Abstract
Pancreatic ductal adenocarcinoma is associated with a poor prognosis: For local disease, the 5-y survival rate is approximately 20% and median survival in locally advanced disease is only about 10 mo. Carcinogenesis is associated with chronic inflammation showing that innate immunity can be a double-edged sword. Here, we discuss recent findings of Ochi et al. in The Journal of Clinical Investigation who described a novel role for TLR7 in the development and progression of pancreatic cancer through interference with cell cycle regulation and by activation of multiple cell signaling pathways. Of note, inhibition of TLR7 signaling in a mouse p48Cre;KrasG12D pancreatic cancer model protected against tumor progression thus paving the road for TLR-blocking strategies to combat tumors.
What is the role of innate immunity in tumor biology? The concept of immunosurveillance, introduced in the seventies,Citation1 suggested that innate and adaptive immunity serve to reduce tumor incidence by recognizing transformed cells as “foreign” resulting in attack and destruction of the latter. In this line, tumor initiation, promotion and progression can be interpreted as failure in induction of a sufficiently strong immune response.Citation2 In turn, strengthening the immune response through targeted activation of the innate and adaptive immune system would be a strategy of tumor immunotherapy.
In contrast, a number of tumors are linked to chronic inflammation, namely those that are associated with chronic microbial infections, e.g., hepatitis B and C virus, Epstein-Barr virus or chronic infection with Helicobacter pylori. Therefore, recent theories tend more to interpret chronic inflammation, especially activation of innate immune effectors, as driver of tumor progression.Citation3 Under-pinning this view, activation of the central signaling module of innate immunity, NFκB, has been linked experimentally to the progression of tumorsCitation4,Citation5 and in this line, tumor immunotherapies could involve strategies that block activation of innate immune responses.
Activation of innate immunity is achieved through stimulation of pattern recognition receptors (PRRs) (reviewed in ref. Citation6). Among those, Toll-like receptors (TLRs) were the first group to be identified and have been studied intensively since their discovery. TLRs are thought to have been developed evolutionary to recognize microbial pathogens. They can be activated by a panel of pathogen-associated molecular patterns (PAMP) including bacterial cell-wall components like lipopolysaccharide and peptidoglycan as well as by microbial DNA and RNA. However, recent results indicate that beside the recognition of PAMPs, TLRs can also sense molecules that are released by necrotic or injured cells—so-called damage-associated molecular patterns (DAMPs)—including heat shock proteins, HMGB-1 and S100 proteins. Thus, TLRs as well as other PRRs can recognize foreign as well as self molecules based upon the context in which recognition occurs. As a net consequence, TLR ligation leads to upregulation of pro-inflammatory cytokines and/or type I interferon involving activation of MAP kinases, NFκB and/or IRFs pathways, respectively.Citation6 With the identification of molecular modes of pattern recognition the development of synthetic agonists as well as antagonists has become feasible, thus allowing for targeted manipulation of innate immune signaling.
In the field of tumor immunology, enhanced expression of TLRs has been described in a variety of different tumors during the past years and research has strongly focused on the question whether targeting of TLR mediated signaling events could have therapeutic implications.Citation7 In fact, a number of experiments suggest that enforcement of innate immunity by targeted TLR activation has beneficial effects to combat tumor growth. For example the TLR7 agonist imiquimod is licensed for therapy of basal cell carcinoma, and TLR3 and TLR9 ligands have been shown to reduce growth of renal cell carcinoma and metastatic colorectal carcinoma. TLR agonists have also been reported to be beneficial in models of mouse mammary tumors and leukemia to name a few. Several TLR-agonistic compounds are currently being investigated in clinical trials (reviewed in ref. Citation8). Modes of action probably include direct effects on tumor cells, e.g., apoptosis, activation of innate immune cells like NK cells and phagocytes as well as enforcement of T-cell immunity (including CTLs) through dendritic cell activation. In general, a plethora of reports proposes that nucleic acid ligands that target endosomal TLRs (TLR3, 7 and 9) might specifically favor antigen cross presentation and subsequent induction of cytotoxic T-cells. Thus, TLR agonists are also evaluated in protocols of dendritic cell based tumor vaccines.Citation9,Citation10 Despite those observations that would argue in favor of a concept supporting an anti-tumorigenic role of TLRs, the fact that about 15% of human tumors are associated with chronic inflammation remains puzzling.
In a recent report by Ochi et al. some of those controversies are now solved based on findings in a model of pancreatic adenocarcinoma. Pancreatic ductal adenocarcinoma is an aggressive cancer that is characterized by pronounced stromal infiltration with inflammatory cells. In fact, the inflammatory environment has long been acknowledged to be of importance for tumor progression. It has been suggested that factors released by the pancreatic tumor stroma play a major role in tumor progression and invasiveness although the precise mechanisms how the stroma interacts with transformed ductal epithelial cells remained poorly defined. Especially, it was unclear if innate immunity mediated inflammation might be involved in pancreatic cancer pathogenesis.
In the present study, the authors focused on the role of Toll-like receptor 7 (TLR7), an endosomally localized TLR that is activated by RNA of viral and bacterial origin.Citation11 Ochi et al. report that normal pancreata of mice and human do not express TLR7. By contrast, in both a mouse p48Cre;KrasG12D pancreatic cancer model as well as in human pancreatic cancer specimen this receptor was heavily upregulated in neoplastic ductal epithelial cells as well as in inflammatory cells within the tumor microenvironment, including macrophages, dendritic cells, neutrophils and surprisingly B and T cells. To gain insights into the functional role of TLR7 in pancreatic cancer, the authors treated p48Cre;KrasG12D mice with ssRNA40, a synthetic ligand for TLR7. Administrating the latter, the authors observed vigorous tumor growth and stromal expansion. Conversely, TLR7 blockade using an inhibitory oligonucleotide prevented malignant progression when administered together with cancer progression accelerator caerulein in Kras mutant mice. On the molecular level, TLR7 ligation in Kras mutated mice caused a dysregulation of proteins involved in cell cycle regulation including p53, p21, p27, cyclin D1 and cyclin B1 in the pancreata. Furthermore, ssRNA40 administration interfered with multiple signaling pathways including MAPK, NFκB and Notch activation and induced loss of PTEN expression. Of note, despite upregulated Notch 1 and 2 and Notch ligands, Hes1 and Hey1, Notch target genes, were downregulated. The authors suggest that Notch signaling is deviated toward inflammatory end products through cooperation with NFκB. This deviation might be characteristic for oncogenic TLR7 activation. Mechanistically the authors observed a strong STAT-3 activation, probably as a consequence of PTEN downregulation. STAT-3 activation has been associated with increased Notch signaling and modulation of anti-apoptotic proteins Bcl-xl and c-Myc.Citation12 Moreover, STAT3 has repeatedly been linked to pancreas cancer progression: STAT3 was shown to be constitutively activated in pancreatic ductal adenocarcinoma and it was demonstrated that IL-6-dependent activation of STAT3 was required for development of pancreatic intraepithelial neoplasia and ductal adenocarcinoma.Citation13-Citation15 Upregulation of STAT3 is not a unique feature of pancreatic cancer but occurs in a variety of human tumors. However, its function in cancer seems to be complex. For example, silencing of STAT3 sensitized A549 lung carcinoma cells to DNA damaging chemotherapeutics, potentially by downregulation of Bcl-xl, but did not affect TNF- and natural killer cell-mediated cytotoxicity. Further, STAT3 silencing was associated with increased NFκB signaling.Citation16
Using bone-marrow chimeric mice, the authors further show that TLR7 expression in the inflammatory stromal cells was required for pancreatic cancer progression as p48Cre;KrasG12D-TLR7−/− chimeric mice were protected from caerulein-induced accelerated neoplasia. Although activation of MAPK, NFκB and Notch signaling occurred in both epithelial and in inflammatory peritumoral cells after ssRNA40 administration in p48Cre;KrasG12D mice, treatment of isolated pancreatic ductal epithelial cells with ssRNA40 in vitro had no effect on proliferation, cell cycle related gene expression, viability and cytokine production. Thus, the authors conclude that carcinogenic changes seen in these cells in vivo after TLR7 ligation are secondary to direct effects of TLR7 activation on the stromal inflammatory cells.
The authors were further able to extend their findings for a proinflammatory role of TLR7 to chronic pancreatitis. Similar to the observation in pancreatic neoplasia, elevated TLR7 expression in epithelial and stromal inflammatory cells was observed in human chronic pancreatitis specimen as well as in a caerulein-induced mouse pancreatitis model in the absence of Kras mutation. Administration of caerulein in combination with ssRNA40 enhanced intrapancreatic inflammation and fibrosis but the authors did not observe dysregulated expression of cell cycle related genes which apparently rely on underlying oncogenic mutations. The authors therefore hypothesize that molecular changes associated with TLR7-induced carcinogenesis require synergistic expression of oncogenic Kras and suggest differences in TLR7 signal transduction based on the cellular baseline.
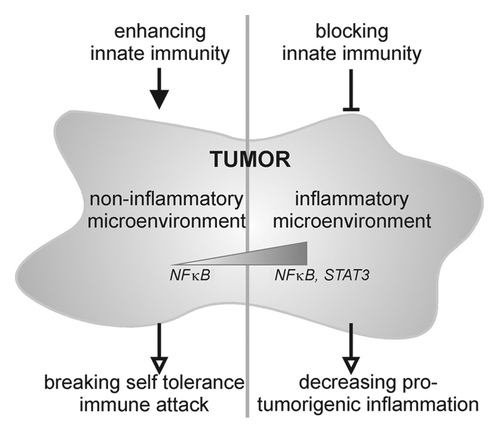
The strong pro-carcinogenic effect of TLR7 observed by Ochi et al. in a mouse model of pancreatic adenocarcinoma was indeed surprising given the fact that TLR7 has been shown to mediate anti-neoplastic effects in other cancer models, as discussed above. Although the findings of Ochi et al. seem contradictory on the first glance, the authors formulate and reinforce an interesting hypothesis unifying the discrepant findings of their and other studies:Citation3 It might be possible that in cancers without primary inflammation, an activation of TLRs might break self-tolerance of the immune system toward the cancer cells and thus promote anti-tumoral immunity. On the other hand, in cancers with a pronounced primary inflammatory component—as is the case for pancreatic adenocarcinoma—TLR activation might rather accelerate carcinogenesis (). This might be associated with differences in the relative strength of NFκB and STAT3 signaling. Confirming the latter notion, TLR4 activation appears to promote the development of colitis-associated colon cancer, another cancer entity characterized by a strong primary inflammatory component.Citation17 In another recent report by Tye et al. a similar concept has been testedCitation18: As gastric cancer is associated with chronic inflammation the authors used a model of gastric cancer induced by chronic STAT3 activation. They observed a strong upregulation of TLR2 and blockade or genetic deletion of TLR2 resulted in a decrease of epithelial proliferation and a parallel increase of apoptosis. Inflammation itself was not affected but the results also support a pro-tumorigenic role of a TLR. STAT3 activation and TLR2 upregulation were associated with poor patient prognosis.
Figure 1. Depending upon the inflammatory nature of the tumor microenvironment which goes along with differences in NFκB and STAT3 activation, different tumor entities require either enhancing or suppressive modulation of innate immune responses.
In the study of Ochi et al. the effective ligand for TLR7 was not further studied. The natural ligand of TLR7 is ssRNA of both viral and bacterial origin whereas self-RNA is usually not recognized. To date, it is unclear how TLR7 is activated in pancreatic cancer. It is possible that under certain circumstances TLR7 might lose its ability to distinguish between self and foreign nucleic acids, leading to activation by endogenous RNA. In fact, TLR7 activation by self-RNA has also been implied in autoimmunity.Citation19-Citation21 Here, it was suggested that an increase of availability of self-RNA due to increased release might overcome the natural self/foreign discrimination. One layer of discrimination is the restriction of nucleic acid recognizing receptors to endosomal compartments where they avoid contact with self-RNA. Increased uptake due to excessive release might overcome this barrier. TLR7 has also been shown to discriminate RNAs based on posttranscriptional modifications.Citation22-Citation25 Currently, there is no information available whether modification patterns might change in tumor cells. Very recently, it was reported that microRNA release through exosomes from tumor cells can trigger TLR7 and TLR8 thus promoting prometastatic inflammation.Citation26 Exosomal microRNA was suggested to shape the tumor microenvironment and might be a candidate for the above observed pro-tumorigenic response of TLR7 in pancreatical ductal adenocarcinoma. Finally, the authors speculate that activation by viral RNA might also play a role as infections with hepatitis B, hepatitis C and TT virus have been correlated with pancreatic cancer; yet the causative link remains elusive.Citation27-Citation29
In summary, the authors present an exciting novel role for TLR7 in pancreatic carcinogenesis in a p48Cre;KrasG12D mouse model which underpins a role of TLRs as tumor promoters. The findings also open the avenue for tumor immunomodulatory therapies through TLR inhibition. In light of the limited therapeutic options for treatment of pancreatic cancer, one can only hope that the promising results in this mouse study can be translated into the human situation. However, although the paper presented here demonstrated that inhibition of TLR7-mediated signaling could prevent pancreatic cancer progression, it is not yet clear if TLR7 antagonism alone is sufficient to induce regression of established adenocarcinoma. It is rather likely that alleviation of pro-inflammatory tumor microenvironment by TLR7 blockade could be used in conjunction with classical anti-proliferative chemotherapeutics. Further, it should be kept in mind that the ssRNA40 used in this study not only activates TLR7 but also acts on human TLR8, a TLR which is not functional in the mouse. Thus, it will be interesting to also explore a potential role for TLR8 in human pancreatic cancer and potential synergistic effects between those two TLRs. Taken together, the discussed publication discovers a central pro-tumorigenic role of TLR7 in pancreas adenocarcinoma biology.
| Abbreviations: | ||
| CTL | = | cytotoxic T-lymphocyte |
| DAMP | = | danger associated molecular pattern |
| PAMP | = | pathogen associated molecular pattern |
| PRR | = | pattern recognition receptor |
| ss | = | single stranded |
| TLR | = | Toll-like receptor |
| Kras | = | Kirsten rat sarcoma viral oncogene homolog |
References
- Burnet FM. The concept of immunological surveillance. Prog Exp Tumor Res 1970; 13:1 - 27; PMID: 4921480
- Pardoll D. Does the immune system see tumors as foreign or self?. Annu Rev Immunol 2003; 21:807 - 39; http://dx.doi.org/10.1146/annurev.immunol.21.120601.141135; PMID: 12615893
- Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 2005; 5:749 - 59; http://dx.doi.org/10.1038/nri1703; PMID: 16175180
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004; 118:285 - 96; http://dx.doi.org/10.1016/j.cell.2004.07.013; PMID: 15294155
- Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004; 431:461 - 6; http://dx.doi.org/10.1038/nature02924; PMID: 15329734
- Kumagai Y, Akira S. Identification and functions of pattern-recognition receptors. J Allergy Clin Immunol 2010; 125:985 - 92; http://dx.doi.org/10.1016/j.jaci.2010.01.058; PMID: 20392481
- Wolska A, Lech-Marańda E, Robak T. Toll-like receptors and their role in carcinogenesis and anti-tumor treatment. Cell Mol Biol Lett 2009; 14:248 - 72; http://dx.doi.org/10.2478/s11658-008-0048-z; PMID: 19096763
- Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med 2007; 13:552 - 9; http://dx.doi.org/10.1038/nm1589; PMID: 17479101
- Prins RM, Craft N, Bruhn KW, Khan-Farooqi H, Koya RC, Stripecke R, et al. The TLR-7 agonist, imiquimod, enhances dendritic cell survival and promotes tumor antigen-specific T cell priming: relation to central nervous system antitumor immunity. J Immunol 2006; 176:157 - 64; PMID: 16365406
- Zheng R, Cohen PA, Paustian CA, Johnson TD, Lee WT, Shu S, et al. Paired Toll-like receptor agonists enhance vaccine therapy through induction of interleukin-12. Cancer Res 2008; 68:4045 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-07-6669; PMID: 18519662
- Eberle F, Sirin M, Binder M, Dalpke AH. Bacterial RNA is recognized by different sets of immunoreceptors. Eur J Immunol 2009; 39:2537 - 47; http://dx.doi.org/10.1002/eji.200838978; PMID: 19662634
- Connolly MK, Bedrosian AS, Malhotra A, Henning JR, Ibrahim J, Vera V, et al. In hepatic fibrosis, liver sinusoidal endothelial cells acquire enhanced immunogenicity. J Immunol 2010; 185:2200 - 8; http://dx.doi.org/10.4049/jimmunol.1000332; PMID: 20639479
- Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, et al. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res 2011; 71:5020 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-11-0908; PMID: 21586612
- Fukuda A, Wang SC, Morris JP 4th, Folias AE, Liou A, Kim GE, et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 2011; 19:441 - 55; http://dx.doi.org/10.1016/j.ccr.2011.03.002; PMID: 21481787
- Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Klöppel G, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011; 19:456 - 69; http://dx.doi.org/10.1016/j.ccr.2011.03.009; PMID: 21481788
- Weronika Kulesza D, Carre T, Chouaib S, Kaminska B. Silencing of the transcription factor STAT3 sensitizes lung cancer cells to DNA damaging drugs, but not to TNFα- and NK cytotoxicity. Exp Cell Res 2013; 319:506 - 16; http://dx.doi.org/10.1016/j.yexcr.2012.11.005; PMID: 23149124
- Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S, et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology 2007; 133:1869 - 81; http://dx.doi.org/10.1053/j.gastro.2007.09.008; PMID: 18054559
- Tye H, Kennedy CL, Najdovska M, McLeod L, McCormack W, Hughes N, et al. STAT3-Driven Upregulation of TLR2 Promotes Gastric Tumorigenesis Independent of Tumor Inflammation. Cancer Cell 2012; 22:466 - 78; http://dx.doi.org/10.1016/j.ccr.2012.08.010; PMID: 23079657
- Ganguly D, Chamilos G, Lande R, Gregorio J, Meller S, Facchinetti V, et al. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med 2009; 206:1983 - 94; http://dx.doi.org/10.1084/jem.20090480; PMID: 19703986
- Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med 2005; 202:1171 - 7; http://dx.doi.org/10.1084/jem.20050630; PMID: 16260486
- Vollmer J, Tluk S, Schmitz C, Hamm S, Jurk M, Forsbach A, et al. Immune stimulation mediated by autoantigen binding sites within small nuclear RNAs involves Toll-like receptors 7 and 8. J Exp Med 2005; 202:1575 - 85; http://dx.doi.org/10.1084/jem.20051696; PMID: 16330816
- Gehrig S, Eberle ME, Botschen F, Rimbach K, Eberle F, Eigenbrod T, et al. Identification of modifications in microbial, native tRNA that suppress immunostimulatory activity. J Exp Med 2012; 209:225 - 33; http://dx.doi.org/10.1084/jem.20111044; PMID: 22312113
- Jöckel S, Nees G, Sommer R, Zhao Y, Cherkasov D, Hori H, et al. The 2′-O-methylation status of a single guanosine controls transfer RNA-mediated Toll-like receptor 7 activation or inhibition. J Exp Med 2012; 209:235 - 41; http://dx.doi.org/10.1084/jem.20111075; PMID: 22312111
- Karikó K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005; 23:165 - 75; http://dx.doi.org/10.1016/j.immuni.2005.06.008; PMID: 16111635
- Koski GK, Karikó K, Xu S, Weissman D, Cohen PA, Czerniecki BJ. Cutting edge: innate immune system discriminates between RNA containing bacterial versus eukaryotic structural features that prime for high-level IL-12 secretion by dendritic cells. J Immunol 2004; 172:3989 - 93; PMID: 15034009
- Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci U S A 2012; 109:E2110 - 6; http://dx.doi.org/10.1073/pnas.1209414109; PMID: 22753494
- El-Serag HB, Engels EA, Landgren O, Chiao E, Henderson L, Amaratunge HC, et al. Risk of hepatobiliary and pancreatic cancers after hepatitis C virus infection: A population-based study of U.S. veterans. Hepatology 2009; 49:116 - 23; http://dx.doi.org/10.1002/hep.22606; PMID: 19085911
- Hassan MM, Li D, El-Deeb AS, Wolff RA, Bondy ML, Davila M, et al. Association between hepatitis B virus and pancreatic cancer. J Clin Oncol 2008; 26:4557 - 62; http://dx.doi.org/10.1200/JCO.2008.17.3526; PMID: 18824707
- Tomasiewicz K, Modrzewska R, Lyczak A, Krawczuk G. TT virus infection and pancreatic cancer: relationship or accidental coexistence. World J Gastroenterol 2005; 11:2847 - 9; PMID: 15884138