Abstract
Sequence-specific binding of STAT1 (signal transducer and activator of transcription 1) transcription factor to palindromic promoter elements, termed γ-activated sites (GAS), and an extended spatial reorientation between two dimer configurations are key events in the interferon signaling pathway. Although the DNA-binding domain of STAT1 is engaged in both processes, how the conformational change from a parallel to an antiparallel dimer configuration affects cytokine-induced target gene activation is unknown. In order to study the impact of the conformational shift on gene expression, we generated a STAT1 point mutant with a structurally altered architecture of the DNA-binding domain and characterized the resulting mutant (F364A) in cells stimulated with interferon-γ. Here, we report that substituting alanine for phenylalanine at position 364 resulted in reduced affinity to GAS sites and, additionally, a decreased dephosphorylation rate by the inactivating Tc45 phosphatase. The mutant had no defect in cooperative DNA binding and displayed normal kinetics of interferon-γ-induced nuclear accumulation, despite its elevated level of tyrosine phosphorylation. By assessing the transcriptional activity of the mutant, we found a strikingly robust expression of known interferon-γ-driven target genes, indicating that an impaired stability of the antiparallel dimer configuration can compensate for a reduced affinity to GAS sites. However, the mutant followed changes in ligand-induced receptor activation more slowly than the wild-type molecule, as demonstrated by its elevated phospho-STAT1 concentration following addition of the kinase inhibitor staurosporine to interferon-pretreated cells. This finding showed that the DNA-binding mutant F364A had partially lost its ability to terminate signal transmission rapidly. Thus, the coupling of high-affinity GAS binding to a rapid exchange from a parallel to an antiparallel dimer conformation is not necessarily required for optimal signal amplification, but rather allows for a dynamic signal response and ensures high adaptability to changes in signal input.
Introduction
Extracellular signaling from the plasma membrane to the nucleus is a hallmark of metazoan animals and frequently entails the activation of transcription factors at ligand-bound receptors.Citation1,Citation2 The activated transcription factors then translocate through nuclear pore complexes to the nucleus and bind sequence-specifically to promoter elements of their target genes. Signal transduction via signal transducers and activators of transcription (STATs) is a paradigmatic example for such direct signaling from cell surface receptors to the nuclear compartment.Citation1-Citation5 Upon stimulation of cells with cytokines, members of the STAT protein family are phosphorylated on a critical tyrosine residue by receptor-associated Janus kinases (JAKs) and subsequently move as transcriptionally active dimers into the nucleus, where they recognize palindromic motifs in the promoter regions of cytokine-driven target genes.Citation6-Citation13 Among the seven human family members, all of which are activated by a diverse set of cytokines and growth factors, the STAT1 pathway transmits interferon (IFN) signals required for numerous immune-modulatory functions. Interferons of type I (IFNα/β) act through binding of STAT1/STAT2/p48 to IFN-stimulated response elements (ISRE), whereas exposure of cells to IFNγ (type II) induces the formation of STAT1 homodimers and their recruitment to GAS (γ-activated sites) sequences.Citation14-Citation16
Although there is evidence of an antiparallel dimer conformation before cytokine exposure, stimulation of cells with IFNγ resulted in the formation of parallel STAT1 homodimers by reciprocal interactions between the SH2 domain on one monomer and the phosphorylated tyrosine residue 701 on the other monomer.Citation8 Beside the carrier-dependent nuclear import of phospho-dimers, also unphosphorylated STAT1 molecules shuttle between the cytoplasm and the nucleus in a process that does not require carriers or metabolic energy, but direct interactions with nucleoporins located in the nuclear pore complexes.Citation17,Citation18 This constitutive nucleocytoplasmic translocation of unphosphorylated STAT1 occurs both in resting and IFNγ-stimulated cells. Tyrosine-phosphorylated STAT1 dimers must first become dephosphorylated by nuclear phosphatases until they are capable of leaving the nuclear compartment to participate in additional rounds of cytosolic reactivation and subsequent nuclear re-import.Citation19 Continuous nucleocytoplasmic shuttling in and out of the nucleus is required for linkage of receptor-associated activation to target gene finding in order to maintain a constant level of transcriptionally active STAT1 in the nucleus. This rather complex regulation of the STAT1 signal pathway ensures that time-dependent changes in receptor-based processes at the plasma membrane are rapidly transmitted into a transcriptional response.
The STAT1 DNA-binding domain plays a key role in linking transport processes to GAS recognition and gene activation. First, it contains a non-classical and non-transferable nuclear localization signal (NLS) with unique features which is exposed only on the surface of activated STAT1 dimers. Binding of importin α5 to the dimer-specific NLS (dsNLS) facilitates nuclear import of phosphorylated STAT1 homodimers in complexation with importin β.Citation17,Citation20-Citation24 Second, a β sheet arrangement running parallel to the long axis of the immunoglobulin fold faces the DNA double helix at one end, where it makes direct contact with the phosphodiester backbone and a limited number of bases involved in GAS recognition.Citation25 Third, in the context of a spatial reorientation of the parallel STAT1 dimer to an antiparallel form, the DNA-binding domain of one monomer interacts reciprocally with the phenylalanine residue 172 in the coiled-coil domain of the other monomer and, thereby, promotes conformational dimer rearrangement.Citation26-Citation29
Given the central role of the DNA-binding domain in the regulation of the activation-inactivation cycle of STAT1, we generated a point mutant with impaired DNA binding and stabilization of the antiparallel dimer configuration and characterized its phenotype in the light of the systemic design of the IFNγ pathway. This mutant allowed us to study the physiological significance of high-affinity GAS binding and a rapid exchange between the two different dimer configurations in terms of signal strength and duration.
Results
Substitution of phenylalanine 364 leads to hyper-phosphorylation of STAT1
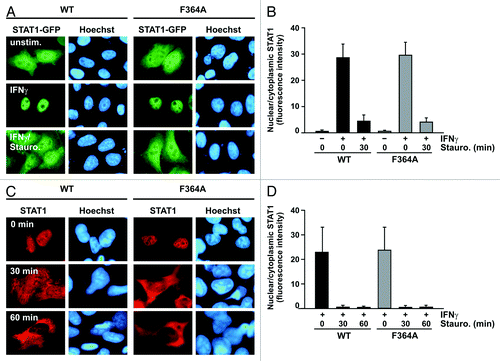
In order to generate DNA-binding mutants of the transcription factor STAT1, we substituted alanine residues for amino acids in the DNA-binding domain and tested the phenotype of the resulting mutants. We identified a conserved hydrophobic amino acid residue in position 364 of the STAT1 molecule, which is a phenylalanine, and in the homologous positions of other STAT family members either isoleucine (in STAT2, STAT3, STAT4, STAT5A and STAT5B) or methionine (in STAT6). The crystal structure of DNA-bound STAT1 revealed that the phenylalanine residue 364 resides underneath a surface pocket in the center of the DNA-binding domain (). Its aromatic side chain appears to contribute to the gross structural alignment of this domain, and is not engaged in direct contacts to DNA. Replacement of the phenylalanine residue 364 by alanine generated a STAT1 mutant, which is expressed at normal levels in transfected HeLa or STAT1-reconstituted U3A cells ( and ). Interestingly, stimulation of cells with 5 ng/ml of interferon-γ (IFNγ) led to an elevated tyrosine phosphorylation of the F364A mutant as compared with the wild-type protein both in HeLa and U3A cells ( and data not shown). Addition of staurosporine, a potent inhibitor of JAK tyrosine kinases, to IFNγ-prestimulated HeLa cells resulted in prolonged and elevated levels of tyrosine-phosphorylated STAT1-F364A. After 45 min of exposure to staurosporine, the percentage of the initial phospho-STAT1/STAT1 ratio was 64.0 ± 23.8% for the mutant vs. 27.1 ± 8.2% for the wild-type molecule (p = 0.026), demonstrating that the F364A mutation cell-independently caused a significant hyper-phosphorylation of STAT1 (). Moreover, in HeLa cells expressing the GFP fusion protein of STAT1-F364A, the non-recombinant endogenous STAT1 was also protected from rapid inactivation and persisted longer in a tyrosine-phosphorylated state than in cells co-expressing the GFP-tagged wild-type protein. There was a 2.3-fold increase in the specific tyrosine phosphorylation rate of native STAT1 in cells co-expressing F364A-GFP as compared with cells co-expressing WT-GFP (standard deviation 2.0, n = 4, p = 0.029, see ).
Figure 1. Localization of phenylalanine residue 364 in the DNA-binding domain of STAT1 transcription factor. (A) Depicted is a ribbon diagram of the crystal structure of a truncated monomeric STAT1 molecule bound to DNA with the DNA-binding domain colored in yellow and the residue F364 in magenta.Citation25 (B) A closer view of the ribbon representation demonstrates the spatial orientation of the aromatic ring of F364 in relation to the residues Q340, G384 and Q408 (colored in blue), which are part of a pocket structure on the surface of the protein required for the interaction with the partner monomer in the antiparallel dimer conformation (not shown).
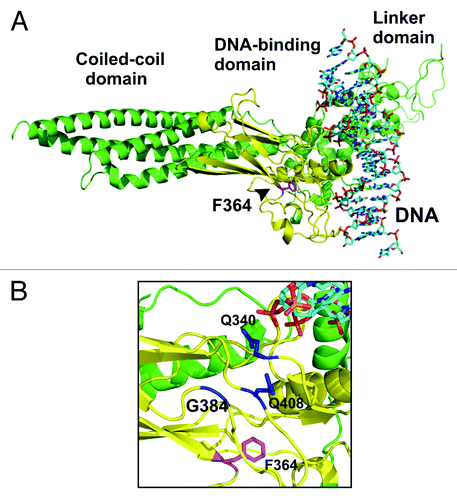
Figure 2. Substitution of alanine for phenylalanine at position 364 resulted in prolonged and elevated tyrosine phosphorylation of mutant STAT1 in cells stimulated with interferon-γ. (A–D) Equal cell numbers of HeLa cells expressing either wild-type (WT) or mutant STAT1-F364A, both tagged with green fluorescent protein (GFP), were exposed to IFNγ (2.5 ng/ml and 5 ng/ml, respectively) for 45 min before the kinase inhibitor staurosporine (500 nM) was added to the cells for the indicated times. Shown are the time courses of tyrosine phosphorylation as examined by western blotting using a STAT1-specific phospho-tyrosine antibody (αp-STAT1, top panel) and the same membranes after stripping and re-probing with the pan-STAT1 antibody C-24 (αSTAT1, bottom panel). Phosphorylation was monitored in whole cell lysates (A–C) as well as in cytosolic and nuclear extracts (D). The top arrowhead marks recombinant GFP-tagged STAT1 (S1-GFP) and the bottom arrowhead indicates endogenous STAT1 (S1). (B) Quantification of four independent western blotting experiments showing the ratio of phospho-STAT1 to total STAT1 normalized to STAT1-WT-expressing cells prior to adding staurosporine. Means and standard deviations for each time point are expressed. (E and F) Hyper-phosphorylation of STAT1-F364A did not result from decreased DNA binding. Cells expressing either F364A or DNAminus (V426D/T427D) were pretreated for 45 min with IFNγ and subsequently exposed to staurosporine for the indicated times. Cellular extracts were normalized to equal ratios of phosphorylated to total cellular STAT1 at baseline and loaded onto a SDS-gel. Shown are a representative western blot result (E) and a diagram depicting the decline in phospho-STAT1/STAT1 ratios for the two mutants [(F), means from two independent experiments]. The upper bands on each blot mark recombinant GFP-tagged STAT1, whereas the lower bands correspond to native STAT1.
![Figure 2. Substitution of alanine for phenylalanine at position 364 resulted in prolonged and elevated tyrosine phosphorylation of mutant STAT1 in cells stimulated with interferon-γ. (A–D) Equal cell numbers of HeLa cells expressing either wild-type (WT) or mutant STAT1-F364A, both tagged with green fluorescent protein (GFP), were exposed to IFNγ (2.5 ng/ml and 5 ng/ml, respectively) for 45 min before the kinase inhibitor staurosporine (500 nM) was added to the cells for the indicated times. Shown are the time courses of tyrosine phosphorylation as examined by western blotting using a STAT1-specific phospho-tyrosine antibody (αp-STAT1, top panel) and the same membranes after stripping and re-probing with the pan-STAT1 antibody C-24 (αSTAT1, bottom panel). Phosphorylation was monitored in whole cell lysates (A–C) as well as in cytosolic and nuclear extracts (D). The top arrowhead marks recombinant GFP-tagged STAT1 (S1-GFP) and the bottom arrowhead indicates endogenous STAT1 (S1). (B) Quantification of four independent western blotting experiments showing the ratio of phospho-STAT1 to total STAT1 normalized to STAT1-WT-expressing cells prior to adding staurosporine. Means and standard deviations for each time point are expressed. (E and F) Hyper-phosphorylation of STAT1-F364A did not result from decreased DNA binding. Cells expressing either F364A or DNAminus (V426D/T427D) were pretreated for 45 min with IFNγ and subsequently exposed to staurosporine for the indicated times. Cellular extracts were normalized to equal ratios of phosphorylated to total cellular STAT1 at baseline and loaded onto a SDS-gel. Shown are a representative western blot result (E) and a diagram depicting the decline in phospho-STAT1/STAT1 ratios for the two mutants [(F), means from two independent experiments]. The upper bands on each blot mark recombinant GFP-tagged STAT1, whereas the lower bands correspond to native STAT1.](/cms/asset/6a49bd16-b723-4fd5-8252-b9d55328ed58/kjks_a_10923576_f0002.gif)
Figure 3. The STAT1 mutant F364A shows unaltered kinetics of interferon-γ-induced nuclear accumulation. (A and B) Treatment of IFNγ-prestimulated HeLa cells with staurosporine led to a collapse of nuclear accumulation of GFP-tagged STAT1-F364A and restored its pancellular resting distribution with similar kinetics to the wild-type protein. Cells were either left untreated (− IFNγ) or treated for 45 min with 5 ng/ml IFNγ (+ IFNγ) before staurosporine (500 nM) was added for additional 0 and 60 min, respectively. (A) Microscopic images show the intracellular localization of STAT1-GFP and the Hoechst-stained nuclei. (B) The corresponding histogram demonstrates the nuclear-to-cytosol fluorescence intensity ratios with bars representing means and standard deviations. (C and D) Treatment with staurosporine for the indicated times resulted in a normal break-down of nuclear accumulation of untagged STAT1, which was independent of the presence of the F364A mutation. Immunocytochemical stainings using a STAT1-specific primary and Cy3-labeled secondary antibody show the rapid collapse of nuclear accumulation in IFNγ-prestimulated reconstituted U3A cells resulting from the exposure to staurosporine. Fluorescence microscopical images (C) demonstrate nuclear and cytoplasmic STAT1 concentrations in representative cells and the histogram (D) shows a quantitative analysis including means and standard deviations. The experiments were performed at least three times with similar results.
When HeLa cells expressing STAT1-F364A-GFP were treated with half of the IFNγ concentration (2.5 ng/ml) used for stimulation of WT-GFP-expressing cells, the tyrosine phosphorylation levels were similar in both variants as judged by western blotting, confirming that the mutation was associated with a hyper-phosphorylated phenotype (). In summary, substitution of a critical phenylalanine residue in the DNA-binding domain not only prolonged tyrosine phosphorylation of the recombinant STAT1-GFP variant, but also partially suppressed dephosphorylation of the co-expressed native STAT1.
Next we investigated whether the increased tyrosine phosphorylation of STAT1-F364A simply resulted from an impaired nuclear import of phospho-dimers which might expose the mutant to the higher kinase and lower phosphatase activities in the cytoplasm as compared with the nucleus. In order to determine the intracellular compartmentalization of hyper-phosphorylated STAT1-F364A in IFNγ-treated cells, we performed cellular fractionation experiments with cytosolic and nuclear extracts. As shown in , there was a detectable difference in the cytoplasmic amount of tyrosine-phosphorylated STAT1 between wild-type and mutant protein, as judged by western blotting using an antibody that specifically recognized tyrosine-phosphorylated STAT1. Similarly also in nuclear extracts, the amount of phosphorylated STAT1 differed substantially, as higher phospho-protein levels were observed for the mutant than for the wild-type molecule. Stripping and re-probing the same membrane with a pan-STAT1 antibody demonstrated that the intracellular pools of STAT1 did not differ very much between the two STAT1 variants. Thus, hyper-phosphorylation of the F364A mutant was associated with both increased cytosolic and nuclear concentrations of phospho-STAT1, thus excluding the possibility that the underlying mechanism of the mutant resulted from a reduced nuclear import rate due to a dysfunctional dsNLS.
Furthermore, we excluded that the elevated phosphorylation level of F364A was the consequence of an altered DNA binding. The DNAminus mutant, which failed to bind to DNA due to the replacement of two DNA-contacting residues by negatively charged aspartic acid (V426D/T427D),Citation19 showed a similar phosphorylation level as the wild-type protein. In contrast, the STAT1-F364A mutant was partially resistant against the inhibitory effect of staurosporine ().
Unaltered cytokine-induced nuclear accumulation kinetics despite elevated tyrosine phosphorylation
It has previously been reported that nuclear accumulation of STAT1 following stimulation of cells with IFNγ reflects the retention of phosphorylated STAT1 dimers on genomic DNA.Citation19 Therefore, we hypothesized that the prolonged and elevated tyrosine phosphorylation of STAT1-F364A was associated with a pronounced cytokine-induced nuclear accumulation. However, we found that the collapse of nuclear accumulation upon staurosporine exposure followed the same kinetics irrespective of the presence of the F364A mutation (). Thirty minutes after addition of the kinase inhibitor to IFNγ-pretreated HeLa cells, the predominant nuclear localization of the two GFP-adducts of STAT1 (mean peak nuclear/cytoplasmic fluorescence intensity for WT 27.5 ± 5.4 vs. F364A 27.0 ± 7.6, p > 0.05) was no longer visible (WT 4.3 ± 2.2 vs. F364A 4.1 ± 1.4, p > 0.05) and the cells had nearly restored their pancellular STAT1 resting distribution (WT 0.6 ± 0.1 vs. F364A 0.6 ± 0.1, p > 0.05). Corroborating this observation, in immunocytochemical staining of reconstituted U3A cells expressing untagged recombinant STAT1, we again detected that the break-down of nuclear accumulation induced by staurosporine did not significantly differ between the two STAT1 variants (WT 0.4 ± 0.2 vs. F364A 0.5 ± 0.2, p > 0.05) (). Furthermore, 45 min after adding IFNγ to the cells, the nuclear build-up of wild-type STAT1 was within the same range as that of the mutant (WT 22.9 ± 10.2 vs. F364A 23.7 ± 9.3, p > 0.05).
Decreased binding affinity of STAT1-F364A to GAS sites
The striking finding that the phenylalanine mutant displayed unaltered nuclear accumulation behavior despite its hyper-phosphorylation suggested an overlooked inhibitory mechanism, which might compensate for the elevated intracellular pool of phosphorylated STAT1. Therefore, we tested for changes in DNA-binding affinity by means of electrophoretic bandshift assays using radioactively labeled DNA probes and whole cell extracts from reconstituted U3A cells. As shown in , the sequence-specific binding of the mutant to a DNA probe containing a single GAS site, termed M67, was significantly lower than for the wild-type protein. When equal amounts of phospho-protein as determined by immunoblotting were densitometrically measured for their binding activity to M67 by means of EMSA, the mutant showed a significantly reduced DNA-binding, which was approximately half that of STAT1-WT (49 ± 5.0%; p = 0.032, ). Furthermore, binding to a single GAS site adjacent to a non-GAS site (GAS-nonGAS) was critically impaired, as, in contrast to its wild-type counterpart, specific binding was hardly detectable (). Competition experiments with excess unlabeled DNA demonstrated that the F364A mutant displayed a rapid dissociation from DNA similar to wild-type STAT1 (). Using labeled oligonucleotides with a tandem array of GAS sites (2 × GAS), tetrameric complexes of both wild-type and mutant STAT1, but not dimeric STAT1 resisted challenge with excess unlabeled M67 (for the mutant the mean ratio of dimeric/tetrametric STAT1 before and after competition was 62.7 ± 8.9% vs. 2.2 ± 0.8%, respectively, p < 0.001), demonstrating that STAT1-F364A exhibited tetramer stability, indicative of normal cooperative DNA binding ().
Figure 4. Decreased DNA-binding affinity of the STAT1 mutant F364A. (A) Gelshift experiment showing the reduced DNA-binding affinity of the mutant as compared with the wild-type protein. Before being loaded onto a native polyacrylamide gel, cell lysates from IFNγ-stimulated U3A cells (5 ng/ml) expressing either wild-type or mutant STAT1 were incubated with radioactively labeled DNA containing M67, GAS-nonGAS or 2 × nonGAS probes. The asterisk marks a non-specific band and the arrowhead at the right margin of the gel corresponds to STAT1. (B) Densitometric assessment of the DNA-binding activity of mutant and wild-type STAT1 normalized to equal amounts of phospho-protein as determined by immunoblotting (p = 0.032). (C and D) Comparison of the dissociation rate between STAT1-WT and F364A on a single STAT binding site (M67) as determined by electrophoretic mobility shift assay (EMSA). Whole cell extracts from IFNγ-prestimulated U3A cells were incubated with radioactively labeled DNA for 15 min and, subsequently, a 750-fold molar excess of unlabeled M67 was added for the durations indicated, before the samples were loaded onto a polyacrylamide gel. In the first lane, anti-STAT1 antibody C-24 was present in the EMSA reaction used for identification of STAT1-DNA complexes which are marked with an arrowhead. (E and F) Normal cooperative DNA binding due to tetramer stabilization of the F364A mutant. Extracts from an equal number of IFNγ-stimulated U3A cells expressing either wild-type or mutant STAT1 (5 μl in each lane) were incubated in vitro with [32P]-labeled DNA containing a tandem GAS site (2 × GAS). The reactions were either left unchallenged (-) or challenged for 30 min with a 750-fold excess of a single, unlabeled GAS site (+ Competition). (E) Autoradiography including, in lane 1, a supershift with 20 ng of STAT1 antibody C-24 added to the reaction. At the margin of the EMSA gel, the positions of tetrameric (T) and dimeric (D) STAT1 are marked with arrowheads. (F) Graphic representation showing the densitometrically measured signal intensities of dimeric-to-tetrameric STAT1 bound to 2 × GAS before (− Comp.) and after (+ Comp.) competition as determined from three independent experiments (p < 0.001). (G and H) Demonstration of the DNA-binding defect of the F364A mutant in HeLa cells expressing recombinant STAT1-NES-GFP, which coded for a transferable nuclear export signal (NES) situated between the cDNAs for full-length STAT1 and GFP. Cells expressing wild-type STAT1-NES-GFP or the respective F364A variant thereof were either left untreated or stimulated with 5 ng/ml of IFNγ in the presence or absence of 10 ng/ml of leptomycin B (LMB). Epifluorescence microscopic images of representative cells under the various stimulation conditions (G) and a quantitative analysis of the cytoplasmic/nuclear signal intensity ratios (H) are shown.
![Figure 4. Decreased DNA-binding affinity of the STAT1 mutant F364A. (A) Gelshift experiment showing the reduced DNA-binding affinity of the mutant as compared with the wild-type protein. Before being loaded onto a native polyacrylamide gel, cell lysates from IFNγ-stimulated U3A cells (5 ng/ml) expressing either wild-type or mutant STAT1 were incubated with radioactively labeled DNA containing M67, GAS-nonGAS or 2 × nonGAS probes. The asterisk marks a non-specific band and the arrowhead at the right margin of the gel corresponds to STAT1. (B) Densitometric assessment of the DNA-binding activity of mutant and wild-type STAT1 normalized to equal amounts of phospho-protein as determined by immunoblotting (p = 0.032). (C and D) Comparison of the dissociation rate between STAT1-WT and F364A on a single STAT binding site (M67) as determined by electrophoretic mobility shift assay (EMSA). Whole cell extracts from IFNγ-prestimulated U3A cells were incubated with radioactively labeled DNA for 15 min and, subsequently, a 750-fold molar excess of unlabeled M67 was added for the durations indicated, before the samples were loaded onto a polyacrylamide gel. In the first lane, anti-STAT1 antibody C-24 was present in the EMSA reaction used for identification of STAT1-DNA complexes which are marked with an arrowhead. (E and F) Normal cooperative DNA binding due to tetramer stabilization of the F364A mutant. Extracts from an equal number of IFNγ-stimulated U3A cells expressing either wild-type or mutant STAT1 (5 μl in each lane) were incubated in vitro with [32P]-labeled DNA containing a tandem GAS site (2 × GAS). The reactions were either left unchallenged (-) or challenged for 30 min with a 750-fold excess of a single, unlabeled GAS site (+ Competition). (E) Autoradiography including, in lane 1, a supershift with 20 ng of STAT1 antibody C-24 added to the reaction. At the margin of the EMSA gel, the positions of tetrameric (T) and dimeric (D) STAT1 are marked with arrowheads. (F) Graphic representation showing the densitometrically measured signal intensities of dimeric-to-tetrameric STAT1 bound to 2 × GAS before (− Comp.) and after (+ Comp.) competition as determined from three independent experiments (p < 0.001). (G and H) Demonstration of the DNA-binding defect of the F364A mutant in HeLa cells expressing recombinant STAT1-NES-GFP, which coded for a transferable nuclear export signal (NES) situated between the cDNAs for full-length STAT1 and GFP. Cells expressing wild-type STAT1-NES-GFP or the respective F364A variant thereof were either left untreated or stimulated with 5 ng/ml of IFNγ in the presence or absence of 10 ng/ml of leptomycin B (LMB). Epifluorescence microscopic images of representative cells under the various stimulation conditions (G) and a quantitative analysis of the cytoplasmic/nuclear signal intensity ratios (H) are shown.](/cms/asset/144a64f5-5110-435e-85f8-b1d6de8b7db5/kjks_a_10923576_f0004.gif)
To confirm that the F364A mutant had indeed a complex phenotype balancing hyper-phosphorylation and impaired DNA-binding affinity, we used a recombinant plasmid coding for a STAT1 mutant with enhanced nuclear export velocity. The resulting transcript exposed a STAT1-specific nuclear export signal (NES) between the STAT1 sequence and the C-terminal fused GFP domain. As expected, the intracellular localization of the resulting STAT1-NES-GFP mutant differed dramatically from wild-type STAT1-GFP in its predominant cytoplasmic distribution before and after 45 min of IFNγ stimulation (, compare with ). Both in the presence and absence of cytokine stimulation, the nuclei of STAT1-NES-expressing cells were depleted of GFP epifluorescence (cytoplasmic/nuclear fluorescence intensity in resting cells expressing WT 11.8 ± 2.0 vs. F364A 9.9 ± 3.8, p > 0.05). However, combined treatment with IFNγ and the CRM1 exportin inhibitor leptomycin B (LMB) completely restored defective nuclear accumulation of STAT1-NES-GFP. In contrast, substitution of an alanine residue in position 364 for phenylalanine generated a STAT1-NES mutant that failed to accumulate in the nucleus upon combined stimulation of cells with IFNγ and LMB (cytoplasmic/nuclear fluorescence intensity 11.9 ± 4.2 as compared with 0.04 ± 0.02 for WT, p < 0.001). Thus, in the context of the NES adduct, the additionally introduced F364A mutation resulted in an opposite accumulation behavior, since the reduced binding affinity of the mutant to genomic DNA could not compete with the artificially enhanced nuclear export rate.
Diminished inactivation of tyrosine-phosphorylated STAT1-F364A by Tc45 phosphatase
The hyper-phosphorylated status of the mutant in cytokine-stimulated cells may result from elevated tyrosine phosphorylation by receptor-associated kinases and/or impaired dephosphorylation by STAT1-inactivating phosphatases. To test these hypotheses, whole cell extracts from untreated U3A cells expressing either wild-type or mutant STAT1 were incubated in vitro with recombinant JAK2 kinase or epidermal growth factor receptor (EGFR), respectively. The results showed that STAT1-F364A was normally phosphorylated by JAK2 () and EGFR (). When we incubated extracts from IFNγ-pretreated U3A cells with purified Tc45, the nuclear isoform of the T-cell protein-tyrosine phosphatase,Citation30 it was found that the mutant was partially protected against the catalytic attack of the inactivating phosphatase (p < 0.034, ). Thus, the inherent ability of STAT1-F364A to become hyper-phosphorylated in response to an interferon-γ stimulus can be explained by a hampered interaction with Tc45 phosphatase.
Figure 5. Phosphorylated STAT1-F364A is partially resistant against inactivation by Tc45 phosphatase. (A–D) In vitro phosphorylation assays demonstrate unaltered tyrosine phosphorylation of STAT1-F364A. Whole cell extracts from reconstituted U3A cells expressing either STAT1-WT or -F364A (10 μl in each reaction) were incubated with 40 ng of recombinant JAK2 kinase (A and B) or 20 ng of EGF receptor [EGFR, (C and D)] and incorporation of phosphate in STAT1 was monitored with time by means of western blotting. Statistical analyses revealed no significant difference in the phosphorylation kinetics between wild-type and mutant STAT1 (p > 0.05). (E and F) STAT1-F364A is partially protected against the attack of the inactivating phosphatase, as revealed by an in vitro dephosphorylation assay. Extracts from IFNγ-prestimulated U3A cells (10 μl each) were incubated with 2 U of the STAT1-specific Tc45 phosphatase and tyrosine dephosphorylation was followed for 30 min. Shown are a representative western blot result (E) and a quantitative depiction (F) of the specific tyrosine phosphorylation (phosphotyrosine signal divided by total STAT1 signal) with bars expressing means and standard deviations. Significant differences between wild-type and mutant STAT1 from five independent experiments are indicated with asterisks (p = 0.034 and p = 0.017, respectively). (G) An electrophoretic mobility shift assay demonstrates the exchange of monomers between STAT1 dimers. Shown is a representative gelshift result using cellular extracts from U3A cells expressing either GFP-tagged or untagged STAT1 bound to M67 DNA. The identity of the bands corresponding to STAT1 (marked with arrowheads) was confirmed by the absence of or reduction in DNA-binding activity in IFNγ-stimulated Y701F- (lane 1) and unstimulated WT-expressing cells (lane 2) as well as from supershift reactions using either a STAT3- (lanes 3 and 5) or STAT1-recognizing antibody (lanes 4 and 6). In lanes 7–10, similar amounts of GFP-tagged and untagged homodimers were either immediately mixed and incubated together for 45 min (lanes 8 and 10) or incubated separately for 45 min before being loaded together onto the gel (lanes 7 and 9). The asterisk at the right margin marks a non-specific band.
![Figure 5. Phosphorylated STAT1-F364A is partially resistant against inactivation by Tc45 phosphatase. (A–D) In vitro phosphorylation assays demonstrate unaltered tyrosine phosphorylation of STAT1-F364A. Whole cell extracts from reconstituted U3A cells expressing either STAT1-WT or -F364A (10 μl in each reaction) were incubated with 40 ng of recombinant JAK2 kinase (A and B) or 20 ng of EGF receptor [EGFR, (C and D)] and incorporation of phosphate in STAT1 was monitored with time by means of western blotting. Statistical analyses revealed no significant difference in the phosphorylation kinetics between wild-type and mutant STAT1 (p > 0.05). (E and F) STAT1-F364A is partially protected against the attack of the inactivating phosphatase, as revealed by an in vitro dephosphorylation assay. Extracts from IFNγ-prestimulated U3A cells (10 μl each) were incubated with 2 U of the STAT1-specific Tc45 phosphatase and tyrosine dephosphorylation was followed for 30 min. Shown are a representative western blot result (E) and a quantitative depiction (F) of the specific tyrosine phosphorylation (phosphotyrosine signal divided by total STAT1 signal) with bars expressing means and standard deviations. Significant differences between wild-type and mutant STAT1 from five independent experiments are indicated with asterisks (p = 0.034 and p = 0.017, respectively). (G) An electrophoretic mobility shift assay demonstrates the exchange of monomers between STAT1 dimers. Shown is a representative gelshift result using cellular extracts from U3A cells expressing either GFP-tagged or untagged STAT1 bound to M67 DNA. The identity of the bands corresponding to STAT1 (marked with arrowheads) was confirmed by the absence of or reduction in DNA-binding activity in IFNγ-stimulated Y701F- (lane 1) and unstimulated WT-expressing cells (lane 2) as well as from supershift reactions using either a STAT3- (lanes 3 and 5) or STAT1-recognizing antibody (lanes 4 and 6). In lanes 7–10, similar amounts of GFP-tagged and untagged homodimers were either immediately mixed and incubated together for 45 min (lanes 8 and 10) or incubated separately for 45 min before being loaded together onto the gel (lanes 7 and 9). The asterisk at the right margin marks a non-specific band.](/cms/asset/cf95beab-4072-4d7d-bb61-cc74cee1792c/kjks_a_10923576_f0005.gif)
Because STAT1 dimers are subjected to a constant oscillation between a parallel and antiparallel conformation,Citation29 we next wondered whether, in the case of dimeric STAT1-F364A, there is also an exchange of monomers. To answer this question, we mixed similar amounts of extracts from transfected U3A cells expressing either GFP-tagged or untagged STAT1 and analyzed by means of EMSA experiments the in vitro formation of STAT1-GFP/STAT1 heterodimers following 45 min of incubation at RT (). In controls, the same extracts were incubated separately for 45 min and mixed immediately before being loaded onto the gel (reaction time 0 min). We observed the de novo formation of heterodimers only in mixed reactions (lanes 8 and 10), but not in extracts that had not been mixed before (lanes 7 and 9). This result confirmed the previous finding of an exchange of monomers between STAT1 dimers, demonstrated here by the occurrence of an additional band corresponding to newly formed heterodimers bound to M67. For the mutant F364A, we also detected a monomer exchange indicative of a transition between the two dimer conformations, which was within the same range as that measured for the wild-type protein. Approximately one fifth of the total STAT1 DNA-binding activity was confined to newly formed heterodimeric STAT1 complexes, irrespective of the presence of the F364A mutation (for both variants 21%).
Transcriptional activity of the F364A DNA-binding mutant
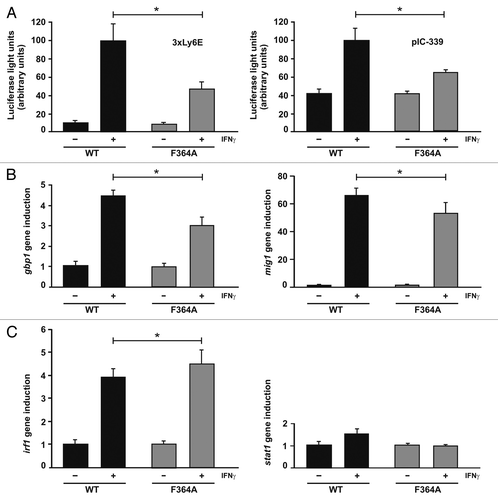
Finally, we tested the transcriptional activity of the F364A mutant by means of luciferase assays () and real-time reverse transcriptase-PCR (). For reporter gene assays, we used two luciferase-encoding plasmids containing either an IFNγ-driven promoter with triple GAS sites (3 × Ly6E) or a promoter containing a 339-bp fragment from the human icam-1 gene (pIC-339). On both reporters, the hyper-phosphorylated STAT1-F364A mutant showed a significantly decreased gene expression as compared with the wild-type (). However, assessment of transcriptional activity on three different STAT1-dependent target genes revealed a differential expression pattern between the mutant and native protein (). Whereas activation of gbp1 and mig1 genes was slightly reduced in the presence of the mutation, the irf1 gene was conversely transcribed at higher rates, underscoring the complex phenotype of the DNA-binding mutant with regard to transcriptional activation.
Figure 6. Transcriptional responses of the STAT1 DNA-binding mutant F364A. (A) Decreased reporter gene activation in U3A cells expressing STAT1-F364A as compared with the wild-type protein. U3A cells were transiently transfected with expression plasmids coding for either of the two STAT1 variants, the luciferase reporter constructs indicated and a constitutively expressed β-galactosidase gene used for normalization. The reporter constructs contained either a triple GAS site from the Ly6E promoter (3 × Ly6E) or a 339 base pair fragment from the native ICAM-1 promoter (pIC-339). On the next day, cells were either left untreated (− IFNγ) or stimulated for 6 h with 5 ng/ml of IFNγ (+ IFNγ) before, in whole cell extracts, luciferase luminescence and β-galactosidase activity were measured in six independent experiments. (B) Activation of three endogenous STAT1 target genes in U3A cells expressing either mutant or wild-type STAT1, as determined by real-time RT-PCR. Expression levels of the irf1, mig1, gbp1 and for control stat1 gene before and after 6 h stimulation with 5 ng/ml of IFNγ are shown. Gene induction was normalized to the expression of the house-keeping gene gapdh. The data are presented as means and standard deviations from at least three independent experiments. Statistical significance between the groups of IFNγ-stimulated cells expressing the indicated STAT1 variants is marked by asterisks.
Discussion
Sequence-specific DNA binding and an extensive spatial dimer reorientation from a DNA-bound parallel to an antiparallel conformation have been identified as basic principles of STAT1 signal transduction.Citation15,Citation27-Citation29 Impeding either of these processes resulted in a substantial decrease in interferon-γ-mediated signal strength, as has been demonstrated from experiments using DNA-binding mutants with reduced affinity to GAS sites or defective transition between dimer conformers due to a critical point mutation in the interacting amino-termini.Citation19,Citation31,Citation32 In the study presented here, we characterized a point mutation in the hydrophobic center of the STAT1 DNA-binding domain, which critically affected the structural integrity of the overall domain architecture and allowed us to analyze the combined phenotype of the mutant with respect to signal propagation.
Whereas all existing DNA-binding mutants with an increased dissociation rate from GAS sites characterized so far have shown normal or even reduced tyrosine-phosphorylation levels upon stimulation of cells with IFNγ,Citation19,Citation33 the structural mutant described here unexpectedly exhibited significantly elevated concentrations of tyrosine-phosphorylated STAT1 in both cytosolic and nuclear extracts from IFNγ-stimulated cells as compared with the wild-type molecule (). The hyper-phosphorylation of the F364A mutant resulted from an impaired stabilization of the antiparallel dimer conformation, in which a pocket composed of Q340, G384 and Q408 in the DNA-binding domain on one monomer interacted with the phenylalanine residue 172 located in the coiled-coil domain of the other monomer.Citation26-Citation28 Mutation of either the pocket residues or the critical F172 residue was shown to lead to persistent tyrosine phosphorylation, indicating that there is an extensive rotation of the monomers from a parallel to an antiparallel orientation, whereas the monomeric core structure is retained virtually intact.Citation27,Citation28
Similar to the pocket mutants affecting the formation of antiparallel dimers, our F364A substitution mutant also showed prolonged and elevated levels of tyrosine phosphorylation (). The structural alterations in the architecture of the DNA-binding domain introduced by this mutation appear to negatively affect the stability of the antiparallel dimer conformation, thereby shifting the conformational equilibrium toward the parallel configuration. The decreased dephosphorylation rate of STAT1-F364A demonstrates that the antiparallel dimer is the preferential substrate of the inactivating phosphatase (). This finding confirms that for the wild-type STAT1 protein there is a high oscillation rate between the two dimer conformations, which is required for both DNA recognition (by the parallel dimer) and dephosphorylation (by the antiparallel dimer). Our in vitro tyrosine dephosphorylation assays with the isolated Tc45 phosphatase demonstrated that a mutation with a shifted conformational equilibrium, such as the F364A substitution, has profound effects on the rate of STAT1 activation. In addition, we found that the exchange rate of monomers between separate dimers was nevertheless unaffected by the mutation, as shown by the rapid de novo formation of STAT1/STAT1-GFP heterodimers from isolated GFP-tagged and untagged homodimers ().
The phenylalanine residue 364 is situated in the hydrophobic interior of the DNA-binding domain and aligns all three connecting loops that constitute the pocket surface required for interaction with the coiled-coil domain of the partner monomer (). Its planar aromatic side chain, with a distance of approximately 9 Å to the bottom of the pocket and less than 12 Å to the surface of the DNA-binding face, is arranged in such a way that it forms two rectangles, nearly perpendicular to each other, which are directed toward the pocket and two threonines at positions 327 and 427, both of which have direct contacts to the DNA. This localization in the hydrophobic interior of the DNA-binding domain is crucial for the stabilization of the reciprocal coiled-coil domain/DNA-binding domain interactions in the antiparallel dimer configuration and, additionally, for the recognition of binding sites on DNA. Given the contribution of this residue to both processes, it is not surprising that a point mutant thereof fails to bind to GAS elements with high affinity and is partially defective in stabilizing the antiparallel dimer conformation.
However, when we tested the substitution mutant F364A for its ability to stimulate IFNγ-driven gene expression by means of real-time RT-PCR, we unexpectedly revealed that it had a well-preserved transcriptional activity that, in the case of the irf1 gene, even surpassed that of the wild-type protein (). Thus, despite two synergistic mechanisms each of which, in isolation, should result in suppressed gene activation, we, nevertheless, found a remarkably high activation of the three endogenous STAT1-regulated target genes studied. This finding suggests that a shift in the equilibrium between the two dimer conformations can compensate for a critical reduction in high-affinity DNA binding and, thereby, restore nearly full transcriptional activity at IFNγ-driven target genes.
The observation that a DNA-binding mutant with decreased sequence-specific DNA binding showed a nearly normal gene induction underlines the physiological significance of the postulated shift between the two dimer conformations.Citation27-Citation29 However, the complex phenotype of STAT1-F364A is achieved at the expense of signal responsiveness, since the mutant showed persistent tyrosine phosphorylation levels upon treating IFNγ-prestimulated cells with staurosporine. The elevated phospho-STAT1 concentrations in the presence of this potent kinase inhibitor, detected both in cytosolic and nuclear extracts of F364A-expressing cells, suggest that the mutant follows time-dependent changes in ligand-induced receptor occupation more slowly than the wild-type protein (). Thus, the F364A DNA-binding mutant partially loses its ability to rapidly terminate signal output. These data demonstrate that the coupling of high-affinity DNA binding and the stabilization of the antiparallel dimer conformation is not required for optimal signal amplification, but rather allows for a dynamic response to decreased levels of receptor occupation. The inherent properties of the multi-functional STAT1 DNA-binding domain ensure both robust transcriptional response and a high adaptability to changing signal inputs.
Materials and Methods
Cell culture
HeLa cells were maintained at 37°C in a humidified 5% CO2 atmosphere in Quantum 101 medium (PAA Laboratories) supplemented with 5% fetal calf serum (FCS; Biochrom), 1% penicillin and 1% streptomycin. U3A cells, first described by Müller et al. (1993) as a fibrosarcoma cell line lacking STAT1 expression,Citation34 were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% FCS and antibiotics. Cells were transfected using the Lipofectamine plus (Invitrogen) method according to the manufacturer’s recommendations. On the next day, transfected cells were either left untreated or stimulated for the indicated times with 2.5 ng/ml and 5 ng/ml of human IFNγ (Biomol), respectively. After 45 min of IFNγ stimulation, the medium was replaced and the cells were subsequently exposed to 500 nM staurosporine (Sigma) for the time periods indicated. In some experiments, cells were additionally treated with 10 ng/ml leptomycin B (LC Laboratories).
Plasmid construction
Expression of green-fluorescent protein (GFP)-tagged STAT1 fusion proteins was achieved by transfecting human cells with the pSTAT1-GFP vector.Citation35 This plasmid coded for full-length human STAT1 (amino acids 1–746) fused C-terminally to a red-shifted variant of GFP. The plasmids coding for dimerization-defective STAT1-Y701F and the DNAminus mutant, which lacks DNA binding due to the replacement of codons V426 and T427 by DD, have been described previously.Citation19 For expression of untagged STAT1, U3A cells were transfected with a pcDNA3.1 expression vector coding for human STAT1. The plasmid pSTAT1-NES-GFP encoded a STAT1 variant with hyperactive nuclear export, which resulted from the presence of a transferable nuclear export signal (NES) activity from amino acids 367–427 of STAT1 situated between the cDNAs for full-length STAT1 and GFP, as was described by Lödige et al.Citation36 Replacement of the native phenylalanine residue at position 364 for alanine was achieved by site-directed point mutagenesis using the QuikChange II kit from Stratagene, as recommended by the manufacturer. The F364A mutation was introduced in all three expression vectors pSTAT1-GFP, pcDNA-STAT1 and pSTAT1-NES-GFP. Mutations were verified by standard didesoxy termination DNA sequencing (Seqlab).
Fluorescence microscopy
The intracellular distribution of GFP-tagged fusions of STAT1 was determined by means of fluorescence microscopy. Transiently transfected cells were treated as described and subsequently fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min at room temperature (RT). Nuclei were stained with 5 μg/ml Hoechst 33258 (Sigma) for 10 min at RT. The samples were finally mounted in fluorescence mounting medium (Southern Biotech) and GFP epifluorescence visualized using a Leica DM5000B microscope. Images were obtained with a CCD camera and further processed with the Leica QWin software.
Immunocytochemistry
Untagged STAT1 was immunocytochemically detected in transfected U3A cells expressing either wild-type or mutant STAT1. Briefly, U3A cells grown on chamber slides were either left untreated or treated with IFNγ for 45 min. Subsequently, cells were either immediately fixed or additionally incubated in the presence of 500 nM staurosporine for an additional 30 and 60 min, respectively. Cells were fixed with methanol for 20 min at −20°C and, after two washes in PBS, permeabilized with 1.0% Triton X-100 in PBS for 20 min at RT. Non-specific binding was blocked by incubation with 25% FCS/PBS for 45 min at RT. STAT1 immunolabeling was performed by incubation with the STAT1-specific antibody C-24 (Santa Cruz) diluted 1:1,000 in 25% FCS/PBS for 45 min at RT. After three washes in PBS, the samples were incubated with Cy3-conjugated secondary antibody (Dianova) diluted 1:500 in 25% FCS/PBS for additional 45 min at RT. Finally, nuclei were stained with Hoechst 33258 and the mounted samples were visualized by fluorescence microscopy using a Leica DM5000B microscope equipped with appropriate fluorescence filters.
Western blotting
Cells grown on 6-well dishes were lysed on ice for 5 min in 35 μl cytoplasmic extraction buffer containing 0.1% Nonidet P40, 10 mM KCl, 1 mM EDTA, 10% glycerol, 20 mM Hepes, pH 7.4, 1 mM vanadate, 3 mM DTT, 0.4 mM Pefabloc and Complete protease inhibitors (Roche). The lysates were spun at 16,000 g for 15 sec at 4°C. The supernatants were recentrifuged at 16,000 g for 5 min and stored at −80°C as cytosolic extracts. The pellets from the first centrifugation step were resuspended in 35 μl nuclear extraction buffer containing 420 mM KCl, 1 mM EDTA, 20% glycerol, 20 mM Hepes, pH 7.4, 1 mM vanadate, 3 mM DTT, 0.4 mM Pefabloc and Complete protease inhibitors. After 30 min on ice, the nuclear extracts were centrifuged for 15 min at 16,000 g and the supernatants from this final centrifugation step were stored. For whole cell preparations, the cytoplasmic and nuclear extracts were combined and boiled in SDS sample buffer. Gel electrophoresis was performed on 10% SDS-polyacrylamide gels and the separated proteins were then transferred to nitrocellulose membranes. The membranes were incubated with a polyclonal antibody specifically recognizing tyrosine-phosphorylated STAT1 (Cell Signaling) and then with a horseradish peroxidase-conjugated secondary antibody (Dako). Bound immunoreactivity was detected using the enhanced chemiluminescence reaction (Pierce). Subsequently, the blots were stripped at 60°C for 60 min in a buffer containing 2% SDS, 0.7% β-mercaptoethanol and 62.5 mM TRIS-HCl, pH 6.8. The blots were then reprobed with polyclonal pan-STAT1 antibody C-24 obtained from Santa Cruz and finally exposed to horseradish peroxidase-conjugated secondary antibodies. STAT1 immunoreactivity was detected using chemiluminescence, as described above.
In vitro phosphorylation and dephosphorylation assays
For in vitro phosphorylation assays, 10 μl of whole cell extract from STAT1-reconstituted U3A cells were mixed with 10 μl of kinase buffer containing 50 mM Hepes, pH 7.4, 3 mM MgCl2, 3 mM MnCl2, 3 µM vanadate, 10 mM DTT, 0.1 mM ATP and either 40 ng of recombinant Janus kinase 2 (JAK2, obtained from Enzo Life Science) or 20 ng of epidermal growth factor receptor (EGFR, obtained from US Biological). In vitro dephosphorylation assays were performed by incubating 10 μl of whole cell extract from reconstituted IFNγ-treated U3A cells with 10 µl of dephosphorylation buffer (25 mM TRIS-HCl, pH 7.5, 0.5 mg/ml BSA, 50 mM KCl, 5 mM EDTA, 20 mM DTT and Complete protease inhibitor) containing 2 U of the T-cell protein-tyrosine phosphatase, obtained from Biomol International. All samples were incubated at 30°C for the indicated times. Dephosphorylation was stopped by adding SDS sample buffer before phospho-STAT1 was detected by western blotting.
Gelshift assays
DNA-binding activity of STAT1 was assessed by means of electrophoretic mobility shift assays (EMSA). Briefly, extracts from U3A cells expressing either wild-type or mutant STAT1 were prepared as described above. Five microliters of each extract were incubated with 1 ng [32P]-labeled duplex oligonucleotide probes, which were generated by an end-filling reaction using Klenow fragment (New England Biolabs). The following duplex oligonucleotides with their respective antisense strands (not listed below) were used (GAS and GAS-like sites are in bold): M67, 5′-CGACATTTCCCGTAAATCTG-´3; 2 × GAS, 5′-CGTTTCCCCGAAATTGACGGATTTCCCCGAAAC-´3; GAS + nonGAS, 5′-CGTTTCCCCGAAATTGACGGATTTACCCCAAC-´3; and 2 × nonGAS, 5′-CGTTTACCCCAAATTGACGGATTTACCCCAAC-´3. For end-filling labeling, the 5′ ends contained 4 bp overhangs (not included). Competition experiments were performed by equilibrating EMSA reactions with a 750-fold molar excess of unlabeled M67 DNA for the indicated times. In supershift assays, 20 ng of the STAT1-recognizing antibody C-24 were present in the shift reaction. The EMSA reactions were loaded on a 4% acrylamide:bisacrylamide (29:1) gel and the DNA-bound proteins separated by electrophoresis at 4°C. Binding activity was visualized in vacuum-dried gels using a phosphoimaging system (BAS-1000, Fujifilm) and the computer programs BAS-reader and TINA version 2.08.
Reporter gene assay
U3A cells grown on 48-well plates were transiently transfected with the following amounts of cDNAs added into a single well: 250 ng of STAT1 expression plasmid, 200 ng of a constitutively expressed β-galactosidase reporter (Stratagene) and 70 ng of a luciferase reporter construct. The IFNγ-sensitive luciferase reporters contained either a triple Ly6E STAT-binding site (termed 3 × Ly6E) or the 5′-region of the human intercellular adhesion molecule 1 (ICAM-1) gene 339 bp relative to the transcription start site (termed pIC-339).Citation33,Citation37 Twenty-four hours post-transfection, cells were either left unstimulated or treated for 6 h with IFNγ. Whole cell extracts were measured for luciferase (Promega) and β-galactosidase activities. For each STAT1 variant and stimulation mode, six independent samples were tested and the experiment was repeated at least three times. Data were normalized for the expression of β-galactosidase.
Real-time reverse transcriptase-PCR
Real-time reverse transcriptase-polymerase chain reactions (RT-PCR) were performed to compare the transcriptional activity of wild-type vs. mutant STAT1. U3A cells were transfected with pcDNA3.1 expression plasmids coding for wild-type or mutant STAT1. Twenty-four hours after transfection, cells were incubated for 15 h with Dulbecco’s modified Eagle’s medium supplemented with 1% FCS. After serum-depletion, cells were either left untreated or stimulated for 6 h with IFNγ. RNA was isolated with the peqGold Total RNA kit (Peqlab) and first-strand cDNA synthesis was performed using the Verso cDNA kit from Thermo Scientific. Real-time PCR reactions were performed in a total volume of 25 µl containing 250 ng cDNA, 70 nM of each specific primer pair and 12.5 µl SYBR Green. Using Primer 3 software (Applied Biosystems), gene-specific primers for three cDNAs from known STAT1 target genes (irf1, mig1 and gbp1) as well as for stat1 and gapdh were designed. In order to amplify fragments of about 200 bp in length, we used the following primer pairs: IRF1F, 5′-AGCTCAGCTGTGCGAGTGTA-′3; IRF1R, 5′-TAGCTGCTGTGGTCATCAGG-′3; MIG1F, 5′-CCACCGAGATCCTTATCGAA-′3; MIG1R, 5′-CTAACCGACTTGGCTGCTTC-′3; GBP1F, 5′-GGTCCAGTTGCTGAAAGAGC-′3; GBP1R, 5′-TGACAGGAAGGCTCTGGTCT-′3; GAPDHF, 5′-GAAGGTGAAGGTCGGAGTC-′3; GAPDHR, 5′-GAAGATGGTGATGGGATTTC-′3; STAT1F, 5′-CCGTTTTCATGACCTCCTGT-′3; and STAT1R, 5′-TGAATATTCCCCGACTGAGC-′3. Polymerase chain reactions were performed by a denaturation step at 95°C for 15 min, which was followed by 40 cycles of denaturation at 95°C for 30 sec, annealing at 55°C for 30 sec and extension at 72°C for 30 sec. After the final amplification step, a melting curve analysis was run via a temperature gradient from 60°C to 95°C in 0.5°C increment steps and fluorescence was measured at each temperature for a period of 10 sec. All reactions were performed in at least triplicate for each sample. The relative expression of a transcript was normalized to the expression level of gapdh, which was determined for each sample. The threshold (Ct) was adjusted to areas of exponential amplification of the traces using the Realplex 1.5 software from Eppendorf. The ΔΔCt-method was used to determine relative expression levels by applying the equation 2−(ΔCt target − ΔCt reference sample).
Statistical analyses
Differences in tyrosine phosphorylation, DNA binding activity, intracellular fluorescence intensity and gene expression between wild-type and mutant STAT1 were assessed using Student’s t-tests and Mann-Whitney-Wilcoxon tests, where appropriate. Means and standard deviations for each variant and stimulation mode were calculated. Statistical significance was defined as p < 0.05.
Acknowledgments
The authors gratefully acknowledge the excellent technical assistance of Anke Gregus and Heike Hühn. We kindly thank Dr Uwe Vinkemeier, University of Nottingham, for valuable reagents and discussions. The research on this subject was funded by a grant from the Deutsche Forschungsgemeinschaft to T.M.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Brivanlou AH, Darnell JE Jr.. Signal transduction and the control of gene expression. Science 2002; 295:813 - 8; http://dx.doi.org/10.1126/science.1066355; PMID: 11823631
- Levy DE, Darnell JE Jr.. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 2002; 3:651 - 62; http://dx.doi.org/10.1038/nrm909; PMID: 12209125
- Darnell JE Jr., Kerr IM, Stark GR. JAK-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994; 264:1415 - 21; http://dx.doi.org/10.1126/science.8197455; PMID: 8197455
- Darnell JE Jr.. STATs and gene regulation. Science 1997; 277:1630 - 5; http://dx.doi.org/10.1126/science.277.5332.1630; PMID: 9287210
- Ihle JN. The Stat family in cytokine signaling. Curr Opin Cell Biol 2001; 13:211 - 7; http://dx.doi.org/10.1016/S0955-0674(00)00199-X; PMID: 11248555
- Shuai K, Schindler C, Prezioso VR, Darnell JE Jr.. Activation of transcription by IFN-gamma: tyrosine phosphorylation of a 91-kD DNA binding protein. Science 1992; 258:1808 - 12; http://dx.doi.org/10.1126/science.1281555; PMID: 1281555
- Shuai K, Stark GR, Kerr IM, Darnell JE Jr.. A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science 1993; 261:1744 - 6; http://dx.doi.org/10.1126/science.7690989; PMID: 7690989
- Shuai K, Horvath CM, Huang LH, Qureshi SA, Cowburn D, Darnell JE Jr.. Interferon activation of the transcription factor Stat91 involves dimerization through SH2-phosphotyrosyl peptide interactions. Cell 1994; 76:821 - 8; http://dx.doi.org/10.1016/0092-8674(94)90357-3; PMID: 7510216
- Greenlund AC, Morales MO, Viviano BL, Yan H, Krolewski J, Schreiber RD. Stat recruitment by tyrosine-phosphorylated cytokine receptors: an ordered reversible affinity-driven process. Immunity 1995; 2:677 - 87; http://dx.doi.org/10.1016/1074-7613(95)90012-8; PMID: 7796299
- Decker T, Kovarik P, Meinke A. GAS elements: a few nucleotides with a major impact on cytokine-induced gene expression. J Interferon Cytokine Res 1997; 17:121 - 34; http://dx.doi.org/10.1089/jir.1997.17.121; PMID: 9085936
- Herrington J, Rui L, Luo G, Yu-Lee LY, Carter-Su C. A functional DNA binding domain is required for growth hormone-induced nuclear accumulation of Stat5B. J Biol Chem 1999; 274:5138 - 45; http://dx.doi.org/10.1074/jbc.274.8.5138; PMID: 9988763
- Sehgal PB. Paradigm shifts in the cell biology of STAT signaling. Semin Cell Dev Biol 2008; 19:329 - 40; http://dx.doi.org/10.1016/j.semcdb.2008.07.003; PMID: 18691663
- Speil J, Baumgart E, Siebrasse JP, Veith R, Vinkemeier U, Kubitscheck U. Activated STAT1 transcription factors conduct distinct saltatory movements in the cell nucleus. Biophys J 2011; 101:2592 - 600; http://dx.doi.org/10.1016/j.bpj.2011.10.006; PMID: 22261046
- Schindler C, Shuai K, Prezioso VR, Darnell JE Jr.. Interferon-dependent tyrosine phosphorylation of a latent cytoplasmic transcription factor. Science 1992; 257:809 - 13; http://dx.doi.org/10.1126/science.1496401; PMID: 1496401
- Horvath CM, Wen Z, Darnell JE Jr.. A STAT protein domain that determines DNA sequence recognition suggests a novel DNA-binding domain. Genes Dev 1995; 9:984 - 94; http://dx.doi.org/10.1101/gad.9.8.984; PMID: 7774815
- Horvath CM, Stark GR, Kerr IM, Darnell JE Jr.. Interactions between STAT and non-STAT proteins in the interferon-stimulated gene factor 3 transcription complex. Mol Cell Biol 1996; 16:6957 - 64; PMID: 8943351
- Meyer T, Begitt A, Lödige I, van Rossum M, Vinkemeier U. Constitutive and IFN-γ-induced nuclear import of STAT1 proceed through independent pathways. EMBO J 2002; 21:344 - 54; http://dx.doi.org/10.1093/emboj/21.3.344; PMID: 11823427
- Marg A, Shan Y, Meyer T, Meissner T, Brandenburg M, Vinkemeier U. Nucleocytoplasmic shuttling by nucleoporins Nup153 and Nup214 and CRM1-dependent nuclear export control the subcellular distribution of latent Stat1. J Cell Biol 2004; 165:823 - 33; http://dx.doi.org/10.1083/jcb.200403057; PMID: 15210729
- Meyer T, Marg A, Lemke P, Wiesner B, Vinkemeier U. DNA binding controls inactivation and nuclear accumulation of the transcription factor Stat1. Genes Dev 2003; 17:1992 - 2005; http://dx.doi.org/10.1101/gad.268003; PMID: 12923054
- Sekimoto T, Imamoto N, Nakajima K, Hirano T, Yoneda Y. Extracellular signal-dependent nuclear import of Stat1 is mediated by nuclear pore-targeting complex formation with NPI-1, but not Rch1. EMBO J 1997; 16:7067 - 77; http://dx.doi.org/10.1093/emboj/16.23.7067; PMID: 9384585
- Fagerlund R, Mélen K, Kinnunen L, Julkunen I. Arginine/lysine-rich nuclear localization signals mediate interactions between dimeric STATs and importin α 5. J Biol Chem 2002; 277:30072 - 8; http://dx.doi.org/10.1074/jbc.M202943200; PMID: 12048190
- McBride KM, Banninger G, McDonald C, Reich NC. Regulated nuclear import of the STAT1 transcription factor by direct binding of importin-α. EMBO J 2002; 21:1754 - 63; http://dx.doi.org/10.1093/emboj/21.7.1754; PMID: 11927559
- Melén K, Fagerlund R, Franke J, Köhler M, Kinnunen L, Julkunen I. Importin α nuclear localization signal binding sites for STAT1, STAT2, and influenza A virus nucleoprotein. J Biol Chem 2003; 278:28193 - 200; http://dx.doi.org/10.1074/jbc.M303571200; PMID: 12740372
- Nardozzi J, Wenta N, Yasuhara N, Vinkemeier U, Cingolani G. Molecular basis for the recognition of phosphorylated STAT1 by importin α5. J Mol Biol 2010; 402:83 - 100; http://dx.doi.org/10.1016/j.jmb.2010.07.013; PMID: 20643137
- Chen X, Vinkemeier U, Zhao Y, Jeruzalmi D, Darnell JE Jr., Kuriyan J. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell 1998; 93:827 - 39; http://dx.doi.org/10.1016/S0092-8674(00)81443-9; PMID: 9630226
- Mao X, Ren Z, Parker GN, Sondermann H, Pastorello MA, Wang W, et al. Structural bases of unphosphorylated STAT1 association and receptor binding. Mol Cell 2005; 17:761 - 71; http://dx.doi.org/10.1016/j.molcel.2005.02.021; PMID: 15780933
- Zhong M, Henriksen MA, Takeuchi K, Schaefer O, Liu B, ten Hoeve J, et al. Implications of an antiparallel dimeric structure of nonphosphorylated STAT1 for the activation-inactivation cycle. Proc Natl Acad Sci U S A 2005; 102:3966 - 71; http://dx.doi.org/10.1073/pnas.0501063102; PMID: 15753310
- Mertens C, Zhong M, Krishnaraj R, Zou W, Chen X, Darnell JE Jr.. Dephosphorylation of phosphotyrosine on STAT1 dimers requires extensive spatial reorientation of the monomers facilitated by the N-terminal domain. Genes Dev 2006; 20:3372 - 81; http://dx.doi.org/10.1101/gad.1485406; PMID: 17182865
- Wenta N, Strauss H, Meyer S, Vinkemeier U. Tyrosine phosphorylation regulates the partitioning of STAT1 between different dimer conformations. Proc Natl Acad Sci U S A 2008; 105:9238 - 43; http://dx.doi.org/10.1073/pnas.0802130105; PMID: 18591661
- ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, Zhu W, Tremblay M, David M, et al. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol 2002; 22:5662 - 8; http://dx.doi.org/10.1128/MCB.22.16.5662-5668.2002; PMID: 12138178
- Yang E, Henriksen MA, Schaefer O, Zakharova N, Darnell JE Jr.. Dissociation time from DNA determines transcriptional function in a STAT1 linker mutant. J Biol Chem 2002; 277:13455 - 62; http://dx.doi.org/10.1074/jbc.M112038200; PMID: 11834743
- Meyer T, Hendry L, Begitt A, John S, Vinkemeier U. A single residue modulates tyrosine dephosphorylation, oligomerization, and nuclear accumulation of stat transcription factors. J Biol Chem 2004; 279:18998 - 9007; http://dx.doi.org/10.1074/jbc.M400766200; PMID: 15010467
- van de Stolpe A, Caldenhoven E, Stade BG, Koenderman L, Raaijmakers JA, Johnson JP, et al. 12-O-tetradecanoylphorbol-13-acetate- and tumor necrosis factor alpha-mediated induction of intercellular adhesion molecule-1 is inhibited by dexamethasone. Functional analysis of the human intercellular adhesion molecular-1 promoter. J Biol Chem 1994; 269:6185 - 92; PMID: 7907090
- Müller M, Laxton C, Briscoe J, Schindler C, Improta T, Darnell JE Jr., et al. Complementation of a mutant cell line: central role of the 91 kDa polypeptide of ISGF3 in the interferon-α and -γ signal transduction pathways. EMBO J 1993; 12:4221 - 8; PMID: 7693454
- Begitt A, Meyer T, van Rossum M, Vinkemeier U. Nucleocytoplasmic translocation of Stat1 is regulated by a leucine-rich export signal in the coiled-coil domain. Proc Natl Acad Sci U S A 2000; 97:10418 - 23; http://dx.doi.org/10.1073/pnas.190318397; PMID: 10973496
- Lödige I, Marg A, Wiesner B, Malecová B, Oelgeschläger T, Vinkemeier U. Nuclear export determines the cytokine sensitivity of STAT transcription factors. J Biol Chem 2005; 280:43087 - 99; http://dx.doi.org/10.1074/jbc.M509180200; PMID: 16195225
- Wen Z, Zhong Z, Darnell JE Jr.. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995; 82:241 - 50; http://dx.doi.org/10.1016/0092-8674(95)90311-9; PMID: 7543024