Abstract
Drosophila is proving to be a valuable model for studying aggressive tumors induced by the combined activation of EGFR and JAK-STAT signaling. Here we summarize some of the most recent data showing that tissue damage and the modulation of common pathway regulators are at the heart tumor progression and metastasis.
Cancer is a complex disease in which a variety of signals contribute to tumor generation and progression. This complexity is increased if we consider how the interaction of the tumor with its cellular environment contributes to the regression or expansion and aggressiveness of the lesion.
Remarkable advances have been made to treat different types of cancer. However, treatments based on the true elimination of tumor cells at early stages of cancer progression would be crucial to improve the yield of successful therapies. Early factors that trigger tumor development can be studied in Drosophila, a genetically tractable model organism.
Given the large evolutionary distances separating Drosophila and humans, flies may seem a bad choice as model system for cancer research. However, experiments done in the last few years proved that tumors can be readily induced in Drosophila by expressing or mutating the same genes involved in human tumors.
More than 68% of the genes involved in human cancer are conserved in Drosophila,Citation1 among others the EGFR and JAK-STAT pathways. The potent Drosophila tool-kit developed to study the involvement of these pathways in normal development is now being used to find out how these genes control cell proliferation and how in some conditions their abnormal regulation induces aggressive tumors. The unparalleled capacity for genetic manipulation in Drosophila permits activating or repressing any gene combination in labeled cells at particular regions. This allows studying how these genes induce over proliferation, metastasis and how the tumorous tissue displaces the normal cells. These techniques also allow the systematic activation of other genes in the tumor in order to identify new molecules that suppress tumor growth and therefore could become drug targets to treat tumors in humans. In this commentary we want to discuss recent developments describing how simultaneous misregulation of the EGFR and the JAK-STAT pathways in Drosophila epithelial cells induce carcinomas, how this carcinomas interact with the cellular microenvironment and how competition between normal and tumorous cells leads to the regression or expansion of the tumor.
It had been observed that activating the EGFR pathway in flies, either by ectopically expressing the receptor or by activating its downstream target Ras (with the constitutively activated RasV12 mutation), can cause benign epithelial tumors. In these tumors the epithelial tissues over proliferate without the cells losing their epithelial character, with clear apical localization of E-cadherin and maintenance of the cell polarityCitation2-Citation4 (). Later work showed that mutation of secondary genes in cells with overactive EGFR pathway could cause over-proliferation and induce aggressive carcinomas that metastasize.Citation2,Citation4 Especially interesting was the interaction found between cell polarity mutations and EGFR pathway activated cells.
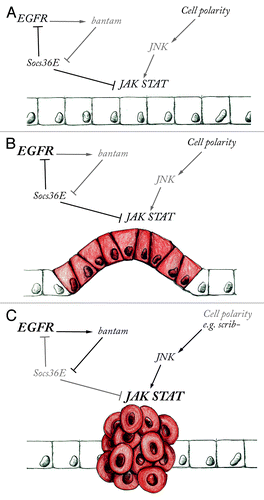
Figure 1. The combined action of EGFR and JAK-STAT signaling results in tumor progression. (A) Epithelial growth in tissues with normal apico-basal cell polarity is controlled by the balanced activity of EGFR, JAK-STAT and other signaling pathways. Negative regulators such as Socs36E control pathway activity by modulating signaling output. (B) When EGFR is over activated (shown in bold), the epithelium proliferates excessively without necessarily causing metastasis. (C) Over proliferation and metastasis are promoted by the combined misregulation of EGFR and high JAK-STAT expression when the signaling balance is broken by the downregulation of Socs36E by bantam miRNA expression or by JNK mediated JAK-STAT activation.
The Scribble (scrib), lethal (2) giant larvae (lgl) and discs-large (dlg) genes encode proteins that form a complex on the basolateral membrane that is necessary to maintain normal epithelial cell polarity. These genes are tumor supressorsCitation5,Citation6 as mutant flies develop epithelial tissue overgrowth that eventually kills the animal (explaining the telling names of these mutations). Surprisingly, instead of forming a tumor, small groups of cells carrying mutations of these epithelial polarity genes are eliminated from the tissue by competition with the surrounding normal cells.
A very different situation arises when mutations on scrib, dlg or lgl occur in Ras activated cells. In this case the double mutant cells survive the competition and become metastaticCitation2,Citation4 proving that, as in humans, tumor aggressiveness results from more than one lesion. To investigate why this gene combination causes such invasiveness, the fly tumors were studied by microarray analysis and it was found that the three upd ligands of the Drosophila JAK-STAT pathway where highly upregulated.Citation7 Studying the functional requirement of JAK-STAT signaling in these tumors was facilitated by the simplicity of the Drosophila pathway that consists of a single receptor (dome), a single JAK kinase and a single STAT transcription factor (Stat92E).Citation8,Citation9 This allowed to find that the tumors caused by activated RasV12 and scrib− mutants were suppressed by expression of a dominant negative JAK-STAT receptor or by mutation of the Stat92E geneCitation7 suggesting that the EGFR tumor becomes much more aggressive by the simultaneous activation of the JAK-STAT pathway. This point was confirmed by the observation that expressing the upd ligands in Ras activated cells results in large metastatic tumors. It was also found that the activation of the upd ligands in scrib− mutant cells with affected polarity was mediated by the JNK pathway activation. Indeed, mutating the Jun kinase gene basket in RasV12 scrib− mutant cells suppressed the metastasis. Interestingly, the same mutation is unable to suppress tumors caused by ectopic upd ligand in RasV12 cells, indicating that the loss of cell polarity activates the JNK pathway that in turn activates JAK-STAT (). Thus, the co-activation of JAK-STAT and the EGFR pathway is ultimately responsible for the aggressive carcinoma as blocking JAK-STAT activation at any level can ameliorate the tumor progression in RasV12 cells.
Although in the above-described experiments the loss of polarity and the activation of the EGFR pathway both happen in the tumor cells, metastatic tumors also appear if the polarity defect is induced in the neighbor cells to those where Ras is active.Citation7 This implies a non-clonal origin of the tumor with cell interactions inducing the metastatic behavior. In this last case the scrib− cells were instrumental for starting the aggressive tumor by inducing the JNK pathway. JNK activation from scrib− mutant cells can spread to the neighboring RasV12 cells that will activate JAK-STAT signaling. Although the scrib− mutant cells eventually disappear due to cell competition, they activated the invasive cocktail of factors that allow the activated RasV12 cells to become metastatic.Citation7
In their recent publication Herranz and collaborators explore how this EGFR JAK-STAT oncogenic activation cocktail may act.Citation10 In this work the authors were investigating Drosophila genes that would increase the proliferation potential of EGFR overexpressing cells. They found that co-expression of EGFR with the bantam microRNA (miRNA), which has been shown to be involved in growth control,Citation11,Citation12 results in massive epithelial overgrowth accompanied by a loss of epithelial polarity that is not observed when the genes are expressed independently. The authors show that expression of EGFR results in activation of Socs36E in the epithelium, and that bantam miRNA exacerbates the EGFR overexpression consequences through the downregulation of Socs36E expression. A similar effect to bantam expression is achieved if the EGFR is coexpressed with a Socs36E RNAi. Thus the activation of Socs36E establishes a brake to EGFR over proliferation and metastasis that is lifted by bantam expression (). In Drosophila, Socs36E is a direct transcriptional target of JAK-STAT and Socs36E has been shown to downregulate EGFR and JAK-STAT signaling.Citation13-Citation16 Herranz and collaborators show that bantam expression or Socs36E downregulation lead to a strong activation of JAK-STAT in EGFR overexpressing cells. Downregulation of the JAK-STAT receptor dome or the Stat92E transcription factor in these metastatic cells results in tumor size normalization, indicating that the EGFR JAK-STAT cocktail is responsible for the aggressive tumorous overgrowth.
Why do EGFR JAK-STAT metastatic tumors displace the normal tissues? A possible explanation comes from the process of cell competition where it has been shown that cells that proliferate faster displace normal neighboring cells that proliferate less.Citation17 Cell competition has been observed to occur among cells with different levels of ribosomal proteins, Myc or Yorkie, but a recent paper provides data suggesting that cells with higher activation of JAK-STAT pathway are more competitive than cells with lower levels.Citation18 The authors showed this in two ways: First, they induced simultaneously a clone of homozygous Stat92E mutant cells and a neighboring clone of wild-type cells and observed that the Stat92E mutant clone gave rise to less daughter cells than the wild type. This was not due to an intrinsic defect in the Stat92E mutant cells, as using established systems that increase cell competitiveness during development (for example when they produce more ribosomal proteins than the wild-type cells) allowed the Stat92E clone to proliferate normally. Second, they showed that activation of the JAK kinase has the complementary effect, making cells proliferate more efficiently and out-compete their wild-type neighbors. Mechanistically, cell competition is due to the JAK activated cells inducing apoptosis on the neighboring wild-type cells, allowing the first to occupy a larger fraction of the tissue. Despite the over-proliferation the final tissue is not overtly aberrant in shape, indicating that the competitive advantage is checked by the normal developmental and homeostatic controls. Although it is not yet clear how the activation of the JAK-STAT pathway transforms cells into super competitors, it is an interesting avenue of research that may help understanding how the EGFR JAK-STAT cocktail induces cells to become highly invasive.
Another recent paper demonstrates that cells require Stat for competitive fitness to eliminate neighboring scrib− cells.Citation19 scrib− clones are eliminated by cell competition when surrounded by normal cells. However, when scrib− cells are abutting Stat92E− cells, the elimination does not occur and instead the scrib− clone overgrows. Thus, these findings show that Stat protects the tissue from invasiveness.
Are these observations relevant for human metastasis? The conservation of the pathways and regulatory elements suggest they are, and some observations indicate there is a deeper conservation in the metastatic processes that goes further than those unveiled by sequence conservation. Although Drosophila Socs36E has got in SOCS5 a human ortholog, there is no homology between bantam miRNA and any human miRNA. SOCS5 behaves as a tumor suppressor in an EGFR/RAS dependent cell transformation assay.Citation10 Recent work shows that a similar genetic interaction to that of bantam and Socs36E, occurs in vertebrate tumor endothelial co-cultures with SOCS5 and the miR-9 miRNA.Citation20 miR-9 is secreted from melanoma cells but not from normal skin melanocites and is taken up by endothelial cells where SOCS5 is downregulated and as a result STAT1 becomes activated. This activation induces the migration of endothelial cells toward the tumor. Downregulation of JAK activation reverts the endothelial growth. This mechanism of regulation reveals a functional similarity between Drosophila and humans even though there are clear sequence differences between the miRNAs involved and even between the target tissues.
These studies and the functional similarities they uncover should make us aware that, as humans, we should keep a selfish interest on the developments in fly tumor research.
References
- Rubin GM, Yandell MD, Wortman JR, Gabor Miklos GL, Nelson CR, Hariharan IK, et al. Comparative genomics of the eukaryotes. Science 2000; 287:2204 - 15; http://dx.doi.org/10.1126/science.287.5461.2204; PMID: 10731134
- Brumby AM, Richardson HE. scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J 2003; 22:5769 - 79; http://dx.doi.org/10.1093/emboj/cdg548; PMID: 14592975
- Karim FD, Rubin GM. Ectopic expression of activated Ras1 induces hyperplastic growth and increased cell death in Drosophila imaginal tissues. Development 1998; 125:1 - 9; PMID: 9389658
- Pagliarini RA, Xu T. A genetic screen in Drosophila for metastatic behavior. Science 2003; 302:1227 - 31; http://dx.doi.org/10.1126/science.1088474; PMID: 14551319
- Bilder D, Li M, Perrimon N. Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science 2000; 289:113 - 6; http://dx.doi.org/10.1126/science.289.5476.113; PMID: 10884224
- Humbert P, Russell S, Richardson H. Dlg, Scribble and Lgl in cell polarity, cell proliferation and cancer. Bioessays 2003; 25:542 - 53; http://dx.doi.org/10.1002/bies.10286; PMID: 12766944
- Wu M, Pastor-Pareja JC, Xu T. Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature 2010; 463:545 - 8; http://dx.doi.org/10.1038/nature08702; PMID: 20072127
- Arbouzova NI, Zeidler MP. JAK/STAT signalling in Drosophila: insights into conserved regulatory and cellular functions. Development 2006; 133:2605 - 16; http://dx.doi.org/10.1242/dev.02411; PMID: 16794031
- Hombría JC, Brown S. The fertile field of Drosophila Jak/STAT signalling. Curr Biol 2002; 12:R569 - 75; http://dx.doi.org/10.1016/S0960-9822(02)01057-6; PMID: 12194841
- Herranz H, Hong X, Hung NT, Voorhoeve PM, Cohen SM. Oncogenic cooperation between SOCS family proteins and EGFR identified using a Drosophila epithelial transformation model. Genes Dev 2012; 26:1602 - 11; http://dx.doi.org/10.1101/gad.192021.112; PMID: 22802531
- Brennecke J, Hipfner DR, Stark A, Russell RB, Cohen SM. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell 2003; 113:25 - 36; http://dx.doi.org/10.1016/S0092-8674(03)00231-9; PMID: 12679032
- Hipfner DR, Weigmann K, Cohen SM. The bantam gene regulates Drosophila growth. Genetics 2002; 161:1527 - 37; PMID: 12196398
- Almudi I, Stocker H, Hafen E, Corominas M, Serras F. SOCS36E specifically interferes with Sevenless signaling during Drosophila eye development. Dev Biol 2009; 326:212 - 23; http://dx.doi.org/10.1016/j.ydbio.2008.11.014; PMID: 19083999
- Karsten P, Häder S, Zeidler MP. Cloning and expression of Drosophila SOCS36E and its potential regulation by the JAK/STAT pathway. Mech Dev 2002; 117:343 - 6; http://dx.doi.org/10.1016/S0925-4773(02)00216-2; PMID: 12204282
- Rawlings JS, Rennebeck G, Harrison SM, Xi R, Harrison DA. Two Drosophila suppressors of cytokine signaling (SOCS) differentially regulate JAK and EGFR pathway activities. BMC Cell Biol 2004; 5:38; http://dx.doi.org/10.1186/1471-2121-5-38; PMID: 15488148
- Stec WJ, Zeidler MP. Drosophila SOCS Proteins. J Signal Transduct 2011; 2011:894510; http://dx.doi.org/10.1155/2011/894510; PMID: 22203896
- Baker NE. Cell competition. Curr Biol 2011; 21:R11 - 5; http://dx.doi.org/10.1016/j.cub.2010.11.030; PMID: 21215926
- Rodrigues AB, Zoranovic T, Ayala-Camargo A, Grewal S, Reyes-Robles T, Krasny M, et al. Activated STAT regulates growth and induces competitive interactions independently of Myc, Yorkie, Wingless and ribosome biogenesis. Development 2012; 139:4051 - 61; http://dx.doi.org/10.1242/dev.076760; PMID: 22992954
- Schroeder MC, Chen CL, Gajewski K, Halder G. A non-cell-autonomous tumor suppressor role for Stat in eliminating oncogenic scribble cells. Oncogene 2012; In press http://dx.doi.org/10.1038/onc.2012.476; PMID: 23108407
- Zhuang G, Wu X, Jiang Z, Kasman I, Yao J, Guan Y, et al. Tumour-secreted miR-9 promotes endothelial cell migration and angiogenesis by activating the JAK-STAT pathway. EMBO J 2012; 31:3513 - 23; http://dx.doi.org/10.1038/emboj.2012.183; PMID: 22773185