Abstract
Autosomal-dominant polycystic kidney disease (ADPKD) is a common genetic disease caused by mutations in the gene coding for polycystin-1 (PC1). PC1 can regulate STAT transcription factors by a novel, dual mechanism. STAT3 and STAT6 are aberrantly activated in renal cysts. Genetic and pharmacological approaches to inhibit STAT3 or STAT6 have led to promising results in ADPKD mouse models. Here, we review current findings that lead to a model of PC1 as a key regulator of STAT signaling in renal tubule cells. We discuss how PC1 may orchestrate appropriate epithelial responses to renal injury, and how this system may lead to aberrant STAT activation in ADPKD thereby causing inappropriate activation of tissue repair programs that culminate in renal cyst growth and fibrosis.
Polycystic Kidney Disease
ADPKD is a very common life-threatening, monogenic disease that affects over 600,000 people in the US alone. Excessive proliferation of renal tubule epithelial cells leads to growth of epithelial-lined cysts, accompanied by fibrosis and accumulation of extracellular matrix. As the disease progresses, this leads to destruction of the normal renal parenchyma, massive renal enlargement, deterioration of renal function and eventually renal failure.Citation1,Citation2 Most patients will require dialysis or kidney transplantation. Unfortunately, despite intensive efforts and several clinical trialsCitation3 there is currently no available treatment to halt or slow disease progression.
The root causes of ADPKD are mutations in the PKD1 or PKD2 genes which encode the proteins polycystin-1 (PC1) and polycystin-2 (PC2), respectively. PC2 is a calcium-permeable channel of the TRP family, and forms a complex with PC1. PC1 is mutated in the majority (85%) of cases of ADPKD and is thought to regulate the channel activity of PC2.Citation1,Citation2,Citation4,Citation5 However, the actual purpose of the PC1/PC2 channel has remained unclear. The picture is further complicated because PC1 can interact with a wide variety of signaling proteins, and regulates numerous signaling pathways including heterotrimeric G proteins, wnt-, integrin-, mTOR- and JAK-STAT-signaling. It has remained unclear which of these numerous proposed functions is most relevant for understanding the molecular mechanisms that leads to renal cyst growth in ADPKD.
The polycystins have been reported to localize to several subcellular compartments in renal epithelial cells, most notably primary cilia, lateral cell-cell junctions and the endoplasmic reticulum. Experimental evidence suggests that the fraction of polycystins that localizes to primary cilia is required for the function of cilia as mechanosensors. Renal epithelial cells possess a single apical cilium that protrudes into the lumen of the renal tubule. Fluid flow of the filtrate is thought to bend primary cilia and trigger an intracellular calcium signal. Disruption of primary cilia or mutations in a large number of cilia-associated proteins leads to the induction of renal epithelial proliferation and the growth of renal cysts in animal models and numerous human genetic diseases.Citation5,Citation6 These diseases are therefore classified as ciliopathies.Citation7 However, it is currently unknown why and how the function of primary cilia, or changes in luminal fluid flow, would be connected to the regulation of proliferation of renal tubule cells.
There are numerous similarities in signaling pathways that are activated both in PKD and in response to kidney injury. This has led to the hypothesis that PKD is a manifestation of aberrant and chronic activation of injury repair pathways that are normally dormant in the healthy kidney but can be activated in response to insults.Citation8 Indeed, different forms of renal injury have been shown to trigger rapid renal cyst growth in experimental animal models.Citation9
Numerous signaling molecules and pathways have been shown to be aberrantly activated in cyst-lining cells in PKD such as Src, Erk and mTOR. Inhibition of many of these pathways leads to significant reductions in renal cyst growth in rodent models of PKD but this has not yet translated into clinical treatments. A case in point are mTOR inhibitors that proved highly effective at high doses in rodent models but were disappointing in subsequent clinical trials.Citation3,Citation10 Recent results from several investigators have indicated that STAT3 and STAT6 are aberrantly activated in PKD, that PC1 can regulate these STATs and that they are promising drug targets for therapy.
In this review we will summarize our current understanding of STAT signaling in normal kidneys vs. PKD, and attempt to develop a model to explain the purpose of the regulation of STAT activity by PC1 and how dysregulation can lead to the pathogenesis of PKD.
STATs
Only the roles of STAT1, 3 and 6 in PKD have been investigated to date, and most available information suggest that STAT3 and STAT6 are involved in renal cyst growth. We briefly cover STAT1 but focus primarily on STAT3 and STAT6. We have recently reviewed the role of STAT3 in PKD.Citation11 Canonical activation of STAT family members occurs via phosphorylation of a single tyrosine residue within the trans-activation domain, which leads to homo- or hetero-dimerization and translocation to the nucleus where STATs bind specific DNA sequences in complex with transcriptional cofactors to regulate gene expression.Citation12 In addition to sequence-specific DNA binding of the STAT protein, the cofactors can provide additional gene specificity. STAT activation often involves their binding to phospho-tyrosine residues on the cytoplasmic tails of activated cytokine or growth factor receptors (such as IL6 family). This is followed by STAT-phosphorylation via receptor-associated tyrosine kinases of the JAK family, receptor tyrosine kinases (such as EGFR and c-Met), or by non-receptor tyrosine kinases such as Src.Citation12
The activities of interferons are largely mediated by STAT1 signaling.Citation13 STAT1 and STAT3 can be co-regulated because they can be activated by some of the same receptors, and they can form hetero-dimers. On the other hand, STAT1 and STAT3 can compete with each other for the same receptorCitation14 and typically have opposing biological effects. While STAT1 activation arrests growth and promotes apoptosis, activated STAT3 can protect cells from apoptosis. STAT3 is considered an oncogene because it confers resistance to apoptosis in many cell types,Citation15,Citation16 it is constitutively activated in many human cancers and its inhibition leads to inhibition of tumor growth.Citation17-Citation22 Intensive efforts are underway to identify STAT3-inhibitory compounds for the development of cancer therapies, but none are yet in clinical use.Citation17,Citation23-Citation25 STAT6 is essential for lymphocyte development, specifically for naïve CD4+ T-cell polarization into Th2 cells.Citation26,Citation27 The primary activators of STAT6 are IL4 and IL13. Th2 cell differentiation is stimulated by IL4, and differentiated Th2 cells secrete IL4 and IL13.Citation27,Citation28 IL4 and IL13 also are responsible for alternative activation of macrophages.Citation29 Diverse target genes of STAT6 have been found, although most have been studied in immune cells.Citation30,Citation31 IL13 plays a critical role in host defense from parasitic nematode infectionsCitation32 and aberrant IL13 signaling is essential for asthma pathology.Citation33 A few other cytokines have been shown to activate STAT6, including IL3/15,Citation34,Citation35 IFNαCitation36,Citation37 and PDGFBB,Citation38 although most of these effects seem to be cell-type specific. STAT6 can also be activated by intracellular pathogens including virusesCitation39 and the parasite Toxoplasma gondii.Citation40
What is the Normal Role of STATs in the Kidney?
STAT1
STAT1 is activated (by phosphorylation on both Y701 and S727) in embryonic rat kidneys but the cell type(s) containing active STAT1 have not been identified.Citation41 In contrast, normal adult kidneys lack active STAT1.Citation41,Citation42 In vitro experiments with embryonic kidney explants suggested that STAT1 activation plays a pro-proliferative role in metanephric mesenchymal cells, and antagonizes epithelial differentiation and tubulogenesis.Citation41 Experiments with MDCK cells as a model of in vitro tubulogenesis yielded conflicting results and suggested that STAT1 is required for tubulogenesis.Citation43 Given that STAT1 null mice do not exhibit a defect in renal developmentCitation41,Citation44,Citation45 it is currently difficult to reconcile these studies or assign any definite role for STAT1 specifically in the kidney (besides its ubiquitous roles during immune responses and inflammation). Mice that are null for the STAT1 inhibitor SOCS1 display a delay in the gross organization of renal medullary tubules into a regular pattern but die perinatally of causes unrelated to the kidney.Citation46 If perinatal death is prevented by simultaneously deleting the IFNγ gene, these double-knockout animals strikingly develop polycystic kidneys later in life.Citation46 However, it is extremely puzzling that usually only one of the kidneys is affected while the other remains normal. Since heavy infiltration of immune cells was also reported in these unilateral polycystic kidneys it is possible that the effect is unrelated to the lack of expression of SOCS1 in the kidney but is rather due to overreaction of the immune system to infection.
STAT3
Due to its early embryonic lethality, STAT3-null mice have been uninformative for a possible role of STAT3 in renal development. STAT3 activity (Y705 phosphorylation) is high in embryonic rat kidneys but undetectable in adult rat kidneys.Citation41 We reported that STAT3 is highly active in renal tubule epithelial cells of mice at postnatal day 7 (d7) when kidneys are actively growing. By d14 STAT3 activity is downregulated, and almost completely undetectable in adult mouse kidneys.Citation42 This time-course of STAT3 inactivation coincides with a developmental switch that occurs in the mouse kidney around d14 and leads to cessation of proliferation and kidney growth.Citation47 In an in vitro culture model with MDCK renal epithelial cells, STAT3 has been shown to be required for hepatocyte growth factor (HGF) induced tubulogenesis.Citation48,Citation49 These studies suggest that STAT3 activity may normally be involved in the regulation of tubule cell proliferation and morphogenesis during renal development.
The fact that STAT3 activity is extremely low in the adult kidney despite high STAT3 expression levelsCitation42 suggests that signaling pathways upstream of STAT3 are not active in the healthy adult kidney and/or that STAT3 activity is strongly suppressed. However, the abundance of STAT3 also suggests that it is ready to be activated at a moment’s notice. Indeed, STAT3 was found to be rapidly activated in renal tubule cells in response to numerous forms of insults. HgCl2-induced acute kidney injury in mice causes STAT3 activation that is due—at least in part—to IL6 trans-signaling, a mechanism that involves IL6 and the shed, soluble form of the IL6-receptor.Citation50 These authors also demonstrated that experimental activation of STAT3 prior to HgCl2 administration dramatically protected animals from AKI and resulted in complete survival. This effect was suggested to involve the induction of reno-protective proteins such as heme oxygenase HO-1.Citation50 Similarly, in cultured proximal tubule cells, STAT3 is activated in response to ATP-depletion as a model of renal ischemic injury.Citation51 Overexpression of constitutively active STAT3 led to increased protection from apoptosis in this system.Citation51 Renal ischemia reperfusion injury causes increased expression of unphosphorylated STAT3, and strong STAT3 activation by tyrosine-phosphorylation but the affected cell types have not been defined.Citation52-Citation55
Unilateral ureteral obstruction (UUO), as a model of obstructive nephropathy, was shown to lead to STAT3 activation. One study found that STAT3 is activated both in tubule epithelial cells and interstitial cellsCitation56 whereas another study reported predominant activation in interstitial fibroblasts.Citation57 Treatment with the STAT3 inhibitor S3I-201 led to inhibition of fibrosis and inflammatory cell infiltration.Citation57 Renal STAT3 activation has also been found in response to adriamycin-induced nephropathy as a model of chronic renal disease but the activated cell types have not been defined.Citation58 Treatment with the JAK2 inhibitor AG490 was shown to inhibit STAT3 and suppress the long-term renal deterioration in this model.Citation58
As an overall conclusion, a model emerges in which STAT3 is rapidly activated in response to several forms of renal insults. STAT3 activity appears to be critical for orchestrating the appropriate responses to such insults such as protection from oxidative stress, recruitment of immune cells and tissue regeneration. However, prolonged renal STAT3 activation appears to play a role in destructive processes such as persistent inflammation and fibrosis.
STAT6
Similar to STAT3, there is very little active STAT6 in the adult kidney, even though there is a high level of STAT6 expressed.Citation59 Upon acute stimulation with systemic IL-4 or IL-13, STAT6 is rapidly activated in renal epithelial cells and interstitial cells.Citation59 A few studies have pointed to STAT6 potentially playing a protective or reparative role in the kidney following kidney injury. Following renal ischemia-reperfusion injury, STAT6−/− mice exhibit more severe tubular injury and worse renal function than in wild-type mice.Citation60 STAT6−/− mice also show enhanced apoptosis and inflammation after unilateral ureteral obstruction vs. wild-type mice.Citation61 In a glomerular disease model, antibody-induced experimental crescentic glomerulonephritis, STAT6−/− mice demonstrate amplified morphological and functional injury.Citation62 IL-13 was shown to be upregulated in patients with lupus nephritis,Citation63 as well as in a rat model of glomerulonephritis.Citation64 Altogether, these studies suggest that activation of renal STAT6, presumably by IL13, in response to renal insults has protective functions and facilitates tissue repair. This is supported by the finding that pre-treatment with systemic IL-13 via gene therapy reduces renal tubulointerstitial damage in a rat model of renal ischemia-reperfusion injury.Citation65
PC1 Can Regulate STAT Activity by a Dual Mechanism: Activation vs. Co-Activation
The initial observation that PC1 can regulate STAT activity was made by Greg Germino’s laboratory.Citation66 These authors showed that overexpression of PC1 causes activation of STAT1 (by phosphorylation at both Ser727 and Tyr701) leading to upregulation of the cyclin-dependent kinase inhibitor, p21waf1, which induces apoptosis as well as cell cycle arrest. PC1 was also found to bind JAK2 suggesting that PC1-mediated regulation of JAK2 activity is responsible for STAT1 activation. Finally, PC1-null mouse embryos at E15.5 almost completely lacked tyrosine-phosphorylated STAT1 and expression of p21waf1 suggesting that PC1 is the master regulator of STAT1/p21waf1 signaling at this developmental stage in the entire embryo. STAT3 was also found to be activated by PC1 overexpression although to a lesser degree, and was not further investigated.
Subsequently, our laboratory discovered that PC1 can also regulate STAT6 activity although the mechanism of regulation differed markedly from the regulation of STAT1 and 3. We found that the C-terminal cytoplasmic tail of the integral membrane protein PC1 is released from the membrane by proteolytic cleavage resulting in C-terminal fragments that undergo nuclear translocation, interact with STAT6 and the transcriptional co-activator P100 and co-activate STAT6-dependent gene expression.Citation67 In contrast, membrane-anchored PC1 inhibited STAT6 activity.Citation67 STAT6 itself was found to translocate between primary cilia and the nucleus depending on apical fluid flow.Citation67 Together with the discovery that cleavage of the PC1 tail is regulated by fluid flowCitation68 these results suggested that PC1-mediated regulation of STAT6 activity plays a role in sensing changes of luminal fluid flow and affecting corresponding changes in gene expression.Citation67 Subsequently, we demonstrated that STAT6 is aberrantly activated in cyst-lining epithelial cells, is part of a positive feedback loop with interleukin 13 and the IL13 receptor, and that inhibition of STAT6 leads to inhibition of renal cyst growth in a PKD mouse model.Citation59 An important mechanistic distinction to STAT1/3 is that membrane-anchored PC1 was not able to “activate” STAT6 by tyrosine-phosphorylation but that instead the soluble, cleaved PC1 tail was able to “co-activate” STAT6 that had previously been “activated” by IL13 cytokine signaling.
To clarify the mechanism of STAT regulation by PC1 we re-investigated the effect on STAT1/3. Although we were unable to detect activation of STAT1 by PC1, we discovered a remarkable dual mechanism of the regulation of STAT3Citation42 (). Membrane-anchored PC1 indeed caused JAK2-dependent activation of STAT3 by tyrosine-phosphorylation, and the membrane-proximal part of the cytoplasmic tail of PC1 was identified as the JAK2 binding site. Kidneys of ADPKD patients accumulate two PC1 cytoplasmic tail fragments (~15 kDa and ~30 kDa).Citation42 The removal of the 15 or 30 kDa fragments from membrane-bound PC1 eliminates its ability to activate STAT3 suggesting that these cleavage events are involved in the downregulation of PC1-induced STAT3 signaling. However, remarkably, the 30 kDa PC1 tail cleavage product was able to co-activate both STAT3 or STAT1 that had been tyrosine-phosphorylated by cytokine signaling.Citation42 This indicated that PC1 can regulate STATs at two levels: First, membrane-anchored, full-length PC1 can act similar to an activated growth factor receptor and activate STAT1 and STAT3 by JAK2-mediated tyrosine-phosphorylation. Second, after PC1 is cleaved—e.g., during renal injury—its cytoplasmic tail can co-activate either STAT1, STAT3 or STAT6 depending on which of these STATs has previously been activated by specific growth factors. Therefore, cleaved PC1 cannot by itself activate STAT signaling but it can amplify STAT signaling in response to the growth factor environment of the cell which can lead to different biological responses including proliferation and apoptosis.Citation42
Figure 1. Model of the regulation of STAT signaling by PC1. During renal development, membrane-anchored, full-length PC1 may cause direct activation of STAT1 and STAT3 via JAK2 that is associated with its C-terminal cytoplasmic tail. Direct STAT1/3 activation by PC1 would be an intrinsic pathway that is independent of growth factors. It is currently unknown how the direct activation of STAT1/3 by full-length PC1 is regulated. It is possible that an—as yet unidentified—extracellular ligand may trigger STAT1/3 activation, or that the extracellular domain of PC1 engages in homotypic interactions. It is also possible that fluid flow may regulate this activity. During renal injury and in PKD, PC1 appears to undergo proteolytic cleavage that releases its cytoplasmic tail into the cytoplasm. This turns “off” the ability of the remaining membrane-anchored portion of PC1 to activate STAT1/3. However, the soluble PC1 tail can now translocate to the nucleus and co-activate STAT3 that has been activated by prior growth factor signaling. In addition to STAT3, the cleaved PC1 tail can also co-activate STAT6 (bottom) and STAT1 (not shown here). Therefore, the cleaved PC1 tail would have the ability to amplify different signaling pathways that lead to different cellular responses depending on the growth factor and cytokine environment.
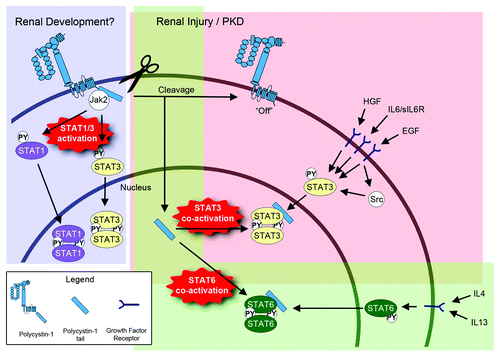
The role in STAT signaling of PC1‘s binding partner, PC2, has not been conclusively elucidated. PC2 was shown to be necessary for PC1-induced activation of JAK2/STAT1 and subsequent p21 expressionCitation66 and cell cycle inhibition.Citation66 PC2 has also been shown to directly inhibit proliferation by interaction with the p21 inhibitor Id2 however any involvement of STAT1 was not investigated.Citation69 Subsequent studies found that PC2 affected proliferation in a STAT1/p21-independent manner.Citation70 Collectively, while it is clear that PC1 activates STAT1 and STAT3, the contribution and/or independent functioning of PC2 in this process is not yet clear.
The Role of STAT3 in Renal Cyst Growth
STAT3 is strongly activated by tyrosine-phosphorylation in cyst-lining cells in human ADPKD kidneys and four different PKD mouse models.Citation42 Independently, two other laboratories also reported strong STAT3 activation in two independent Pkd1 mouse models.Citation71,Citation72 Importantly, attempts to inhibit STAT3 in PKD mouse models have led to promising results. Treatment of Pkd1 mice with high doses of the natural compound curcumin led to inhibition of renal cyst growth.Citation71 Curcumin has an extremely broad spectrum of molecular targets including Ser/Thr-kinases (including mTOR), Tyr-kinases, growth factor and cytokine receptors, inflammatory enzymes and several transcription factors including STAT3.Citation73 It is possible that the beneficial effect of curcumin observed in Pkd1 mice may be partially due to inhibition of STAT3. Curcumin was also recently found to inhibit cyst growth in an in vitro cell culture system and in embryonic kidney culture but any possible role of STAT3 was not investigated.Citation74 Another group identified the anti-parasitic compound pyrimethamine as a novel STAT3 inhibitor and showed that it inhibits renal STAT3 activity and renal cyst growth in a Pkd1 mouse model.Citation72 Similar results were obtained using another STAT3 inhibitor, S3I-201.Citation72 Even though the specificity toward STAT3 of these compounds is either poor or not well established, altogether these studies suggest that STAT3 may be a highly promising therapeutic target for treatment of PKD. More specific inhibitors or genetic approaches are needed to define the contribution of STAT3 as a driver of renal cyst growth.
What are the possible upstream activators of STAT3 in PKD? Since PC1, the protein affected in most cases of ADPKD, regulates STAT3 (see above) it is reasonable to assume that PC1 may play a role in the aberrant activation of STAT3 in renal cysts. However, the picture is complicated by paradoxical situations. Two pathogenic patient mutations were identified that altered the ability of membrane-anchored PC1 to activate STAT3, however, one mutation increased STAT3 activation while another mutation diminished it.Citation42 More strikingly, the effect of PC1 on ADPKD is altogether paradoxical because renal cyst growth can be caused by both reducing/eliminating the expression of PC1 (e.g., in conditional KO models or by hypomorphic alleles) and also by overexpression of PC1.Citation75,Citation76 While ADPKD has traditionally been viewed as resulting from the loss of PC1, kidneys of ADPKD patients have actually consistently been found to overexpress PC1.Citation42,Citation67,Citation77,Citation78 Indeed, the cleaved, C-terminal tail of PC1 is strongly overexpressed in kidneys from ADPKD patientsCitation42 and a PKD mouse model.Citation68 PC1 expression is also increased after renal injury.Citation79 Since the cleaved PC1 tail has the ability to co-activate STAT signaling in response to cytokine/growth factor activityCitation42 it is possible that the observed STAT3 activation in PKD is due to a combination of STAT3-activating cytokines and the signal-amplifying property of the cleaved PC1 tail.
Several growth factors and upstream activators of STAT3 have been implicated in PKD including epidermal growth factor (EGF) and its receptor (EGFR), HGF and its receptor c-Met, and Src. EGFCitation80 and HGFCitation81 are both elevated in PKD kidneys and found in cyst fluid, the EGFR is overexpressed and mis-targeted to the apical plasma membrane in cyst-lining cells,Citation80 and overexpression of c-Met leads to polycystic kidneys.Citation82 Treatment of PKD mouse models with EGFR inhibitorsCitation83 and treatment of Pkd1-null embryos with a c-Met inhibitorCitation84 reduce renal cyst growth. A possible link between PC1 and c-Met/EGFR signaling has been uncovered when it was found that the loss of PC1 leads to a trafficking defect of the E3-ubiquitin ligase c-Cbl which is required for the downregulation of MET and EGFR after receptor activation.Citation84 Furthermore, Src—a tyrosine kinase that can activate STAT3 directly—is aberrantly activated in PKD, and the Src inhibitor SKI-606 reduces renal cyst growth in PKD mice.Citation85
The immune system may also play a likely role as a source of STAT3-activating cytokines in PKD. For example, IL6 is secreted by T cells and macrophages, and IL6-trans-signaling has been shown to activate STAT3 in renal tubule cells in response to AKI.Citation50 Macrophages were recently shown to promote cyst growth in PKD.Citation86 Interestingly, cystic epithelial cells secrete macrophage chemoattractants including MCP-1Citation86 whose expression is known to be driven by STAT3.Citation87 An interesting speculation is that STAT3-dependent expression of macrophage chemoattractants by tubule epithelial cells leads to macrophage recruitment which, in turn, further activate STAT3 in these cells by cytokine signaling. Such an interplay between renal epithelial cells and immune cells could lead to a vicious cycle of mutual positive feedback stimulation that causes persistent STAT3 activation and eventually cyst growth and fibrosis.
In addition to activation by tyrosine-phosphorylation, STAT3 is regulated by Ser727-phosphorylation by mTOR- and ERK-dependent pathwaysCitation88 which are known to be activated in PKD.Citation89,Citation90 However, the situation is complicated by the fact that Ser727 phosphorylation can lead both to increased and decreased nuclear STAT3 activity.Citation91,Citation92 Unphosphorylated STAT3 (U-STAT3) can also regulate gene expression which leads to a more sustained effect than the canonical effects of Tyr-phosphorylated STAT3.Citation93 Increased expression of U-STAT3 has been observed in PKD mouse models.Citation42,Citation71,Citation72 U-STAT3 has been suggested to play a role in increased expression of pro-fibrotic/inflammatory genes in acute kidney injury.Citation53 U-STAT3 can also increase the expression of c-MetCitation94 which could potentially be involved in the observed upregulation of c-Met-signaling in PKD.
Given the abundance of over-activated pathways that are known to signal via STAT3 it is surprising that the role of STAT3 in PKD has only very recently been investigated. It is currently unknown which of the biological effects of STAT3-dependent gene activation may be most relevant to the pathogenesis of PKD. Based on the known roles of STAT3 in promoting proliferation, survival and resistance to apoptosis in cancer cells, these are obvious candidates. But other effects should not be discounted. For example, STAT3 drives the expression of heme oxygenase (HO) in response to kidney injury which is thought to lead to protection from oxidative stress and increased cell survival.Citation50 A role of HO activity in the regulation of renal cyst growth has recently been identified.Citation95 Furthermore, STAT3 is required for HIF-1α RNA expression under both hypoxia and growth signaling conditions.Citation96 HIF-1α is an important regulator of tumor growth and angiogenesis and has been found to be upregulated in the renal cyst-lining epithelium and implicated in the progression of PKD.Citation97
The Role of STAT6 in Renal Cyst Growth
Our group reported high levels of activated STAT6 in cyst-lining cells in two different PKD mouse models.Citation59 This aberrant STAT6 activation appears to be in part due to persistent signaling in a positive feedback loop involving overexpression of IL13 and the IL13 receptor in cyst-lining cells, both of which are under positive transcriptional control by STAT6 itself.Citation59 Genetic removal of STAT6, by crossing the Bpk polycystic kidney mouse model with a STAT6 knockout mouse, led to a significant improvement in kidney function and decrease in cyst size.Citation59 Treatment of cystic mice with teriflunomide, the active metabolite of the clinically approved rheumatoid arthritis drug leflunomide, also decreased cystic disease.Citation59 The main mechanism of action of teriflunomide—responsible for its efficacy in rheumatoid arthritis—is as a pyrimidine synthesis inhibitor.Citation98,Citation99 However, it is not a very specific drug and has also been reported to act as a tyrosine-kinase inhibitor and to inhibit STAT6 activation.Citation100 Indeed, teriflunomide treatment of Bpk mice led to inhibition of STAT6 activity in renal cyst lining cellsCitation59 suggesting that its beneficial effect was due to this mechanism. Altogether, these results suggest that aberrant STAT6 activation is a partial driving force of renal cyst growth in PKD.
Several effects of STAT6 activation in tubular epithelial cells could play a role in PKD including fibrosis and the immune system. Fibrosis is a major contributing factor to severity of disease in ADPKD.Citation101 TGFβ plays a central role in regulating renal fibrosisCitation102 and has been shown to be upregulated in PKD.Citation103 In other tissues it has been shown that TGFβ and IL-13 regulate each other’s expressionCitation104,Citation105 and that IL-13 alone can activate fibrosis pathways.Citation106 However, the role of IL-13 and STAT6 in fibrosis has yet to be studied specifically in the kidney. IL-4 and IL-13 can induce the secretion of periostin, a protein implicated in integrin activation, cell adhesion and proliferation.Citation107 Periostin is overexpressed in ADPKD and was found to stimulate proliferation of cystic epithelial cells via its receptor αv-integrin.Citation108 Hence, periostin may contribute to renal cyst growth in an auto/paracrine fashion as a mitogen downstream of STAT6. Components of the immune system that may be involved in STAT6-mediated effects on renal cyst growth are macrophages. After renal ischemia-reperfusion injury, macrophages mediate tissue repair in a delicate balance between the M1 and M2 phenotypes, with more M1 macrophages active in the beginning injury phase shifting to more M2 macrophages later in the repair phase.Citation109 Notably, M2 macrophages (also called alternatively activated) are major IL4- and IL13-secreting (and responsive) cells and are thought to be part of general innate and rapid responses to tissue injury.Citation29,Citation110 Importantly, macrophages have recently been shown to be a significant factor in PKD. In two different mouse models of PKD, an abundance of M2 macrophages was found to surround cysts.Citation86 Strikingly, depletion of macrophages resulted in reduced disease severity in these mouse models.Citation86 It is plausible that IL4/13 secreted by these renal M2 macrophages results in aberrant STAT6 activation of tubule epithelial cells—at least as an initiating or sustaining event—and promotes their proliferation and cyst growth.
Conclusions
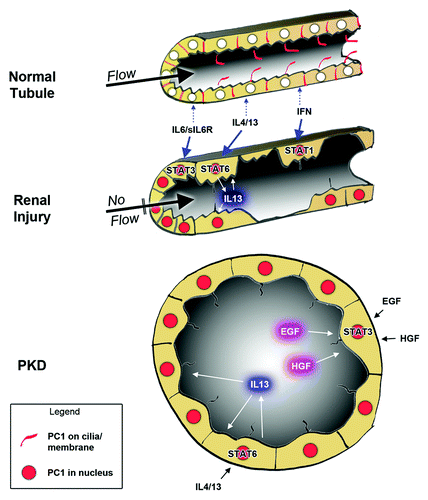
Independent work from several laboratories has established that PC1 can regulate the activity of several STAT transcription factors, that STAT3 and STAT6 are aberrantly activated in cyst-lining cells in PKD, and that they appear to be driving forces of renal cyst growth. Numerous STAT3-activating growth factors and signaling pathways have already been known to be involved in renal cyst growth in PKD for some time. However, these factors/pathways do not exclusively signal through STAT3, and any involvement of STAT3 had not been considered until very recently. Mounting evidence is beginning to lead to a model in which PC1 plays a key role in orchestrating cellular responses to growth factors that may originate from the immune system and the cyst-lining epithelial cells themselves. The purpose of the STAT-regulation by PC1 may be to sensitize cells to the cytokine environment in response to renal injury. Under these conditions—perhaps triggered by the lack of luminal fluid flow—PC1 appears to be overexpressed, proteolytically cleaved and its cytoplasmic tail accumulates in the nucleus where it can co-activate STAT proteins ( and ). In contrast, unaffected, normal tubules would be relatively insensitive to the same cytokines that would trigger repair responses in damaged tubules (). In ADPKD, this system would be permanently activated, leading to continuous “tissue repair” in the absence of actual damage.
Figure 2. Model on how the cleaved, nuclear PC1 tail can lead to sensitization to STAT-activating growth factors. In normal renal tubule cells, PC1 expression is low and its localization to restricted to primary cilia and cell junctions (red). Renal injury leads to increased PC1 expression, cleavage of its cytoplasmic tail that then traffics to the nuclei (red) where it can co-activate STAT transcription factors. Depending on the growth factor/cytokine environment, different STATs will be activated. In injured tubules, this will lead to amplified STAT activity whereas intact tubules should be relatively insensitive (due to lack of nuclear PC1 tail). Different cytokines will lead to different responses. For example, IL13 secretion by M2 macrophages would cause STAT6 activation in tubule cells which may activate tissue regeneration. In contrast, interferon secretion by M1 macrophages may trigger cell death by activation of STAT1 in damaged tubule cells. In PKD, overexpression of (mutated) PC1 may lead to constitutively high levels of the nuclear PC1 tail which will hyper-sensitize tubule cells to cytokine signaling leading to inappropriate responses. Because factors secreted into cyst lumens cannot escape, positive feedback loops establish themselves which lead to persistent activation of STAT6 and STAT3.
Much still needs to be learned about the exact molecular mechanisms that lead from the initial PC1 gene mutation to the growth of renal cysts and eventually kidney failure. However, since STAT3 and STAT6 have emerged as likely key players in the progression of PKD, they already represent promising drug targets for attempts at therapy. STAT3 is a hotly pursued target for the treatment of numerous types of cancer and it is likely that clinically useful drugs will emerge in the future. Likewise, intensive research efforts have been focused on inhibiting the IL-4/IL-13/STAT6 signaling pathway due to its involvement in asthma. Antagonistic antibodies against IL4/13 and their receptor chains are being developed and some have already shown promising results in clinical trials.
Besides numerous other proposed functions, the ability of PC1 to regulate STAT proteins may turn out to be a key function that may ultimately lead to therapeutic approaches by targeting the aberrantly activated STAT3 and STAT6 pathways.
Acknowledgments
Supported by a National Institutes of Health grant (DK62338) and a Santa Barbara Cottage Hospital—UCSB, Special Research Award for Biomedical Science to T.W.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med 2009; 60:321 - 37; http://dx.doi.org/10.1146/annurev.med.60.101707.125712; PMID: 18947299
- Gallagher AR, Germino GG, Somlo S. Molecular advances in autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis 2010; 17:118 - 30; http://dx.doi.org/10.1053/j.ackd.2010.01.002; PMID: 20219615
- Huber TB, Walz G, Kuehn EW. mTOR and rapamycin in the kidney: signaling and therapeutic implications beyond immunosuppression. Kidney Int 2011; 79:502 - 11; http://dx.doi.org/10.1038/ki.2010.457; PMID: 21085109
- Hofherr A, Köttgen M. TRPP channels and polycystins. Adv Exp Med Biol 2011; 704:287 - 313; http://dx.doi.org/10.1007/978-94-007-0265-3_16; PMID: 21290302
- Zhou J. Polycystins and primary cilia: primers for cell cycle progression. Annu Rev Physiol 2009; 71:83 - 113; http://dx.doi.org/10.1146/annurev.physiol.70.113006.100621; PMID: 19572811
- Berbari NF, O’Connor AK, Haycraft CJ, Yoder BK. The primary cilium as a complex signaling center. Curr Biol 2009; 19:R526 - 35; http://dx.doi.org/10.1016/j.cub.2009.05.025; PMID: 19602418
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med 2011; 364:1533 - 43; http://dx.doi.org/10.1056/NEJMra1010172; PMID: 21506742
- Weimbs T. Polycystic kidney disease and renal injury repair: common pathways, fluid flow, and the function of polycystin-1. Am J Physiol Renal Physiol 2007; 293:F1423 - 32; http://dx.doi.org/10.1152/ajprenal.00275.2007; PMID: 17715262
- Weimbs T. Third-hit signaling in renal cyst formation. J Am Soc Nephrol 2011; 22:793 - 5; http://dx.doi.org/10.1681/ASN.2011030284; PMID: 21493772
- Watnick T, Germino GG. mTOR inhibitors in polycystic kidney disease. N Engl J Med 2010; 363:879 - 81; http://dx.doi.org/10.1056/NEJMe1006925; PMID: 20581393
- Weimbs T, Talbot JJ. STAT3 Signaling in Polycystic Kidney Disease. Drug Discov Today Dis Mech 2013; In press http://dx.doi.org/10.1016/j.ddmec.2013.03.001
- Levy DE, Darnell JE Jr.. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 2002; 3:651 - 62; http://dx.doi.org/10.1038/nrm909; PMID: 12209125
- Battle TE, Frank DA. The role of STATs in apoptosis. Curr Mol Med 2002; 2:381 - 92; http://dx.doi.org/10.2174/1566524023362456; PMID: 12108949
- Qing Y, Stark GR. Alternative activation of STAT1 and STAT3 in response to interferon-gamma. J Biol Chem 2004; 279:41679 - 85; http://dx.doi.org/10.1074/jbc.M406413200; PMID: 15284232
- Stepkowski SM, Chen W, Ross JA, Nagy ZS, Kirken RA. STAT3: an important regulator of multiple cytokine functions. Transplantation 2008; 85:1372 - 7; http://dx.doi.org/10.1097/TP.0b013e3181739d25; PMID: 18497672
- Avalle L, Pensa S, Regis G, Novelli F, Poli V. STAT1 and STAT3 in tumorigenesis: A matter of balance. JAK-STAT 2012; 1:65 - 72; http://dx.doi.org/10.4161/jkst.20045
- Debnath B, Xu S, Neamati N. Small molecule inhibitors of signal transducer and activator of transcription 3 (Stat3) protein. J Med Chem 2012; 55:6645 - 68; http://dx.doi.org/10.1021/jm300207s; PMID: 22650325
- Schlessinger K, Levy DE. Malignant transformation but not normal cell growth depends on signal transducer and activator of transcription 3. Cancer Res 2005; 65:5828 - 34; http://dx.doi.org/10.1158/0008-5472.CAN-05-0317; PMID: 15994959
- Lin L, Hutzen B, Zuo M, Ball S, Deangelis S, Foust E, et al. Novel STAT3 phosphorylation inhibitors exhibit potent growth-suppressive activity in pancreatic and breast cancer cells. Cancer Res 2010; 70:2445 - 54; http://dx.doi.org/10.1158/0008-5472.CAN-09-2468; PMID: 20215512
- Onimoe GI, Liu A, Lin L, Wei CC, Schwartz EB, Bhasin D, et al. Small molecules, LLL12 and FLLL32, inhibit STAT3 and exhibit potent growth suppressive activity in osteosarcoma cells and tumor growth in mice. Invest New Drugs 2012; 30:916 - 26; http://dx.doi.org/10.1007/s10637-011-9645-1; PMID: 21340507
- Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U S A 2007; 104:7391 - 6; http://dx.doi.org/10.1073/pnas.0609757104; PMID: 17463090
- Darnell JE. Validating Stat3 in cancer therapy. Nat Med 2005; 11:595 - 6; http://dx.doi.org/10.1038/nm0605-595; PMID: 15937466
- Zhao M, Jiang B, Gao FH. Small molecule inhibitors of STAT3 for cancer therapy. Curr Med Chem 2011; 18:4012 - 8; http://dx.doi.org/10.2174/092986711796957284; PMID: 21824090
- Lavecchia A, Di Giovanni C, Novellino E. STAT-3 inhibitors: state of the art and new horizons for cancer treatment. Curr Med Chem 2011; 18:2359 - 75; http://dx.doi.org/10.2174/092986711795843218; PMID: 21568920
- Page BDG, Ball DP, Gunning PT. Signal transducer and activator of transcription 3 inhibitors: a patent review. Expert Opin Ther Pat 2011; 21:65 - 83; http://dx.doi.org/10.1517/13543776.2011.539205; PMID: 21114420
- Shimoda K, van Deursen J, Sangster MY, Sarawar SR, Carson RT, Tripp RA, et al. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature 1996; 380:630 - 3; http://dx.doi.org/10.1038/380630a0; PMID: 8602264
- Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity 1996; 4:313 - 9; http://dx.doi.org/10.1016/S1074-7613(00)80439-2; PMID: 8624821
- Hebenstreit D, Wirnsberger G, Horejs-Hoeck J, Duschl A. Signaling mechanisms, interaction partners, and target genes of STAT6. Cytokine Growth Factor Rev 2006; 17:173 - 88; http://dx.doi.org/10.1016/j.cytogfr.2006.01.004; PMID: 16540365
- Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol 2009; 27:451 - 83; http://dx.doi.org/10.1146/annurev.immunol.021908.132532; PMID: 19105661
- Elo LL, Järvenpää H, Tuomela S, Raghav S, Ahlfors H, Laurila K, et al. Genome-wide profiling of interleukin-4 and STAT6 transcription factor regulation of human Th2 cell programming. Immunity 2010; 32:852 - 62; http://dx.doi.org/10.1016/j.immuni.2010.06.011; PMID: 20620947
- Wei L, Vahedi G, Sun HW, Watford WT, Takatori H, Ramos HL, et al. Discrete roles of STAT4 and STAT6 transcription factors in tuning epigenetic modifications and transcription during T helper cell differentiation. Immunity 2010; 32:840 - 51; http://dx.doi.org/10.1016/j.immuni.2010.06.003; PMID: 20620946
- Urban JF Jr., Schopf L, Morris SC, Orekhova T, Madden KB, Betts CJ, et al. Stat6 signaling promotes protective immunity against Trichinella spiralis through a mast cell- and T cell-dependent mechanism. J Immunol 2000; 164:2046 - 52; PMID: 10657657
- Wills-Karp M. Interleukin-13 in asthma pathogenesis. Immunol Rev 2004; 202:175 - 90; http://dx.doi.org/10.1111/j.0105-2896.2004.00215.x; PMID: 15546393
- Quelle FW, Shimoda K, Thierfelder W, Fischer C, Kim A, Ruben SM, et al. Cloning of murine Stat6 and human Stat6, Stat proteins that are tyrosine phosphorylated in responses to IL-4 and IL-3 but are not required for mitogenesis. Mol Cell Biol 1995; 15:3336 - 43; PMID: 7760829
- Masuda A, Matsuguchi T, Yamaki K, Hayakawa T, Kubo M, LaRochelle WJ, et al. Interleukin-15 induces rapid tyrosine phosphorylation of STAT6 and the expression of interleukin-4 in mouse mast cells. J Biol Chem 2000; 275:29331 - 7; http://dx.doi.org/10.1074/jbc.M910290199; PMID: 10882748
- Gupta S, Jiang M, Pernis AB. IFN-alpha activates Stat6 and leads to the formation of Stat2:Stat6 complexes in B cells. J Immunol 1999; 163:3834 - 41; PMID: 10490982
- Fasler-Kan E, Pansky A, Wiederkehr M, Battegay M, Heim MH. Interferon-alpha activates signal transducers and activators of transcription 5 and 6 in Daudi cells. Eur J Biochem 1998; 254:514 - 9; http://dx.doi.org/10.1046/j.1432-1327.1998.2540514.x; PMID: 9688261
- Hua K, Deng J, Harp JB. Interleukin-4 inhibits platelet-derived growth factor-induced preadipocyte proliferation. Cytokine 2004; 25:61 - 7; http://dx.doi.org/10.1016/j.cyto.2003.09.008; PMID: 14693161
- Chen H, Sun H, You F, Sun W, Zhou X, Chen L, et al. Activation of STAT6 by STING is critical for antiviral innate immunity. Cell 2011; 147:436 - 46; http://dx.doi.org/10.1016/j.cell.2011.09.022; PMID: 22000020
- Denkers EY, Bzik DJ, Fox BA, Butcher BA. An inside job: hacking into Janus kinase/signal transducer and activator of transcription signaling cascades by the intracellular protozoan Toxoplasma gondii. Infect Immun 2012; 80:476 - 82; http://dx.doi.org/10.1128/IAI.05974-11; PMID: 22104110
- Wang H, Yang Y, Sharma N, Tarasova NI, Timofeeva OA, Winkler-Pickett RT, et al. STAT1 activation regulates proliferation and differentiation of renal progenitors. Cell Signal 2010; 22:1717 - 26; http://dx.doi.org/10.1016/j.cellsig.2010.06.012; PMID: 20624457
- Talbot JJ, Shillingford JM, Vasanth S, Doerr N, Mukherjee S, Kinter MT, et al. Polycystin-1 regulates STAT activity by a dual mechanism. Proc Natl Acad Sci U S A 2011; 108:7985 - 90; http://dx.doi.org/10.1073/pnas.1103816108; PMID: 21518865
- Kim M, O’Brien LE, Kwon SH, Mostov KE. STAT1 is required for redifferentiation during Madin-Darby canine kidney tubulogenesis. Mol Biol Cell 2010; 21:3926 - 33; http://dx.doi.org/10.1091/mbc.E10-02-0112; PMID: 20861313
- Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 1996; 84:443 - 50; http://dx.doi.org/10.1016/S0092-8674(00)81289-1; PMID: 8608598
- Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell 1996; 84:431 - 42; http://dx.doi.org/10.1016/S0092-8674(00)81288-X; PMID: 8608597
- Metcalf D, Mifsud S, Di Rago L, Nicola NA, Hilton DJ, Alexander WS. Polycystic kidneys and chronic inflammatory lesions are the delayed consequences of loss of the suppressor of cytokine signaling-1 (SOCS-1). Proc Natl Acad Sci U S A 2002; 99:943 - 8; http://dx.doi.org/10.1073/pnas.022628499; PMID: 11782537
- Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG. A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med 2007; 13:1490 - 5; http://dx.doi.org/10.1038/nm1675; PMID: 17965720
- Boccaccio C, Andò M, Tamagnone L, Bardelli A, Michieli P, Battistini C, et al. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature 1998; 391:285 - 8; http://dx.doi.org/10.1038/34657; PMID: 9440692
- Santos OF, Barros EJ, Yang XM, Matsumoto K, Nakamura T, Park M, et al. Involvement of hepatocyte growth factor in kidney development. Dev Biol 1994; 163:525 - 9; http://dx.doi.org/10.1006/dbio.1994.1169; PMID: 8200486
- Nechemia-Arbely Y, Barkan D, Pizov G, Shriki A, Rose-John S, Galun E, et al. IL-6/IL-6R axis plays a critical role in acute kidney injury. J Am Soc Nephrol 2008; 19:1106 - 15; http://dx.doi.org/10.1681/ASN.2007070744; PMID: 18337485
- Wang J, Ouyang C, Chen X, Fu B, Lu Y, Hong Q. STAT3 inhibits apoptosis of human renal tubular epithelial cells induced by ATP depletion/recovery. Nephron Exp Nephrol 2008; 108:e11 - 8; http://dx.doi.org/10.1159/000112557; PMID: 18097150
- Arany I, Megyesi JK, Nelkin BD, Safirstein RL. STAT3 attenuates EGFR-mediated ERK activation and cell survival during oxidant stress in mouse proximal tubular cells. Kidney Int 2006; 70:669 - 74; http://dx.doi.org/10.1038/sj.ki.5001604; PMID: 16788692
- Arany I, Reed DK, Grifoni SC, Chandrashekar K, Booz GW, Juncos LA. A novel U-STAT3-dependent mechanism mediates the deleterious effects of chronic nicotine exposure on renal injury. Am J Physiol Renal Physiol 2012; 302:F722 - 9; http://dx.doi.org/10.1152/ajprenal.00338.2011; PMID: 22169004
- Ogata K, Shimamura Y, Hamada K, Hisa M, Bun M, Okada N, et al. Upregulation of HNF-1β during experimental acute kidney injury plays a crucial role in renal tubule regeneration. Am J Physiol Renal Physiol 2012; 303:F689 - 99; http://dx.doi.org/10.1152/ajprenal.00086.2012; PMID: 22759397
- Yang N, Luo M, Li R, Huang Y, Zhang R, Wu Q, et al. Blockage of JAK/STAT signalling attenuates renal ischaemia-reperfusion injury in rat. Nephrol Dial Transplant 2008; 23:91 - 100; http://dx.doi.org/10.1093/ndt/gfm509; PMID: 17670769
- Kuratsune M, Masaki T, Hirai T, Kiribayashi K, Yokoyama Y, Arakawa T, et al. Signal transducer and activator of transcription 3 involvement in the development of renal interstitial fibrosis after unilateral ureteral obstruction. Nephrology (Carlton) 2007; 12:565 - 71; http://dx.doi.org/10.1111/j.1440-1797.2007.00881.x; PMID: 17995582
- Pang M, Ma L, Gong R, Tolbert E, Mao H, Ponnusamy M, et al. A novel STAT3 inhibitor, S3I-201, attenuates renal interstitial fibroblast activation and interstitial fibrosis in obstructive nephropathy. Kidney Int 2010; 78:257 - 68; http://dx.doi.org/10.1038/ki.2010.154; PMID: 20520592
- Li R, Yang N, Zhang L, Huang Y, Zhang R, Wang F, et al. Inhibition of Jak/STAT signaling ameliorates mice experimental nephrotic syndrome. Am J Nephrol 2007; 27:580 - 9; http://dx.doi.org/10.1159/000108102; PMID: 17823504
- Olsan EE, Mukherjee S, Wulkersdorfer B, Shillingford JM, Giovannone AJ, Todorov G, et al. Signal transducer and activator of transcription-6 (STAT6) inhibition suppresses renal cyst growth in polycystic kidney disease. Proc Natl Acad Sci U S A 2011; 108:18067 - 72; http://dx.doi.org/10.1073/pnas.1111966108; PMID: 22025716
- Yokota N, Burne-Taney M, Racusen L, Rabb H. Contrasting roles for STAT4 and STAT6 signal transduction pathways in murine renal ischemia-reperfusion injury. Am J Physiol Renal Physiol 2003; 285:F319 - 25; PMID: 12709397
- Yukawa K, Kishino M, Goda M, Liang XM, Kimura A, Tanaka T, et al. STAT6 deficiency inhibits tubulointerstitial fibrosis in obstructive nephropathy. Int J Mol Med 2005; 15:225 - 30; PMID: 15647835
- Summers SA, Phoon RK, Odobasic D, Dewage L, Kitching AR, Holdsworth SR. Signal transducer and activation of transcription 6 (STAT6) regulates T helper type 1 (Th1) and Th17 nephritogenic immunity in experimental crescentic glomerulonephritis. Clin Exp Immunol 2011; 166:227 - 34; http://dx.doi.org/10.1111/j.1365-2249.2011.04437.x; PMID: 21985369
- Chen X, Zhang Z, Jiang L, Ye F, Wang J, Wu P. Elevated interleukin-13 in patients with active lupus nephritis. Chin Med J (Engl) 2001; 114:1022 - 5; PMID: 11677758
- Lakkis FG, Cruet EN. Cloning of rat interleukin-13 (IL-13) cDNA and analysis of IL-13 gene expression in experimental glomerulonephritis. Biochem Biophys Res Commun 1993; 197:612 - 8; http://dx.doi.org/10.1006/bbrc.1993.2523; PMID: 7916615
- Sandovici M, Henning RH, van Goor H, Helfrich W, de Zeeuw D, Deelman LE. Systemic gene therapy with interleukin-13 attenuates renal ischemia-reperfusion injury. Kidney Int 2008; 73:1364 - 73; http://dx.doi.org/10.1038/ki.2008.18; PMID: 18354382
- Bhunia AK, Piontek K, Boletta A, Liu L, Qian F, Xu PN, et al. PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell 2002; 109:157 - 68; http://dx.doi.org/10.1016/S0092-8674(02)00716-X; PMID: 12007403
- Low SH, Vasanth S, Larson CH, Mukherjee S, Sharma N, Kinter MT, et al. Polycystin-1, STAT6, and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Dev Cell 2006; 10:57 - 69; http://dx.doi.org/10.1016/j.devcel.2005.12.005; PMID: 16399078
- Chauvet V, Tian X, Husson H, Grimm DH, Wang T, Hiesberger T, et al. Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 C terminus. J Clin Invest 2004; 114:1433 - 43; PMID: 15545994
- Li X, Luo Y, Starremans PG, McNamara CA, Pei Y, Zhou J. Polycystin-1 and polycystin-2 regulate the cell cycle through the helix-loop-helix inhibitor Id2. Nat Cell Biol 2005; 7:1202 - 12; http://dx.doi.org/10.1038/ncb1326; PMID: 16311606
- Felekkis KN, Koupepidou P, Kastanos E, Witzgall R, Bai CX, Li L, et al. Mutant polycystin-2 induces proliferation in primary rat tubular epithelial cells in a STAT-1/p21-independent fashion accompanied instead by alterations in expression of p57KIP2 and Cdk2. BMC Nephrol 2008; 9:10; http://dx.doi.org/10.1186/1471-2369-9-10; PMID: 18721488
- Leonhard WN, van der Wal A, Novalic Z, Kunnen SJ, Gansevoort RT, Breuning MH, et al. Curcumin inhibits cystogenesis by simultaneous interference of multiple signaling pathways: in vivo evidence from a Pkd1-deletion model. Am J Physiol Renal Physiol 2011; 300:F1193 - 202; http://dx.doi.org/10.1152/ajprenal.00419.2010; PMID: 21345977
- Takakura A, Nelson EA, Haque N, Humphreys BD, Zandi-Nejad K, Frank DA, et al. Pyrimethamine inhibits adult polycystic kidney disease by modulating STAT signaling pathways. Hum Mol Genet 2011; 20:4143 - 54; http://dx.doi.org/10.1093/hmg/ddr338; PMID: 21821671
- Kunnumakkara AB, Anand P, Aggarwal BB. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett 2008; 269:199 - 225; http://dx.doi.org/10.1016/j.canlet.2008.03.009; PMID: 18479807
- Gao J, Zhou H, Lei T, Zhou L, Li W, Li X, et al. Curcumin inhibits renal cyst formation and enlargement in vitro by regulating intracellular signaling pathways. Eur J Pharmacol 2011; 654:92 - 9; http://dx.doi.org/10.1016/j.ejphar.2010.12.008; PMID: 21187084
- Harris PC. What is the role of somatic mutation in autosomal dominant polycystic kidney disease?. J Am Soc Nephrol 2010; 21:1073 - 6; http://dx.doi.org/10.1681/ASN.2010030328; PMID: 20488953
- Lantinga-van Leeuwen IS, Dauwerse JG, Baelde HJ, Leonhard WN, van de Wal A, Ward CJ, et al. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum Mol Genet 2004; 13:3069 - 77; http://dx.doi.org/10.1093/hmg/ddh336; PMID: 15496422
- Ward CJ, Turley H, Ong AC, Comley M, Biddolph S, Chetty R, et al. Polycystin, the polycystic kidney disease 1 protein, is expressed by epithelial cells in fetal, adult, and polycystic kidney. Proc Natl Acad Sci U S A 1996; 93:1524 - 8; http://dx.doi.org/10.1073/pnas.93.4.1524; PMID: 8643665
- Lanoix J, D’Agati V, Szabolcs M, Trudel M. Dysregulation of cellular proliferation and apoptosis mediates human autosomal dominant polycystic kidney disease (ADPKD). Oncogene 1996; 13:1153 - 60; PMID: 8808689
- Prasad S, McDaid JP, Tam FW, Haylor JL, Ong AC. Pkd2 dosage influences cellular repair responses following ischemia-reperfusion injury. Am J Pathol 2009; 175:1493 - 503; http://dx.doi.org/10.2353/ajpath.2009.090227; PMID: 19729489
- Sweeney WE Jr., Avner ED. Molecular and cellular pathophysiology of autosomal recessive polycystic kidney disease (ARPKD). Cell Tissue Res 2006; 326:671 - 85; http://dx.doi.org/10.1007/s00441-006-0226-0; PMID: 16767405
- Horie S, Higashihara E, Nutahara K, Mikami Y, Okubo A, Kano M, et al. Mediation of renal cyst formation by hepatocyte growth factor. Lancet 1994; 344:789 - 91; http://dx.doi.org/10.1016/S0140-6736(94)92344-2; PMID: 7916076
- Takayama H, LaRochelle WJ, Sabnis SG, Otsuka T, Merlino G. Renal tubular hyperplasia, polycystic disease, and glomerulosclerosis in transgenic mice overexpressing hepatocyte growth factor/scatter factor. Lab Invest 1997; 77:131 - 8; PMID: 9274855
- Torres VE, Sweeney WE Jr., Wang X, Qian Q, Harris PC, Frost P, et al. EGF receptor tyrosine kinase inhibition attenuates the development of PKD in Han:SPRD rats. Kidney Int 2003; 64:1573 - 9; http://dx.doi.org/10.1046/j.1523-1755.2003.00256.x; PMID: 14531789
- Qin S, Taglienti M, Nauli SM, Contrino L, Takakura A, Zhou J, et al. Failure to ubiquitinate c-Met leads to hyperactivation of mTOR signaling in a mouse model of autosomal dominant polycystic kidney disease. J Clin Invest 2010; 120:3617 - 28; http://dx.doi.org/10.1172/JCI41531; PMID: 20852388
- Sweeney WE Jr., von Vigier RO, Frost P, Avner ED. Src inhibition ameliorates polycystic kidney disease. J Am Soc Nephrol 2008; 19:1331 - 41; http://dx.doi.org/10.1681/ASN.2007060665; PMID: 18385429
- Karihaloo A, Koraishy F, Huen SC, Lee Y, Merrick D, Caplan MJ, et al. Macrophages promote cyst growth in polycystic kidney disease. J Am Soc Nephrol 2011; 22:1809 - 14; http://dx.doi.org/10.1681/ASN.2011010084; PMID: 21921140
- Chatterjee PK, Al-Abed Y, Sherry B, Metz CN. Cholinergic agonists regulate JAK2/STAT3 signaling to suppress endothelial cell activation. Am J Physiol Cell Physiol 2009; 297:C1294 - 306; http://dx.doi.org/10.1152/ajpcell.00160.2009; PMID: 19741199
- Yokogami K, Wakisaka S, Avruch J, Reeves SA. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr Biol 2000; 10:47 - 50; http://dx.doi.org/10.1016/S0960-9822(99)00268-7; PMID: 10660304
- Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A 2006; 103:5466 - 71; http://dx.doi.org/10.1073/pnas.0509694103; PMID: 16567633
- Calvet JP. MEK inhibition holds promise for polycystic kidney disease. J Am Soc Nephrol 2006; 17:1498 - 500; http://dx.doi.org/10.1681/ASN.2006040353; PMID: 16687624
- Chung J, Uchida E, Grammer TC, Blenis J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol 1997; 17:6508 - 16; PMID: 9343414
- Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009; 323:793 - 7; http://dx.doi.org/10.1126/science.1164551; PMID: 19131594
- Yang J, Stark GR. Roles of unphosphorylated STATs in signaling. Cell Res 2008; 18:443 - 51; http://dx.doi.org/10.1038/cr.2008.41; PMID: 18364677
- Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, et al. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res 2005; 65:939 - 47; PMID: 15705894
- Zhou J, Ouyang X, Schoeb TR, Bolisetty S, Cui X, Mrug S, et al. Kidney injury accelerates cystogenesis via pathways modulated by heme oxygenase and complement. J Am Soc Nephrol 2012; 23:1161 - 71; http://dx.doi.org/10.1681/ASN.2011050442; PMID: 22518005
- Niu G, Briggs J, Deng J, Ma Y, Lee H, Kortylewski M, et al. Signal transducer and activator of transcription 3 is required for hypoxia-inducible factor-1alpha RNA expression in both tumor cells and tumor-associated myeloid cells. Mol Cancer Res 2008; 6:1099 - 105; http://dx.doi.org/10.1158/1541-7786.MCR-07-2177; PMID: 18644974
- Bernhardt WM, Wiesener MS, Weidemann A, Schmitt R, Weichert W, Lechler P, et al. Involvement of hypoxia-inducible transcription factors in polycystic kidney disease. Am J Pathol 2007; 170:830 - 42; http://dx.doi.org/10.2353/ajpath.2007.060455; PMID: 17322369
- Greene S, Watanabe K, Braatz-Trulson J, Lou L. Inhibition of dihydroorotate dehydrogenase by the immunosuppressive agent leflunomide. Biochem Pharmacol 1995; 50:861 - 7; http://dx.doi.org/10.1016/0006-2952(95)00255-X; PMID: 7575649
- Williamson RA, Yea CM, Robson PA, Curnock AP, Gadher S, Hambleton AB, et al. Dihydroorotate dehydrogenase is a high affinity binding protein for A77 1726 and mediator of a range of biological effects of the immunomodulatory compound. J Biol Chem 1995; 270:22467 - 72; http://dx.doi.org/10.1074/jbc.270.38.22467; PMID: 7673235
- Siemasko K, Chong AS, Jäck HM, Gong H, Williams JW, Finnegan A. Inhibition of JAK3 and STAT6 tyrosine phosphorylation by the immunosuppressive drug leflunomide leads to a block in IgG1 production. J Immunol 1998; 160:1581 - 8; PMID: 9469413
- Norman J. Fibrosis and progression of autosomal dominant polycystic kidney disease (ADPKD). Biochim Biophys Acta 2011; 1812:1327 - 36; http://dx.doi.org/10.1016/j.bbadis.2011.06.012; PMID: 21745567
- Liu Y. Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int 2006; 69:213 - 7; http://dx.doi.org/10.1038/sj.ki.5000054; PMID: 16408108
- Hassane S, Leonhard WN, van der Wal A, Hawinkels LJ, Lantinga-van Leeuwen IS, ten Dijke P, et al. Elevated TGFbeta-Smad signalling in experimental Pkd1 models and human patients with polycystic kidney disease. J Pathol 2010; 222:21 - 31; PMID: 20549648
- Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med 2006; 12:99 - 106; http://dx.doi.org/10.1038/nm1332; PMID: 16327802
- Scharl M, Frei S, Pesch T, Kellermeier S, Arikkat J, Frei P, et al. Interleukin-13 and transforming growth factor beta synergise in the pathogenesis of human intestinal fistulae. Gut 2013; 62:63 - 72; http://dx.doi.org/10.1136/gutjnl-2011-300498; PMID: 22287592
- Kaviratne M, Hesse M, Leusink M, Cheever AW, Davies SJ, McKerrow JH, et al. IL-13 activates a mechanism of tissue fibrosis that is completely TGF-beta independent. J Immunol 2004; 173:4020 - 9; PMID: 15356151
- Takayama G, Arima K, Kanaji T, Toda S, Tanaka H, Shoji S, et al. Periostin: a novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. J Allergy Clin Immunol 2006; 118:98 - 104; http://dx.doi.org/10.1016/j.jaci.2006.02.046; PMID: 16815144
- Wallace DP, Quante MT, Reif GA, Nivens E, Ahmed F, Hempson SJ, et al. Periostin induces proliferation of human autosomal dominant polycystic kidney cells through alphaV-integrin receptor. Am J Physiol Renal Physiol 2008; 295:F1463 - 71; http://dx.doi.org/10.1152/ajprenal.90266.2008; PMID: 18753297
- Iwata Y, Boström EA, Menke J, Rabacal WA, Morel L, Wada T, et al. Aberrant macrophages mediate defective kidney repair that triggers nephritis in lupus-susceptible mice. J Immunol 2012; 188:4568 - 80; http://dx.doi.org/10.4049/jimmunol.1102154; PMID: 22467656
- Loke P, Gallagher I, Nair MG, Zang X, Brombacher F, Mohrs M, et al. Alternative activation is an innate response to injury that requires CD4+ T cells to be sustained during chronic infection. J Immunol 2007; 179:3926 - 36; PMID: 17785830