Abstract
Primary tumors can affect organ functions, either mechanically when they grow to a considerable size or via production of hormones. However, mortality of cancer patients is in most cases due to formation of secondary growths.Citation1,Citation2 Consequently, various drugs are currently employed in clinical trials to impair specific steps of cancer metastasis such as mesenchymal or amoeboid cell migration, intravasation and/or colonization.Citation2 From the clinical point of view, targeting late metastatic processes such as extravasation or colonization might be required for cancer patients that bear already dormant micrometastases in their capillaries which have left behind earlier metastatic steps. Development of such drugs needs characterization of molecular targets implicated in distinct steps of cancer metastasis.
Extravasation of tumor cells resembles diapedesis of leukocytes which penetrate blood vessel walls at sites of inflammation. Similar to leukocytes, tumor cells attach to endothelial cells through adhesion molecules such as selectins.Citation2 More ingenious, instead of inventing extravasation anew, tumor cells can attract neutrophils via chemotactic factors and use them as carriage horses to leave the capillaries.Citation3 A related role has been attributed to platelets that are exploited by tumor cells for attachment to blood vessel endothelia. Attachment is mediated by a fibrinogen coat that can shield tumor cells from shear stress or lytic NK cell attack. Although platelets do not guide tumor cells through the blood vessel wall, they secrete factors such as TGFβ1 that promote epithelial to mesenchymal transition and extravasation.Citation4 Additional support for extravasation is provided by vascular permeability factors such as vascular endothelial growth factor (VEGF). The founding member of the VEGF family, VEGF-A, was initially also termed “vascular permeability factor” (VPF) based on its ability to enhance the leakiness of blood vessels to plasma and plasma proteins. VEGF is produced by stromal cells but many cancer cells (including cells of hematologic malignancies) have acquired the ability to contribute to VEGF production.Citation5 Thus, VEGF does not only promote angiogenesis and vascular permeability in primary tumors but also extravasation of disseminated micrometastases.Citation6 Alternative factors that modulate endothelial barrier integrity are extracellular matrix proteins such as TGFβ-induced (TGFBI)Citation7 or the protease thrombinCitation8 that act on endothelial cells and result in cytoskeletal rearrangements ().
Figure 1. Disseminated tumor cells use different extravasation mechanisms. (A) Tumor cells can activate the coagulation system and generate thrombin from its precursor, prothrombin, at sites of metastasis.Citation23 Activation of the thrombin receptor PAR-1 on endothelial cells leads to G protein-mediated activation of RhoA and RhoA downstream signaling components that converge on the increase of actomyosin contractility and cell-cell junction remodeling. Adapted from reference Citation8. (B) VEGF, derived from tumor cells at the site of metastasis, activates focal adhesion kinase (FAK) in endothelial cells that subsequently binds to the cytoplasmic tail of VE-cadherin (VE-Cad). At this location, β-Catenin (β-Cat) is phosphorylated by FAK and dissociates from VE-cadherin leading to endothelial cell junctional breakdown.Citation6 Additional modes how VEGF affects vascular permeability have also been described.Citation24 (C) Plateletes can support metastasis in several ways: Platelet-derived TGFβ1 activates SMADs in tumor cells. Simultaneously, NFκB is activated by platelet-tumor cell contacts. Both synergize to induce epithelial-mesenchymal transition (EMT) thereby promoting extravasation. Adapted from reference Citation4. (D) Human melanoma cells readily extravasate when co-injected with human neutrophils into recipient mice. This effect is mediated by melanoma-derived IL-8 which attracts neutrophils and induces expression of β2-Integrin (β2-Int). Neutrophils anchor tumors cells to endothelial cells via contacts of β2-Integrin to extracellular matrix compounds and the cell adhesion molecule ICAM-1.Citation3 (E) Tumor cell-derived CCL2 can activate corresponding CCR2 receptors on endothelial cells thereby inducing JAK2/STAT5 and p38MAPK signaling pathways. Both pathways synergize to increase vascular permeability. p38MAPK induces expression of E-Selectin (E-Sel) in endothelial cells which promotes attachment of tumor cells. Simultaneously, monocytes are attracted by CCL2 which support extravasation of tumor cells through walls of capillaries with reduced barrier function. Adapted from reference Citation9.
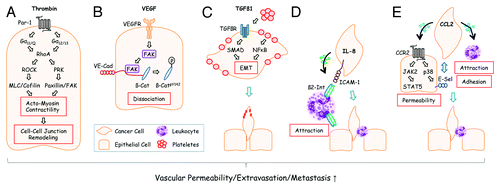
In a recent issue of Cancer Cell, Wolf and coworkers have reported about a novel mechanism in tumor cell metastasis that involves the chemokine CCL2.Citation9 This mechanism goes beyond the well-characterized pro-metastatic functions of CCL2/CCR2-signaling in primary tumorsCitation10 and provides already disseminated cancer cells with an enhanced potential for extravasation and colonization. Chemokines are mediators of heterotypic interactions between tumor cells and their microenvironment. These interactions are essential for acquisition of invasive and metastatic behaviors. Chemokines bind to extracellular matrix compounds such as heparin and proteoglycans. They activate signaling in target cells via 7-span transmembrane chemokine receptors and trimeric G proteins. The activity of chemokines in metastasis is pleiotropic: they are implicated in many metastatic steps such as local invasiveness, intravasation, anoikis, extravasation, chemotaxis and colonization.Citation10 The chemokine CCL2 (also termed monocyte chemotactic protein 1 or MCP-1) is implicated in immunosuppression and tumor angiogenesis.Citation4,Citation11 Moreover, CCL2 represents a major chemotactic factor for attraction of myeloid-derived suppressor cells (MDSCs), circulating monocytes and other immune cells to the tumor stroma.Citation11,Citation12 Consequently, CCL2 receptors CCR2 and CCR4 are expressed on immune cells (monocytes, macrophages, dendritic cells, NK cells and T cells). CCR2 is also expressed on epithelial and endothelial cells whereas CCR4 is not. Apart from the well characterized pro-metastatic activities in tumors (e.g., activation in endothelial cells promotes tumor neovascularization via matrix metalloproteinase MMP-14 activityCitation13), Wolf and coworkers discovered a distinct function of CCL2/CCR2-signaling in distant endothelia of capillaries that is triggered by CCL2-producing tumor cells. They used lung colonization by MC38 colorectal cancer cells in mice as model for identification of molecular events triggered by CCL2/CCR2 during tumor cell extravasation. The C57BL/6-syngeneic MC38 colonization model is not suitable for investigation of initial events of cancer metastasis such as local invasiveness or intravasation. However, it offers the advantage to appropriately manipulate gene expression in transplanted cancer cells, a task that is more difficult to achieve in primary tumor models. CCR2 functions were selectively ablated, either in metastatic target tissues (by using CCR2-deficient recipient mice) or in tumor cells (by shRNA-mediated knock-down of CCR2). Moreover, reciprocal bone marrow reconstitution experiments were performed to evaluate the implication of monocytic CCL2/CCR-signaling in tumor cell extravasation. These experiments demonstrated that tumor cell-derived CCL2 promotes extravasation by a dual mechanism: (1) chemoattraction of CCR2+ Ly6Chigh monocytes (which might be used by tumor cells as carriers through the vessel wall) and (2) enhancement of local vascular permeability. Of note, the permeabilizing effect of CCL2 on the endothelial compartment in this metastasis model turned out to be more important for extravasation than chemoattraction of leukocytes. CCL2 exerted this permeabilizing effect in a transient and non-systemic manner. Vascular endothelia became impermeable again after tumor cells have found their way through the vessel walls and established secondary growths in metastatic niches. The relevant CCL2/CCR2 downstream signaling pathways for extravasation were identified in vitro using tumor cell transmigration in the presence and absence of specific inhibitors. These experiments, together with additional experiments performed in mice, revealed the JAK2/STAT5 and p38MAPK pathways downstream of CCL2/CCR2 as crucial for induction of vascular permeability. p38MAPK is known to promote extravasation in a non-tumor cell-autonomous manner as reported by Matsuo and coworkers. They used heterozygous p38α+/− mice as recipients for tumor cell transplantation studies and observed impaired lung colonization of B16 melanoma cells when compared with p38α+/+ recipients. At the molecular level, p38α was shown to regulate expression of P-selectin and E-selectin in endothelial cells thereby promoting endothelial adhesion of tumor cells.Citation14 In contrast, the role of JAK-STAT activation in cellular functions leading to extravasation is not well defined. G-proteins such as Gαs, Gαi, Gαq and Gα12 have been reported to activate STAT1 and STAT3 via several signaling intermediates such as JAKs, Src-kinase or Ras/Raf/MEK/ERK. These activation events are implicated in molecular processes leading to cell transformation, neurite outgrowth or STAT-mediated transactivation of platelet-derived growth factor α.Citation15 JAK2/STAT5 enhances adhesiveness of cancer cells in cooperation with Ras/ERK signaling which might contribute to metastasis.Citation16 In contrast, STAT1 inhibits Ras/MAPK signaling, cell-cell adhesion and transcriptional activity of STAT3/5 which depends on STAT1 Y701 and S727 phosphorylation.Citation17 Besides these cell-autonomous activities, non-tumor cell autonomous functions of JAK-STAT in extravastation and its crosstalk with other signaling pathways have to be further addressed. For example, JAKs can phosphorylate tyrosines on receptor tyrosine kinases (RTKs) thereby influencing docking of SH2-containing adaptor proteins for activation of Ras and PI3K pathways.Citation18 It would be interesting to evaluate extravasation of tail vein-injected tumor cells in mice with conditional deletion of JAK2 or STAT5 in vascular endothelial cells.
The implication of CCL2/CCR2-signaling in early and late metastatic steps identifies it as an attractive target for anti-metastatic therapies. As a proof of concept, CCL2-neutralizing antibodies have been successfully used to block dissemination of prostate and breast cancer cells in mouse transplantation models.Citation19-Citation21 Moreover, the small molecular weight anti-inflammatory compound bindarit, which interferes with CCL2 production, seems to operate as anti-metastatic drug at least in mouse xenograft models.Citation22 The findings by Wolf and coworkers support continuation of therapeutic approaches to block CCL2/CCR2-signaling. Moreover, they suggest new approaches that target JAK2/STAT5 and p38MAPK in the endothelial compartment at micrometastatic sites. Additional studies are needed to clarify if CCL2/CCR2-signaling is generally used by cancer cells for extravasation or if other chemokine/chemokine receptor pairs have similar pro-metastatic activities at distant sites. These studies might provide novel biomarkers for metastasis and increase the number of potential molecular targets for anti-metastatic therapies.
References
- Rampetsreiter P, Casanova E, Eferl R. Genetically modified mouse models of cancer invasion and metastasis. Drug Discov Today Dis Models 2011; 8:67 - 74; http://dx.doi.org/10.1016/j.ddmod.2011.05.003; PMID: 22577462
- Spano D, Heck C, De Antonellis P, Christofori G, Zollo M. Molecular networks that regulate cancer metastasis. Semin Cancer Biol 2012; 22:234 - 49; http://dx.doi.org/10.1016/j.semcancer.2012.03.006; PMID: 22484561
- Huh SJ, Liang S, Sharma A, Dong C, Robertson GP. Transiently entrapped circulating tumor cells interact with neutrophils to facilitate lung metastasis development. Cancer Res 2010; 70:6071 - 82; http://dx.doi.org/10.1158/0008-5472.CAN-09-4442; PMID: 20610626
- Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011; 20:576 - 90; http://dx.doi.org/10.1016/j.ccr.2011.09.009; PMID: 22094253
- Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol 2002; 20:4368 - 80; http://dx.doi.org/10.1200/JCO.2002.10.088; PMID: 12409337
- Chen XL, Nam JO, Jean C, Lawson C, Walsh CT, Goka E, et al. VEGF-induced vascular permeability is mediated by FAK. Dev Cell 2012; 22:146 - 57; http://dx.doi.org/10.1016/j.devcel.2011.11.002; PMID: 22264731
- Ma C, Rong Y, Radiloff DR, Datto MB, Centeno B, Bao S, et al. Extracellular matrix protein betaig-h3/TGFBI promotes metastasis of colon cancer by enhancing cell extravasation. Genes Dev 2008; 22:308 - 21; http://dx.doi.org/10.1101/gad.1632008; PMID: 18245446
- Gavard J, Gutkind JS. Protein kinase C-related kinase and ROCK are required for thrombin-induced endothelial cell permeability downstream from Galpha12/13 and Galpha11/q. J Biol Chem 2008; 283:29888 - 96; http://dx.doi.org/10.1074/jbc.M803880200; PMID: 18713748
- Wolf MJ, Hoos A, Bauer J, Boettcher S, Knust M, Weber A, et al. Endothelial CCR2 signaling induced by colon carcinoma cells enables extravasation via the JAK2-Stat5 and p38MAPK pathway. Cancer Cell 2012; 22:91 - 105; http://dx.doi.org/10.1016/j.ccr.2012.05.023; PMID: 22789541
- Mukaida N, Baba T. Chemokines in tumor development and progression. Exp Cell Res 2012; 318:95 - 102; http://dx.doi.org/10.1016/j.yexcr.2011.10.012; PMID: 22036649
- Huang B, Lei Z, Zhao J, Gong W, Liu J, Chen Z, et al. CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett 2007; 252:86 - 92; http://dx.doi.org/10.1016/j.canlet.2006.12.012; PMID: 17257744
- Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011; 475:222 - 5; http://dx.doi.org/10.1038/nature10138; PMID: 21654748
- Gálvez BG, Genís L, Matías-Román S, Oblander SA, Tryggvason K, Apte SS, et al. Membrane type 1-matrix metalloproteinase is regulated by chemokines monocyte-chemoattractant protein-1/ccl2 and interleukin-8/CXCL8 in endothelial cells during angiogenesis. J Biol Chem 2005; 280:1292 - 8; http://dx.doi.org/10.1074/jbc.M408673200; PMID: 15516694
- Matsuo Y, Amano S, Furuya M, Namiki K, Sakurai K, Nishiyama M, et al. Involvement of p38alpha mitogen-activated protein kinase in lung metastasis of tumor cells. J Biol Chem 2006; 281:36767 - 75; http://dx.doi.org/10.1074/jbc.M604371200; PMID: 17028194
- Ho MK, Su Y, Yeung WW, Wong YH. Regulation of transcription factors by heterotrimeric G proteins. Curr Mol Pharmacol 2009; 2:19 - 31; PMID: 20021442
- Shi Z, Hodges VM, Dunlop EA, Percy MJ, Maxwell AP, El-Tanani M, et al. Erythropoietin-induced activation of the JAK2/STAT5, PI3K/Akt, and Ras/ERK pathways promotes malignant cell behavior in a modified breast cancer cell line. Mol Cancer Res 2010; 8:615 - 26; http://dx.doi.org/10.1158/1541-7786.MCR-09-0264; PMID: 20353997
- Wang S, Koromilas AE. Stat1 is an inhibitor of Ras-MAPK signaling and Rho small GTPase expression with implications in the transcriptional signature of Ras transformed cells. Cell Cycle 2009; 8:2070 - 9; http://dx.doi.org/10.4161/cc.8.13.8891; PMID: 19502801
- Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci 2004; 117:1281 - 3; http://dx.doi.org/10.1242/jcs.00963; PMID: 15020666
- Lu X, Kang Y. Chemokine (C-C motif) ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J Biol Chem 2009; 284:29087 - 96; http://dx.doi.org/10.1074/jbc.M109.035899; PMID: 19720836
- Loberg RD, Ying C, Craig M, Yan L, Snyder LA, Pienta KJ. CCL2 as an important mediator of prostate cancer growth in vivo through the regulation of macrophage infiltration. Neoplasia 2007; 9:556 - 62; http://dx.doi.org/10.1593/neo.07307; PMID: 17710158
- Loberg RD, Ying C, Craig M, Day LL, Sargent E, Neeley C, et al. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res 2007; 67:9417 - 24; http://dx.doi.org/10.1158/0008-5472.CAN-07-1286; PMID: 17909051
- Zollo M, Di Dato V, Spano D, De Martino D, Liguori L, Marino N, et al. Targeting monocyte chemotactic protein-1 synthesis with bindarit induces tumor regression in prostate and breast cancer animal models. Clin Exp Metastasis 2012; 29:585 - 601; http://dx.doi.org/10.1007/s10585-012-9473-5; PMID: 22484917
- Han N, Jin K, He K, Cao J, Teng L. Protease-activated receptors in cancer: A systematic review. Oncol Lett 2011; 2:599 - 608; PMID: 22848234
- Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol 2006; 8:1223 - 34; http://dx.doi.org/10.1038/ncb1486; PMID: 17060906