and macrophages. Th cells become activated when they are stimulated with antigens presented by MHC class II molecules that are expressed on the surface of antigen presenting cells (APCs) such as dendritic cells. Once activated, they proliferate rapidly and secrete small proteins called cytokines that regulate or assist the active immune response. These cells can differentiate into one of several subtypes (e.g., Th1, Th2 and Th17), which secrete cytokines to regulate immune response.Citation26
The expression levels of Th1 cell genes coding for Interferon-γ (IFNG), TAP1 and Granzyme B (GZMB) are significantly higher in colorectal tumors than in normal tissue. This high expression of Th1 cytotoxic genes was associated with significantly improved disease-free survival rates and patients with low expression of those genes had earlier recurrence.Citation27 Tbet, the master transcriptional regulator for Th1 differentiation is induced by T cell receptors (TCRs) and IL-12 stimulation.Citation28 In contrast, GATA3 is the master control for Th2 differentiation after stimulation with IL-4.Citation29 IL-27, a member of the IL-6/IL-12 family produced by macrophages and dendritic cells, favors the Th1/Th2 differentiation balance toward Th1 by upregulating Tbet (independently of STAT1), downregulating GATA3 (through a STAT1-dependent mechanism) and suppressing proinflammatory cytokine production such as IL-2, IL-4, and IL-13.Citation30 Even if IL-27 activates STAT3, all those events were STAT3-independent. In this context, only the IL-27 dependent CD4+ T cell proliferation was mediated by STAT3 (with an upregulation of c-myc and pim-1 transcription) and not by STAT1.Citation30,Citation31
Th17
Th17 cells are CD4+ T cells induced by TCR triggering with IL-6 and transforming growth factor (TGF)-β stimulation. After induction, IL-23, an IL-12 family member, maintains Th17 cell polarization.Citation32 Th17 cells have emerged as key driver of a wide range of autoimmune disorders, including inflammatory bowel disease, psoriasis and ankylosing spondylitis.Citation33 Th17 cell expansion was observed in human cancers such as ovarian, melanoma, breast or colon cancers.Citation34 In colorectal cancer, patients with low expression of Th17 genes seemed to have a prolonged disease-free survival.Citation27 Despite these early observations, the role of Th17 cells in cancer immunity remains controversial.
Th17 are characterized by the expression of the transcription factors RORγt and RORα.Citation35 In addition, STAT3 is also indispensable for Th17 cell differentiation, because STAT3 ablation in CD4 cells in mice results in an absence of Th17 differentiation.Citation36 Moreover, many in vitro studies have shown that STAT3 can be activated downstream of receptors for several pro-inflammatory cytokines including IL-6, IL-21 and IL-23 leading to the regulation of RORγt, RORα, IL-21, IL-23R and IL-17 expression along with the development and the stabilization of Th17 cells.Citation37-Citation41 In the absence of STAT3, other signaling pathways are engaged, such as STAT1 pathway, leading to the induction of the Th1 cytokine IFNγ.Citation38
Our team found that in vitro Th17 cells generated with IL-6 and TGF-β and in vivo tumor-infiltrating Th17 cells express CD39 and CD73 ectonucleotidases. This ectonucleotidase catalytic machinery leads to the cleavage of extracellular ATP into the immunosuppressive molecule adenosine which could suppress effector T cells. The expression of ectonucleotidases is dependent on STAT3 and the zinc finger protein growth factor independent-1 (Gfi-1), which is a transcription repressor. IL-6-driven STAT3 binds to the promoter regions of CD39 and CD73 and TGF-β-mediated downregulation of Gfi-1 removes its transcriptional inhibition during Th17 cell differentiation, both leading to the transcription of ectonucleotidases. CD39 expression on Th17 cells promotes tumor growth, suggesting that expression of ectonucleotidases dictates the immunosuppressive fate of Th17 cells in cancer.Citation42 The generation of Th17 cells from naive T cells activated with IL-1β, IL-6 and IL-23 in the absence of TGF-β has been reported. Unlike Th17 cells generated with TGF-β and IL-6, these Th17 cells generated without TGF-β were highly pathogenic in vivoCitation43 and failed to express ectonucleotidases.Citation42 These observations suggest that STAT3 and Gfi-1 determine the effector immunoregulatory fate of Th17 cells through regulation of ectonucleotidase expression.Citation42
Treg
Regulatory T cells (Treg) are suppressor T cells that maintain peripheral immune tolerance.Citation44,Citation45 Conversion of naive CD4 T cells in CD4+/CD25+ Treg cells can be achieved through co-stimulation with TCRs and TGF-β. Such stimulation induce the expression of FOXP3 the master transcription factor for Tregs.Citation46 These T cells accumulate in tumors and in the peripheral blood of patients with cancerCitation47 and the increased Treg frequency has been generally considered as a marker of poor prognosis in cancer presumably because of Treg-mediated suppression of anti-tumor immunity, which benefits the tumor.Citation48-Citation50
The role of STAT3 in regulating Foxp3 expression by Tregs appears to be context-dependent. In vitro, IL-2 induces the binding of STAT3 and STAT5 to a highly conserved STAT-binding site located within the first intron of the Foxp3 gene leading to the upregulation of FOXP3 expression in purified CD4+CD25+ T cells but not in CD4+CD25− cells.Citation51
In tumor-infiltrating Tregs both STAT3 and STAT5 can bind to a STAT consensus site in the Foxp3 promoter and enhances FOXP3 expression which seems to be important in maintaining Tregs' inhibitory functions.Citation51-Citation53 Low-dose IL-2 treatment of patients with metastatic cancer or chronic myelogenous leukemia after allogeneic hematopoietic stem cell transplantation resulted in an increase in the frequency of CD4+CD25+ cells in peripheral blood as well as FOXP3 expression in CD3+ T cells.Citation51 In Treg cells, activated STAT3 and FOXP3 interact together and co-operatively regulate IL6 and TGFβ1 genes, which likely endow Treg cells with the ability to suppress Th17 cell-mediated inflammation and fatal colitis.Citation54 In addition, IL-21, which activates STAT3, does not activate STAT5 and has no effect on Treg viability, activation or function.Citation55 On the contrary, in naive T cells induced to differentiate into Tregs in vitro, IL-6 or IL-27 inhibit the differentiation to the Treg lineage in a STAT3-dependent and STAT1-independent manner.Citation40,Citation56 In another context, STAT3 binds to a silencer element within the Foxp3 locusCitation57 and could also inhibits STAT5 binding to its binding element in Foxp3 promoter,Citation58 both inhibiting FOXP3 expression. Moreover, the modulation of STAT3 activity in Tregs by molecular compounds could lead to an inhibition of their activity. Thus, WP1066 (an inhibitor of STAT3 signaling) enhances T cell cytotoxicity against melanoma through inhibition of FOXP3+ Tregs, suggesting that STAT3 is required for immunosuppressive functions of Tregs.Citation59
STAT3 and Myeloid Cells
APCs lie at the center of this critical decision of the immune system, since these cells have been shown to capture antigens in the periphery, migrate to the lymphoid organs and present processed peptides to T cells in a way that may lead to either priming tolerance induction.Citation60
Macrophages
Macrophages can differentiate into two subsets, M1 and M2 macrophages, based on their capacity to produce IL-12 or IL-10, respectively. M1 has potent microbicidal properties and promotes Th1 responses, whereas M2 supports Th2-associated effector functions.Citation61,Citation62 M2 macrophages are further subdivided into several subtypes including tumor-associated macrophages (TAMs). TAMs accumulate at the tumor site by tumor-derived signals, such as macrophage colony-stimulating factor (M-CSF/CSF-1) and monocyte chemoattractant protein-1 (MCP-1) or chemokine (C-C motif) ligand-2 (CCL2). They actively contribute to tumor growth by releasing proangiogenic cytokines and growth factors, such as vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), colony stimulating factor-1 (CSF-1), platelet-derived growth factor (PDGF) and basic fibroblast growth factor (βFGF). They also produce arginase-1, IL-10 and TGF-β, which inhibit the antitumor function of T cells and natural killer cells leading to tumor tolerance and the impairment of antitumor immunotherapies efficacy.Citation63-Citation65
IL-6 inhibition of macrophage colony-stimulating factor (M-CSF)-induced colony formation observed in mice was abolished in mice mutated for the gp130-STAT1/3 signaling, suggesting that the IL-6/STAT3 pathway could regulate macrophage homeostasis.Citation66
Cheng et al. were the first to show that disruption of STAT3 signaling in either macrophages or bone marrow-derived dendritic cells (DCs) renders these APCs capable of restoring the responsiveness of tolerant T cells from tumor-bearing mice suggesting an important implication for cancer immunotherapy. The finding that peritoneal elicited macrophages with a targeted disruption of STAT3 have a constitutively activated phenotype and are more prone to produce inflammatory mediators in response to LPS (such as RANTES, MIP-1α, MIP-1β, MIP-2, IP-10, IL-6 or IL-12) points to STAT3 signaling as a negative regulatory pathway in these cells. This could be due to an increased STAT1 activity (leading to high production of inflammatory factors e.g., RANTES or IL-12) or a lack of IL-10 production.Citation67 The same team showed that macrophages derived from conditional STAT3 knockout mice are superior to wild-type macrophages in terms of their ability to prime cognate CTL responses, and to cross-present tumor-derived antigen to CTLs in vitro leading to a stronger proliferation of CTLs and an increased production of IFN-γ and tumor necrosis factor (TNF)-α. In the same way, ablating STAT3 in hematopoietic cells results in rapid activation of innate immunity by CpG (a TLR9 ligand), with enhanced production of IFN-γ, TNF-α and IL-12, and activation of macrophages, neutrophils and natural killer cells (which present strong STAT1 activation) and with an eradication of B16 melanoma tumors.Citation68 Targeting STAT3 signaling represents therefore an enticing strategy to augment CTL responses in the tumor-bearing host.Citation69 Immunosuppressive activities of TAMs correlate with over-activated STAT3 signaling of the cells and disruption of STAT3 activity of TAMs can enhance rat immune response to breast cancer.Citation70 In glioblastoma, tumor-infiltrating macrophages were shown to be predominantly STAT3-positive M2 macrophages which is associated with a poor prognostic for those patients.Citation71 The same team proposed corosolic acid and oleanic acid as new compounds for tumor prevention and therapy through their capacity to suppress M2 polarization of macrophages and tumor cell proliferation by inhibiting STAT3 activation and IL-10 production.Citation72,Citation73 In intrahepatic cholangiocarcinoma (ICC), patients with high counts of CD163+ M2 macrophages showed poor disease-free survival. Tumor cell supernatant from ICC cell lines (such as HuCCT1) induced the production of IL-10 and VEGF-A by macrophages through activation of STAT3 and polarization toward the M2 phenotype.Citation74 This was confirmed by the fact that macrophages isolated from mouse tumors displayed activated STAT3 and induced angiogenesis in an in vitro tube formation assay via STAT3 induction of angiogenic factors, including VEGF and βFGF.Citation75 STAT3 signaling within the tumor microenvironment induces a procarcinogenic cytokine, IL-23, via direct transcriptional activation of the IL-23/p19 gene in tumor-associated macrophages, while inhibiting a central anticarcinogenic cytokine, IL-12, thereby shifting the balance of tumor immunity toward carcinogenesis.Citation53 The M-CSF-inducible DC-SIGN (dendritic cell-specific ICAM-3-grabbing nonintegrin or CD209) expression along monocyte-to-macrophage differentiation is dependent on JNK and STAT3 activation. This effect is potentiated by STAT3-activating cytokines IL-6 and IL-10 produced by STAT3-activated tumor cells. DC-SIGN contributes to the release of factors (IL-10) that would maintain STAT3 activation in tumor cells, thus implying that DC-SIGN favors the maintenance of an activated STAT3 context in the tumor stroma, which would compromise the ability of tumor-associated macrophages (and DCs) to generate effective antitumor responses and maintain an immunosuppressive environment.Citation76
However an anti-tumoral role of STAT3 in macrophages has been proposed. This came from studies that investigated the importance of STAT3 in macrophages through an indirect manner, using SOCS3 conditional knockout mice in macrophage. These macrophages presented a prolonged activation of STAT3 (but also a reduced activation of STAT1) and simultaneously exerted anti-inflammatory (e.g., through IL-6 and TNFα downregulation) as well as anti-tumor effects (through MCP2/CCL8-mediated anti-metastatic effect).Citation77 In that respect Lipoxin A4, by inducing the phosphorylation of STAT3, mediates monocyte differentiation into M2 subtypes that present anti-tumorigenic activities.Citation78 The discrepancies between these studies could be explained by the fact that SOCS3 and lipoxin signaling should regulate other pathways such as NFκB.
Tumor-STAT3 could also modulate macrophage fate. For example, in murine melanomas, natural STAT3 activity is associated with tumor growth and reduction of lymphocytes, NK cells, neutrophils and macrophages infiltration. In addition, blocking STAT3 triggers tumor cells to produce TNF-α and IFN-β capable of activating macrophage production of NO and RANTES in vitro and in vivo, leading to macrophage-mediated, nitrite-dependent cytostatic activity against tumor cells.Citation79,Citation80 Finally the inhibition of phagocytosis and the secretion of IL-10 induced by glioma cancer stem cells conditioned medium were reversed when the STAT3 pathway was blocked in the glioma cancer stem cells.Citation81
Dendritic cells
Dendritic cells represent a terminally differentiated stage of monocytes and are the key antigen-presenting cells of the immune system. DCs play a main role as immune sentinels in the initiation of T-cell responses to microbial pathogens, tumors and inflammation.Citation82,Citation83
The first evidence that STAT3 is important for DC fate was observed in mice lacking expression of STAT3 in hematopoietic progenitors. These animals present profound deficiency in the DC compartment and abrogated Flt3L effects on DC development.Citation84 However, the same mice bearing a tumor present enhanced function of dendritic cells, T cells, NK cells and neutrophils, and a decreased tumor progression.Citation85 DCs derived from LysMcre/STAT3(flox/flox) mice displayed higher cytokine production (such as IL-12 and TNFα) in response to TLR stimulation, activated more efficiently T cells and intratumoral administration of these DCs significantly inhibited MC38 tumor growth.Citation86 The more important production of IL-12 in STAT3 deficient cells has been attributed to the capacity of STAT3 to suppress NFκB/c-Rel mediated IL-12 transcription.Citation53 One could speculate that it is also true for TNFα, a known NFκB target gene. Moreover, ablating STAT3 in myeloid cells increases CpG-induced dendritic cell maturation (which present high levels of activated STAT1), T-cell activation, generation of tumor antigen-specific T cells and long-lasting antitumor immunity in B16 melanoma tumor model.Citation68 In the same way, CpG-bound STAT3 siRNA were used to downregulate STAT3 expression specifically in myeloid and B cells. Then silencing STAT3 in these cells significantly augments dendritic cell engagement and effector functions of adoptively transferred CD8+ T cells in vivo, with an upregulation of effector molecules such as perforin, granzyme B and IFN-γ.Citation87 The mechanism showing that STAT3-deficient DCs could activate more efficiently T cells than wild-type DCs was completed by Kortylewski et al.Citation85 Indeed mice lacking STAT3 in myeloid compartment of tumor stroma, including DCs and macrophages, showed reduced numbers of tumor-infiltrating CD4+CD25+/Foxp3+/Lag-3+ Tregs, which was accompanied by increased CD8+ effector T cells. Reduced expression of MHC class II and costimulatory molecules due to constitutive STAT3 activation in tumor-residing DCs contribute to the expansion of tumor-infiltrating FOXP3+ T cells.Citation85 Finally IL-6 has been shown to be a potent suppressor of bone marrow-derived DC activation/maturation and a regulator of DC differentiation in vivo, through STAT3 phosphorylation. Then an IL-6-gp130-STAT3 amplification loop controls DC differentiation/maturation and may represent a critical target for controlling T cell-mediated immune responses.Citation88 In this way, mammary tumor-derived exosomes block the differentiation of murine myeloid precursor cells into DCs in vitro. This was associated with an increased level of IL-6 and phosphorylated STAT3 and was blocked when bone marrow cells come from IL-6 knockout mice, suggesting that tumor cells could dampen DC differentiation through an autocrine STAT3 activation by IL-6.Citation89 IL-6 could also be produced by cancer cells and impact on STAT3 activation in DCs. Thus tumor cell conditioned medium from murine colon carcinoma or fibrosarcoma cells induced activation of JAK2 and STAT3 (but not STAT1 or STAT5), which was associated with an accumulation of immature myeloid cells and an inhibition of their differentiation into mature dendritic cells.Citation90-Citation92 More precisely, this observation was confirmed by the fact that soluble factors released by pancreatic cancer cells, IL-6 and G-CSF, are responsible for inhibiting DC differentiation and activation respectively in a STAT3-dependent manner.Citation93
In humans, STAT3-depleted DCs with adenoviral STAT3 short hairpin RNA (shRNA) were also capable of producing more cytokines under TLR stimulation (such as IL-12 and TNF-α), and they induced tumor Ag-specific T cells more efficiently than control DCs.Citation86
The production by tumor cells of molecules regulating DC activation could be induced by STAT3. Blocking STAT3 in tumor cells (by anti-sense oligonucleotides or STAT3β to displace STAT3α DNA binding) increases expression of proinflammatory cytokines (such as IFN-β, TNF-α and IL-6) and chemokines (RANTES and IP-10) that activate innate immunity and dendritic cells, leading to tumor-specific T-cell responses.Citation80 On the contrary, constitutive STAT3 activity (by transformation with v-src or transfection with a constitutively active STAT3 i.e., STAT3C) reduces production of pleiotropic factors (e.g., IL-6 and RANTES) and inhibits dendritic cell functional maturation. It is surprising that STAT3 negatively regulates IL-6 production by tumor cells in this context, because IL-6 has been largely described to be a target of STAT3 (for reviews, see refs. Citation14 and Citation94). Nevertheless, tumor-derived factors (that were not defined in this study) inhibit dendritic cell maturation through STAT3 activation in progenitor cells.Citation80
MDSCs
MDSCs have been identified in humans and mice as a population of immature myeloid cells with the ability to suppress T cell activation.Citation95 In tumor-bearing mice, these cells have been shown to markedly expand in lymphoid organ and blood when mice are inoculated with transplantable tumor cells or when tumors spontaneously develop in transgenic mice with tissue-restricted oncogene expression.Citation96 In addition, an increased MDSC frequency was detected in the blood of patients with different types of cancers.Citation97,Citation98 In mice and humans, MDSCs from tumor bearers induce antigen-specific MHC class I restricted tolerance of CD8+ T cells and are one of the major suppressors of antitumor immunity.Citation99
STAT3 is probably one of the main transcription factors that regulate MDSC function. MDSCs from tumor-bearing mice have markedly increased levels of phosphorylated STAT3 compared with immature myeloid cells from naive mice.Citation91 Moreover, ablation of STAT3 expression through the use of conditional knockout mice or selective STAT3 inhibitor (JSI-124 that did not affect pSTAT1, pSTAT5 or pSTAT6) markedly reduced the expansion of MDSCs, promoted accumulation of dendritic cells and increased T-cell responses in tumor-bearing mice.Citation85,Citation90,Citation91 Then, activated STAT3 has been proposed to be the main regulator of MDSC expansion. We have shown in contrast that tumor-derived exosome (TDE)-associated HSP72 triggered STAT3 activation in MDSCs in a TLR2/MyD88-dependent manner through autocrine production of IL-6. This mechanism is also relevant in cancer patients, as TDEs from a human tumor cell line activated human MDSCs and triggered their suppressive function but not MDSC expansion in an HSP72/TLR2-dependent manner.Citation100 How to explain these discrepancies? It is well known that in myeloid cells, STAT3 signaling drives the expression of Bcl-xL, c-myc, cyclin D1 or survivin, which prevents cell apoptosis, promotes cell proliferation, and prevents differentiation to mature cell types.Citation14,Citation101 Moreover, in vivo STAT3 ablation or inhibition mediates the differentiation of MDSCs into DCs, leading to the observation of an absence of MDSCs and to the conclusion that STAT3 mediates MDSC expansion. In our hands, tumor cell supernatant triggers two distinct molecular pathways in MDSCs: tumor-derived soluble factors trigger the activation of ERK, which results in the expansion of MDSCs, while TDEs trigger the activation of STAT3 without promoting MDSC expansion.Citation100 Nevertheless, STAT3 controlled the G-CSF-responsive induction of C/EPBβ (CCAAT-enhancer-binding protein β) expression in myeloid progenitor cells.Citation102 The transcription factor C/EBPβ was reported to play a crucial role in controlling the differentiation of myeloid precursors to functional MDSCs, with a correlation between C/EBPβ expression and CD11b+ Gr-1+ MDSC accumulation in response to G-CSF.Citation103,Citation104 Moreover, when C/EBPβ is deleted in bone-marrow cells they lose the ability to differentiate in vitro into functional MDSCs. It is conceivable that STAT3 can at least partially induce MDSC generation via upregulation of C/EBPβ.Citation102
Finally, recent works have highlighted the importance of signaling pathways downstream of STAT3 that are responsible for MDSC differentiation. As mentioned above for macrophages, MDSCs isolated from mouse tumors displayed activated STAT3 and induced angiogenesis in an in vitro tube formation assay via STAT3-dependent induction of angiogenic factors, including VEGF and βFGF.Citation75 In MDSCs, STAT3 controls the NADPH oxidase NOX2 expression leading to the production of ROS responsible for MDSC-induced immune suppression in murine mammary, colon, lung carcinoma, thymoma, sarcoma models and in patients with head and neck cancer.Citation105 In mice, the myeloid-related protein S100A9, a direct target of STAT3 is responsible for an enhanced MDSC production in EL4 or CT26 tumor-bearing mice. Mice lacking this protein mounted potent antitumor immune responses and rejected implanted tumors an effect reversed by administration of wild-type MDSCs from tumor-bearing mice to S100A9-null mice.Citation106
Conclusion
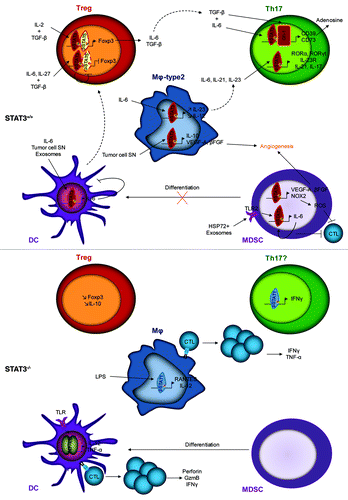
STAT3 activation within immune cells is responsible for the accumulation and the activation of immunosuppressive cells, such as Treg, Th17 and MDSCs, the differentiation of macrophages toward the M2 phenotype and the absence of functional DCs. On the contrary the deletion or the inhibition of STAT3 in these cells leads to the inhibition of Treg and Th17 cells, the differentiation of MDSC, a good antigen presentation by macrophages and DCs leading to an anti-tumor immune response mediated by CTL activation (). Thus, because STAT3 also regulates genes involved in biological functions of cancer cells, it makes this pathway a promising therapeutic target. Then STAT3 could be inhibited directly with peptides or natural compounds or indirectly by blocking upstream signaling pathways such as IL-6 and JAK2 pathways (for review, see ref. Citation107).
Figure 1. Effects of STAT3 presence (STAT3+/+) or absence (STAT3−/−) within immune cells. STAT3 activation leads to the activation of Treg and Th17 cells, the differentiation of M2 macrophages, the accumulation of MDSCs and the absence of functional DCs. STAT3 deletion or inhibition leads to the inhibition of Treg and Th17 cells, the differentiation of MDSCs into DCs, a good antigen presentation to CTLs by macrophages and DCs leading to an anti-tumor immune response.
| Abbreviations: | ||
| APC | = | antigen presenting cell |
| C/EBP | = | CCAAT-enhancer-binding protein |
| CD | = | cluster differentiation |
| CDK | = | cyclin dependent kinase |
| CSF-1 | = | colony stimulating factor-1 |
| CTL | = | cytotoxic T lymphocyte |
| DC | = | dendritic cell |
| DC-SIGN | = | dendritic cell-specific ICAM-3 grabbing nonintegrin |
| DNA | = | deoxyribonucleic acid |
| EGF | = | epidermal growth factor |
| ERK | = | extracellular signaling regulated kinase |
| FGF | = | fibroblast growth factor |
| FOXP3 | = | forkhead box P3 |
| G-CSF | = | granulocyte colony stimulating factor |
| GFI-1 | = | growth factor independent-1 |
| GM-CSF | = | granulocyte macrophage colony stimulating factor |
| HSP | = | heat shock protein |
| ICAM | = | intracellular adhesion molecule |
| IFN | = | interferon |
| IL | = | interleukin |
| JAK | = | Janus activated kinase |
| LPS | = | lipopolysaccharide |
| MCP-1 | = | monocyte chemoattractant protein-1 |
| MDSC | = | myeloid derived suppressor cell |
| MHC | = | major histocompatibility complex |
| NADPH | = | nicotinamide adenine dinucleotide phosphate hydrogen |
| NFκB | = | nuclear factor-κB |
| NK | = | natural killer |
| NO | = | nitric oxide |
| PDGF | = | platelet-derived growth factor |
| PI3K | = | phosphatidylinositol 3-kinase |
| PIAS | = | protein inhibitor of activated STAT |
| RANTES | = | regulated on activation normal T cell expressed and secreted |
| RAR | = | retinoic acid receptor |
| RNA | = | ribonucleic acid |
| ROR | = | RAR related orphan receptor |
| ROS | = | radical oxygen species |
| shRNA | = | short hairpin RNA |
| siRNA | = | small interfering RNA |
| SOCS | = | suppressor of cytokine signaling |
| STAT | = | signal transducer and activator of transcription |
| TAM | = | tumor associated macrophages |
| TCR | = | T cell receptor |
| TDE | = | tumor-derived exosome |
| TGF | = | transforming growth factor |
| Th | = | T helper cell |
| TLR | = | Toll-like receptor |
| TNF | = | tumor necrosis factor |
| Treg | = | regulatory T cell |
| VEGF | = | vascular endothelial growth factor |
Acknowledgments
C.R. was supported by Ligue contre le cancer comité Grand-Est, F.V. by fellowships from Ligue contre le cancer, H.B. by fellowships from the Conseil Régional Bourgogne/INSERM and F.G. by Fondation ARC, Fondation pour la recherche Médicale, INCA, Ligue contre le Cancer.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol 2007; 7:454 - 65; http://dx.doi.org/10.1038/nri2093; PMID: 17525754
- Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 2000; 19:2548 - 56; http://dx.doi.org/10.1038/sj.onc.1203551; PMID: 10851053
- Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005; 307:269 - 73; http://dx.doi.org/10.1126/science.1105166; PMID: 15653507
- Alexander WS, Hilton DJ. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu Rev Immunol 2004; 22:503 - 29; http://dx.doi.org/10.1146/annurev.immunol.22.091003.090312; PMID: 15032587
- Shuai K, Liu B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol 2005; 5:593 - 605; http://dx.doi.org/10.1038/nri1667; PMID: 16056253
- Sun S, Steinberg BM. PTEN is a negative regulator of STAT3 activation in human papillomavirus-infected cells. J Gen Virol 2002; 83:1651 - 8; PMID: 12075083
- Irie-Sasaki J, Sasaki T, Matsumoto W, Opavsky A, Cheng M, Welstead G, et al. CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature 2001; 409:349 - 54; http://dx.doi.org/10.1038/35053086; PMID: 11201744
- Migone TS, Cacalano NA, Taylor N, Yi T, Waldmann TA, Johnston JA. Recruitment of SH2-containing protein tyrosine phosphatase SHP-1 to the interleukin 2 receptor; loss of SHP-1 expression in human T-lymphotropic virus type I-transformed T cells. Proc Natl Acad Sci U S A 1998; 95:3845 - 50; http://dx.doi.org/10.1073/pnas.95.7.3845; PMID: 9520455
- Schaper F, Gendo C, Eck M, Schmitz J, Grimm C, Anhuf D, et al. Activation of the protein tyrosine phosphatase SHP2 via the interleukin-6 signal transducing receptor protein gp130 requires tyrosine kinase Jak1 and limits acute-phase protein expression. Biochem J 1998; 335:557 - 65; PMID: 9794795
- Krasilnikov M, Ivanov VN, Dong J, Ronai Z. ERK and PI3K negatively regulate STAT-transcriptional activities in human melanoma cells: implications towards sensitization to apoptosis. Oncogene 2003; 22:4092 - 101; http://dx.doi.org/10.1038/sj.onc.1206598; PMID: 12821943
- Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang Y, Deng J, et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc Natl Acad Sci U S A 2007; 104:16158 - 63; http://dx.doi.org/10.1073/pnas.0702596104; PMID: 17911267
- Daino H, Matsumura I, Takada K, Odajima J, Tanaka H, Ueda S, et al. Induction of apoptosis by extracellular ubiquitin in human hematopoietic cells: possible involvement of STAT3 degradation by proteasome pathway in interleukin 6-dependent hematopoietic cells. Blood 2000; 95:2577 - 85; PMID: 10753837
- Bromberg J, Darnell JE Jr.. The role of STATs in transcriptional control and their impact on cellular function. Oncogene 2000; 19:2468 - 73; http://dx.doi.org/10.1038/sj.onc.1203476; PMID: 10851045
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 2009; 9:798 - 809; http://dx.doi.org/10.1038/nrc2734; PMID: 19851315
- Darnell JE. Validating Stat3 in cancer therapy. Nat Med 2005; 11:595 - 6; http://dx.doi.org/10.1038/nm0605-595; PMID: 15937466
- Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 2009; 15:91 - 102; http://dx.doi.org/10.1016/j.ccr.2009.01.002; PMID: 19185844
- Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009; 15:103 - 13; http://dx.doi.org/10.1016/j.ccr.2009.01.001; PMID: 19185845
- Aggarwal BB, Kunnumakkara AB, Harikumar KB, Gupta SR, Tharakan ST, Koca C, et al. Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship?. Ann N Y Acad Sci 2009; 1171:59 - 76; http://dx.doi.org/10.1111/j.1749-6632.2009.04911.x; PMID: 19723038
- Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 2011; 331:1565 - 70; http://dx.doi.org/10.1126/science.1203486; PMID: 21436444
- Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011; 480:480 - 9; http://dx.doi.org/10.1038/nature10673; PMID: 22193102
- Shiku H. Importance of CD4+ helper T-cells in antitumor immunity. Int J Hematol 2003; 77:435 - 8; http://dx.doi.org/10.1007/BF02986610; PMID: 12841380
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006; 313:1960 - 4; http://dx.doi.org/10.1126/science.1129139; PMID: 17008531
- Marrogi AJ, Munshi A, Merogi AJ, Ohadike Y, El-Habashi A, Marrogi OL, et al. Study of tumor infiltrating lymphocytes and transforming growth factor-beta as prognostic factors in breast carcinoma. Int J Cancer 1997; 74:492 - 501; http://dx.doi.org/10.1002/(SICI)1097-0215(19971021)74:5<492::AID-IJC3>3.0.CO;2-Z; PMID: 9355970
- Hiraoka K, Miyamoto M, Cho Y, Suzuoki M, Oshikiri T, Nakakubo Y, et al. Concurrent infiltration by CD8+ T cells and CD4+ T cells is a favourable prognostic factor in non-small-cell lung carcinoma. Br J Cancer 2006; 94:275 - 80; http://dx.doi.org/10.1038/sj.bjc.6602934; PMID: 16421594
- Tüting T, Storkus WJ, Lotze MT. Gene-based strategies for the immunotherapy of cancer. J Mol Med (Berl) 1997; 75:478 - 91; http://dx.doi.org/10.1007/s001090050133; PMID: 9253711
- Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother 2005; 54:721 - 8; http://dx.doi.org/10.1007/s00262-004-0653-2; PMID: 16010587
- Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res 2011; 71:1263 - 71; http://dx.doi.org/10.1158/0008-5472.CAN-10-2907; PMID: 21303976
- Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 2000; 100:655 - 69; http://dx.doi.org/10.1016/S0092-8674(00)80702-3; PMID: 10761931
- Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 1997; 89:587 - 96; http://dx.doi.org/10.1016/S0092-8674(00)80240-8; PMID: 9160750
- Owaki T, Asakawa M, Morishima N, Mizoguchi I, Fukai F, Takeda K, et al. STAT3 is indispensable to IL-27-mediated cell proliferation but not to IL-27-induced Th1 differentiation and suppression of proinflammatory cytokine production. J Immunol 2008; 180:2903 - 11; PMID: 18292512
- Lucas S, Ghilardi N, Li J, de Sauvage FJ. IL-27 regulates IL-12 responsiveness of naive CD4+ T cells through Stat1-dependent and -independent mechanisms. Proc Natl Acad Sci U S A 2003; 100:15047 - 52; http://dx.doi.org/10.1073/pnas.2536517100; PMID: 14657353
- Chen Z, O’Shea JJ. Th17 cells: a new fate for differentiating helper T cells. Immunol Res 2008; 41:87 - 102; http://dx.doi.org/10.1007/s12026-007-8014-9; PMID: 18172584
- van den Berg WB, Miossec P. IL-17 as a future therapeutic target for rheumatoid arthritis. Nat Rev Rheumatol 2009; 5:549 - 53; http://dx.doi.org/10.1038/nrrheum.2009.179; PMID: 19798029
- Su X, Ye J, Hsueh EC, Zhang Y, Hoft DF, Peng G. Tumor microenvironments direct the recruitment and expansion of human Th17 cells. J Immunol 2010; 184:1630 - 41; http://dx.doi.org/10.4049/jimmunol.0902813; PMID: 20026736
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006; 126:1121 - 33; http://dx.doi.org/10.1016/j.cell.2006.07.035; PMID: 16990136
- Harris TJ, Grosso JF, Yen HR, Xin H, Kortylewski M, Albesiano E, et al. Cutting edge: An in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol 2007; 179:4313 - 7; PMID: 17878325
- Nishihara M, Ogura H, Ueda N, Tsuruoka M, Kitabayashi C, Tsuji F, et al. IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int Immunol 2007; 19:695 - 702; http://dx.doi.org/10.1093/intimm/dxm045; PMID: 17493959
- Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol 2007; 8:967 - 74; http://dx.doi.org/10.1038/ni1488; PMID: 17581537
- Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem 2007; 282:9358 - 63; http://dx.doi.org/10.1074/jbc.C600321200; PMID: 17277312
- Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 2007; 26:371 - 81; http://dx.doi.org/10.1016/j.immuni.2007.02.009; PMID: 17363300
- Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 2008; 28:29 - 39; http://dx.doi.org/10.1016/j.immuni.2007.11.016; PMID: 18164222
- Chalmin F, Mignot G, Bruchard M, Chevriaux A, Végran F, Hichami A, et al. Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity 2012; 36:362 - 73; http://dx.doi.org/10.1016/j.immuni.2011.12.019; PMID: 22406269
- Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature 2010; 467:967 - 71; http://dx.doi.org/10.1038/nature09447; PMID: 20962846
- Sakaguchi S. Regulatory T cells: key controllers of immunologic self-tolerance. Cell 2000; 101:455 - 8; http://dx.doi.org/10.1016/S0092-8674(00)80856-9; PMID: 10850488
- Shevach EM. CD4+ CD25+ suppressor T cells: more questions than answers. Nat Rev Immunol 2002; 2:389 - 400; PMID: 12093005
- Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med 2003; 198:1875 - 86; http://dx.doi.org/10.1084/jem.20030152; PMID: 14676299
- Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer 2010; 127:759 - 67; PMID: 20518016
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004; 10:942 - 9; http://dx.doi.org/10.1038/nm1093; PMID: 15322536
- Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, et al. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol 2006; 24:5373 - 80; http://dx.doi.org/10.1200/JCO.2006.05.9584; PMID: 17135638
- Perrone G, Ruffini PA, Catalano V, Spino C, Santini D, Muretto P, et al. Intratumoural FOXP3-positive regulatory T cells are associated with adverse prognosis in radically resected gastric cancer. Eur J Cancer 2008; 44:1875 - 82; http://dx.doi.org/10.1016/j.ejca.2008.05.017; PMID: 18617393
- Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 2006; 108:1571 - 9; http://dx.doi.org/10.1182/blood-2006-02-004747; PMID: 16645171
- Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature 2007; 445:766 - 70; http://dx.doi.org/10.1038/nature05479; PMID: 17220876
- Kortylewski M, Xin H, Kujawski M, Lee H, Liu Y, Harris T, et al. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 2009; 15:114 - 23; http://dx.doi.org/10.1016/j.ccr.2008.12.018; PMID: 19185846
- Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science 2009; 326:986 - 91; http://dx.doi.org/10.1126/science.1172702; PMID: 19797626
- Wuest TY, Willette-Brown J, Durum SK, Hurwitz AA. The influence of IL-2 family cytokines on activation and function of naturally occurring regulatory T cells. J Leukoc Biol 2008; 84:973 - 80; http://dx.doi.org/10.1189/jlb.1107778; PMID: 18653463
- Huber M, Steinwald V, Guralnik A, Brüstle A, Kleemann P, Rosenplänter C, et al. IL-27 inhibits the development of regulatory T cells via STAT3. Int Immunol 2008; 20:223 - 34; http://dx.doi.org/10.1093/intimm/dxm139; PMID: 18156621
- Xu L, Kitani A, Stuelten C, McGrady G, Fuss I, Strober W. Positive and negative transcriptional regulation of the Foxp3 gene is mediated by access and binding of the Smad3 protein to enhancer I. Immunity 2010; 33:313 - 25; http://dx.doi.org/10.1016/j.immuni.2010.09.001; PMID: 20870174
- Laurence A, Amarnath S, Mariotti J, Kim YC, Foley J, Eckhaus M, et al. STAT3 transcription factor promotes instability of nTreg cells and limits generation of iTreg cells during acute murine graft-versus-host disease. Immunity 2012; 37:209 - 22; http://dx.doi.org/10.1016/j.immuni.2012.05.027; PMID: 22921119
- Kong LY, Wei J, Sharma AK, Barr J, Abou-Ghazal MK, Fokt I, et al. A novel phosphorylated STAT3 inhibitor enhances T cell cytotoxicity against melanoma through inhibition of regulatory T cells. Cancer Immunol Immunother 2009; 58:1023 - 32; http://dx.doi.org/10.1007/s00262-008-0618-y; PMID: 19002459
- Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev 2008; 222:162 - 79; http://dx.doi.org/10.1111/j.1600-065X.2008.00602.x; PMID: 18364001
- Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol 2000; 164:6166 - 73; PMID: 10843666
- Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci 2008; 13:453 - 61; http://dx.doi.org/10.2741/2692; PMID: 17981560
- Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 2004; 4:71 - 8; http://dx.doi.org/10.1038/nrc1256; PMID: 14708027
- Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer 2006; 42:717 - 27; http://dx.doi.org/10.1016/j.ejca.2006.01.003; PMID: 16520032
- Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res 2006; 66:605 - 12; http://dx.doi.org/10.1158/0008-5472.CAN-05-4005; PMID: 16423985
- Jenkins BJ, Grail D, Inglese M, Quilici C, Bozinovski S, Wong P, et al. Imbalanced gp130-dependent signaling in macrophages alters macrophage colony-stimulating factor responsiveness via regulation of c-fms expression. Mol Cell Biol 2004; 24:1453 - 63; http://dx.doi.org/10.1128/MCB.24.4.1453-1463.2004; PMID: 14749363
- Cheng F, Wang HW, Cuenca A, Huang M, Ghansah T, Brayer J, et al. A critical role for Stat3 signaling in immune tolerance. Immunity 2003; 19:425 - 36; http://dx.doi.org/10.1016/S1074-7613(03)00232-2; PMID: 14499117
- Kortylewski M, Kujawski M, Herrmann A, Yang C, Wang L, Liu Y, et al. Toll-like receptor 9 activation of signal transducer and activator of transcription 3 constrains its agonist-based immunotherapy. Cancer Res 2009; 69:2497 - 505; http://dx.doi.org/10.1158/0008-5472.CAN-08-3031; PMID: 19258507
- Brayer J, Cheng F, Wang H, Horna P, Vicente-Suarez I, Pinilla-Ibarz J, et al. Enhanced CD8 T cell cross-presentation by macrophages with targeted disruption of STAT3. Immunol Lett 2010; 131:126 - 30; http://dx.doi.org/10.1016/j.imlet.2010.03.004; PMID: 20346983
- Sun Z, Yao Z, Liu S, Tang H, Yan X. An oligonucleotide decoy for Stat3 activates the immune response of macrophages to breast cancer. Immunobiology 2006; 211:199 - 209; http://dx.doi.org/10.1016/j.imbio.2005.11.004; PMID: 16530087
- Komohara Y, Ohnishi K, Kuratsu J, Takeya M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol 2008; 216:15 - 24; http://dx.doi.org/10.1002/path.2370; PMID: 18553315
- Fujiwara Y, Komohara Y, Ikeda T, Takeya M. Corosolic acid inhibits glioblastoma cell proliferation by suppressing the activation of signal transducer and activator of transcription-3 and nuclear factor-kappa B in tumor cells and tumor-associated macrophages. Cancer Sci 2011; 102:206 - 11; http://dx.doi.org/10.1111/j.1349-7006.2010.01772.x; PMID: 21073634
- Fujiwara Y, Komohara Y, Kudo R, Tsurushima K, Ohnishi K, Ikeda T, et al. Oleanolic acid inhibits macrophage differentiation into the M2 phenotype and glioblastoma cell proliferation by suppressing the activation of STAT3. Oncol Rep 2011; 26:1533 - 7; PMID: 21922144
- Hasita H, Komohara Y, Okabe H, Masuda T, Ohnishi K, Lei XF, et al. Significance of alternatively activated macrophages in patients with intrahepatic cholangiocarcinoma. Cancer Sci 2010; 101:1913 - 9; http://dx.doi.org/10.1111/j.1349-7006.2010.01614.x; PMID: 20545696
- Kujawski M, Kortylewski M, Lee H, Herrmann A, Kay H, Yu H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest 2008; 118:3367 - 77; http://dx.doi.org/10.1172/JCI35213; PMID: 18776941
- Domínguez-Soto A, Sierra-Filardi E, Puig-Kröger A, Pérez-Maceda B, Gómez-Aguado F, Corcuera MT, et al. Dendritic cell-specific ICAM-3-grabbing nonintegrin expression on M2-polarized and tumor-associated macrophages is macrophage-CSF dependent and enhanced by tumor-derived IL-6 and IL-10. J Immunol 2011; 186:2192 - 200; http://dx.doi.org/10.4049/jimmunol.1000475; PMID: 21239715
- Hiwatashi K, Tamiya T, Hasegawa E, Fukaya T, Hashimoto M, Kakoi K, et al. Suppression of SOCS3 in macrophages prevents cancer metastasis by modifying macrophage phase and MCP2/CCL8 induction. Cancer Lett 2011; 308:172 - 80; http://dx.doi.org/10.1016/j.canlet.2011.04.024; PMID: 21624767
- Li Y, Cai L, Wang H, Wu P, Gu W, Chen Y, et al. Pleiotropic regulation of macrophage polarization and tumorigenesis by formyl peptide receptor-2. Oncogene 2011; 30:3887 - 99; http://dx.doi.org/10.1038/onc.2011.112; PMID: 21499310
- Burdelya L, Kujawski M, Niu G, Zhong B, Wang T, Zhang S, et al. Stat3 activity in melanoma cells affects migration of immune effector cells and nitric oxide-mediated antitumor effects. J Immunol 2005; 174:3925 - 31; PMID: 15778348
- Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med 2004; 10:48 - 54; http://dx.doi.org/10.1038/nm976; PMID: 14702634
- Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol 2010; 12:1113 - 25; http://dx.doi.org/10.1093/neuonc/noq082; PMID: 20667896
- Crowley M, Inaba K, Steinman RM. Dendritic cells are the principal cells in mouse spleen bearing immunogenic fragments of foreign proteins. J Exp Med 1990; 172:383 - 6; http://dx.doi.org/10.1084/jem.172.1.383; PMID: 1694226
- Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 1991; 9:271 - 96; http://dx.doi.org/10.1146/annurev.iy.09.040191.001415; PMID: 1910679
- Laouar Y, Welte T, Fu XY, Flavell RA. STAT3 is required for Flt3L-dependent dendritic cell differentiation. Immunity 2003; 19:903 - 12; http://dx.doi.org/10.1016/S1074-7613(03)00332-7; PMID: 14670306
- Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med 2005; 11:1314 - 21; http://dx.doi.org/10.1038/nm1325; PMID: 16288283
- Iwata-Kajihara T, Sumimoto H, Kawamura N, Ueda R, Takahashi T, Mizuguchi H, et al. Enhanced cancer immunotherapy using STAT3-depleted dendritic cells with high Th1-inducing ability and resistance to cancer cell-derived inhibitory factors. J Immunol 2011; 187:27 - 36; http://dx.doi.org/10.4049/jimmunol.1002067; PMID: 21632716
- Herrmann A, Kortylewski M, Kujawski M, Zhang C, Reckamp K, Armstrong B, et al. Targeting Stat3 in the myeloid compartment drastically improves the in vivo antitumor functions of adoptively transferred T cells. Cancer Res 2010; 70:7455 - 64; http://dx.doi.org/10.1158/0008-5472.CAN-10-0736; PMID: 20841481
- Park SJ, Nakagawa T, Kitamura H, Atsumi T, Kamon H, Sawa S, et al. IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J Immunol 2004; 173:3844 - 54; PMID: 15356132
- Yu S, Liu C, Su K, Wang J, Liu Y, Zhang L, et al. Tumor exosomes inhibit differentiation of bone marrow dendritic cells. J Immunol 2007; 178:6867 - 75; PMID: 17513735
- Nefedova Y, Huang M, Kusmartsev S, Bhattacharya R, Cheng P, Salup R, et al. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol 2004; 172:464 - 74; PMID: 14688356
- Nefedova Y, Nagaraj S, Rosenbauer A, Muro-Cacho C, Sebti SM, Gabrilovich DI. Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res 2005; 65:9525 - 35; http://dx.doi.org/10.1158/0008-5472.CAN-05-0529; PMID: 16230418
- Nefedova Y, Cheng P, Gilkes D, Blaskovich M, Beg AA, Sebti SM, et al. Activation of dendritic cells via inhibition of Jak2/STAT3 signaling. J Immunol 2005; 175:4338 - 46; PMID: 16177074
- Bharadwaj U, Li M, Zhang R, Chen C, Yao Q. Elevated interleukin-6 and G-CSF in human pancreatic cancer cell conditioned medium suppress dendritic cell differentiation and activation. Cancer Res 2007; 67:5479 - 88; http://dx.doi.org/10.1158/0008-5472.CAN-06-3963; PMID: 17545630
- Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev 2010; 21:11 - 9; http://dx.doi.org/10.1016/j.cytogfr.2009.11.005; PMID: 20018552
- Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res 2007; 67:425 - , author reply 426; http://dx.doi.org/10.1158/0008-5472.CAN-06-3037; PMID: 17210725
- Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008; 14:408 - 19; http://dx.doi.org/10.1016/j.ccr.2008.10.011; PMID: 18977329
- Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol 2001; 166:678 - 89; PMID: 11123353
- Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother 2009; 58:49 - 59; http://dx.doi.org/10.1007/s00262-008-0523-4; PMID: 18446337
- Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med 2007; 13:828 - 35; http://dx.doi.org/10.1038/nm1609; PMID: 17603493
- Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest 2010; 120:457 - 71; PMID: 20093776
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9:162 - 74; http://dx.doi.org/10.1038/nri2506; PMID: 19197294
- Zhang H, Nguyen-Jackson H, Panopoulos AD, Li HS, Murray PJ, Watowich SS. STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood 2010; 116:2462 - 71; http://dx.doi.org/10.1182/blood-2009-12-259630; PMID: 20581311
- Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity 2010; 32:790 - 802; http://dx.doi.org/10.1016/j.immuni.2010.05.010; PMID: 20605485
- Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno S, Imanishi J, et al. C/EBPbeta is required for ‘emergency’ granulopoiesis. Nat Immunol 2006; 7:732 - 9; http://dx.doi.org/10.1038/ni1354; PMID: 16751774
- Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol 2009; 182:5693 - 701; http://dx.doi.org/10.4049/jimmunol.0900092; PMID: 19380816
- Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med 2008; 205:2235 - 49; http://dx.doi.org/10.1084/jem.20080132; PMID: 18809714
- Mankan AK, Greten FR. Inhibiting signal transducer and activator of transcription 3: rationality and rationale design of inhibitors. Expert Opin Investig Drugs 2011; 20:1263 - 75; http://dx.doi.org/10.1517/13543784.2011.601739; PMID: 21751940