Abstract
Janus kinase (JAK)-signal transducers and activators of transcription (STAT) signaling pathways are critical for the maintenance of homeostatic and developmental processes; however, deregulation and chronic activation of JAK-STAT3 results in numerous diseases. Among others, obesity is currently being intensively studied. In obesity, chronic JAK-STAT3 is activated by the CNS by increased circulating leptin levels leading to the development of leptin resistance, whereas in the peripheral organs chronic IL-6-induced JAK-STAT3 impairs insulin action. We report the consequences of chronic JAK-STAT3 induced signaling as present under obese conditions in the main metabolic organs.
The evolutionary conserved Janus kinase (JAK)-signaling transducers and activators of transcription (STAT) signaling pathway is used by a variety of cytokines, hormones and growth factors to regulate numerous developmental and homeostatic processes, including hematopoiesis, immune cell development, stem cell maintenance and organismal growth.Citation1 However, chronic activation of JAK-STAT underlies various diseases or disorders, such as cancer or obesity.Citation2-Citation4
Upon binding of the ligand to its receptor, two or more receptor-associated JAKs are brought into close proximity through receptor oligomerization, which leads to their autophosphorylation and/or transphosphorylation by the opposing JAK. Specifically, JAKs possess two nearly identical phosphate transfer domains, one domain exhibits the kinase activity, and the second inhibits the kinase activity of the first. Four members of the JAK family have been identified to date, namely JAK1, 2, 3, and TYK2.Citation5-Citation11 The activated JAKs phosphorylate signature tyrosine residues in the cytoplasmic region of the receptors to create docking sites for STATs. The mammalian STAT protein family comprises seven members, namely STAT 1, 2, 3, 4, 5A, 5B and 6, which bind to phosphorylated tyrosine residues with their Src homology 2 (SH2) domain.Citation12-Citation18 Upon docking to the receptor, the JAKs phosphorylate conserved tyrosine residues within the STAT protein that are located between the SH3 domain and the C-terminal transactivation domain (TAD). This phosphorylation results in the formation of parallel STAT dimers, which are stabilized by reciprocal phosphotyrosine and SH2 domain interactions.Citation19,Citation20 Dimerization of STATs induces their nuclear translocation, whereupon the STAT dimer or more complex STAT oligomers bind to specific palindromic enhancer sequences in their target genes, ultimately controlling the expression of these genes.Citation19-Citation25
The JAK-STAT signaling pathway transcriptionally regulates its own suppressor. Suppressors of cytokine signaling (SOCS) molecules act as a negative feedback signal by inhibiting JAK and STAT activation and phosphorylation.Citation26,Citation27 The SOCS family includes eight family members, SOCS1–7 and cytokine-inducible SH2-containing protein (CIS).Citation28,Citation29 Another known suppressor of JAK-STAT is the phosphotyrosine phosphatase 1B (PTP1B), which can directly inactivate JAK and STAT and/or prevent JAK interaction with the tyrosine residue of the receptors.Citation30,Citation31 Moreover, the transcriptional activity of STAT in the nucleus can also be controlled by protein inhibitors of activated STAT (PIAS), which blocks the DNA-binding activity of STAT.Citation32
This review focuses on the chronic JAK-STAT3-SOCS3 signaling that is induced by accelerated leptin and interleukin-6 (IL-6) as present in obesity.
Chronic JAK-STAT3-SOCS3 in the CNS Causes Leptin Resistance and Obesity
Obesity is a steadily increasing health burden that affects more than one-third of Western populations. This disorder develops either as a result of increased nutritional intake, of decreased physical activity or both. These processes of energy uptake and expenditure are tightly controlled via leptin and insulin, hormones that act on specific neuronal populations in the central nervous system (CNS).Citation33,Citation34 Leptin is secreted from white adipocytes proportional to the amount of fat stored in the white adipose tissue and acts in the CNS under normal conditions to suppress food intake and increase energy expenditure.Citation35 Leptin binds to the leptin receptor (LepR), of which multiple isoforms exist. In mice, six isoforms of the lepr protein have been identified (LepRa–LepRf) that can be divided into three classes with distinct functions: four short isoforms with shortened intracellular tails, one secreted (LepRe) and one long isoform (LepRb).Citation36 The LepRb consists of 1,162 amino acids and is the only isoform with a clearly demonstrated signaling capacity.Citation37 LepRb belongs to the family of class 1 cytokine receptors, which contain an extracellular ligand-binding, a transmembrane spanning and a cytoplasmic signaling domain.Citation37 Similar to other cytokine receptors, LepRb does not possess intrinsic enzymatic activity. Instead this receptor signals via JAK2, a non-covalently associated tyrosine kinase of the JAK family.Citation38-Citation40 The autophosphorylation of JAK2 results in its activation and further phosphorylation of the tail of LepRb, which is then bound by the SH2 domain of STAT3.Citation41 In this way, LepRb-induced gene expression mediates the effects of leptin on energy homeostasis and neuroendocrine function.
The absence of LepRb in db/db mice results in obesity, impaired growth, infertility and diabetes mellitus.Citation37 These mice display a phenotype that is indistinguishable from that of db3J/ db3J mice, which lack all three LepR isoforms, and comparable to that of leptin-deficient ob/ob animals.Citation41 Leptin administration to leptin-deficient ob/ob mice reduces the development of obesity and results in normal and lean mice.Citation42 Pioneering studies using the Cre-loxP system revealed that leptin exerts its physiological effects in neurons of the CNS, given that mice carrying a specific LepR inactivation in Nestin-expressing neurons developed obesity comparable to that of ob/ob and db/db mice.Citation43 Furthermore, complementation of LepRb expression specifically in the CNS of db/db mice resulted in lean mouse mutants, further underlining leptin’s function in the CNS.Citation44 In line with these experiments, intracerebroventricular leptin administration reduces food intake in lean and ob/ob mice.Citation45 Interestingly, conditional inactivation of STAT3 in neurons of the CNS phenocopied the obese phenotype of LepR-deficient mice, which indicates that LepR-induced signaling is mediated through downstream activation of STAT3.Citation46
LepR expression is widely distributed throughout the brain, and identification of neuronal populations that mediate leptin’s effects on energy expenditure and caloric intake is important to understand the physiological relevance of leptin action. Early dissectional studies have demonstrated that specific areas of the hypothalamus, such as the arcuate nucleus, are crucial for the central control of energy expenditure and food ingestion. In line with this notion, experiments with conditional LepR mice demonstrated that neurons which express the anorexigenic pro-opiomelanocortin (POMC) and those expressing orexigenic agouti-related peptide (AgRP) and neuropeptide Y (NPY)—two neuronal populations in the arcuate nucleus—are primary sites within the brain that control the aforementioned processes through further regulation of second-order neurons.Citation47 Specifically, LepR-induced signal transduction via STAT3 directly activates POMC expression, which is post-translationally processed into the active peptide α melanocyte-stimulating hormone (MSH).Citation48 The secreted α-MSH activates second-order neurons that express the receptors for α-MSH namely melanocortin 3 and 4 receptors (MC3R and MC4R).Citation35 On the other hand, leptin reduces AgRP expression, which inhibits MC3R and MC4R induced signaling.Citation49 In line with this evidence, defined inactivation of either LepR or STAT3 in POMC neurons reduced POMC expression and energy expenditure and increased food intake, thereby causing mild obesity in the knockout animals; however, the obesity was less severe compared with that of db/db mice, which implies that leptin targets neuronal populations other than POMC neurons in the arcuate nucleus.Citation47,Citation48
The importance of these knockout studies for our current understanding of the molecular mechanisms of leptin action in specific neuronal populations of the CNS is invaluable; however, exogenous leptin administration shows no therapeutic effect in most patients with obesity or diet-induced obese mice. This fact arises owing to the development of leptin resistance, a phenomenon that underlies the majority of obesity cases rather than the rare described mutations in the genes encoding leptin or LepR. In obese individuals with leptin resistance, leptin levels are high due to the drastically elevated fat load and expansion of the white adipose tissue; the protein is, however, unable to convey its biological effects. Two mechanisms have been proposed that might account for the development of leptin resistance: (1) the saturation of leptin transport across the blood-brain barrier in obesity or (2) the chronic leptin signal transduction caused by high leptin levels increases expression of negative feedback regulators, thereby making LepR-expressing neurons resistant to leptin. Although the first assumption has been addressed by some reports,Citation50 the current scientific view favors the second argument.
Leptin-induced signaling via STAT3 rapidly activates the negative feedback regulator SOCS3, which inhibits leptin-induced signal transduction. It is tempting to speculate that, under obese conditions, increased basal circulating leptin might increase the basal activity of LepRb/STAT3 signaling in neurons of the CNS. This process would in turn elevate the otherwise silent SOCS3 expression levels, thus further impairing LepRb sensitivity. Leptin resistance therefore manifests mainly as the inability of acute leptin action to suppress food intake and increase energy expenditure.Citation51 A function of central SOCS3 in limiting LepRb action has been implicated by the finding of largely accelerated SOCS3 expression in numerous animal models of obesity.Citation52 Moreover, mice lacking one allele of SOCS3 are protected from the development of diet-induced obesity and maintain central leptin sensitivity.Citation53 Of note, the complete inactivation of SOCS3 is embryonically lethal owing to the fact that SOCS3 limits signaling of numerous cytokine receptors during development and physiology.Citation54,Citation55 Whereas mice with a CNS-specific inactivation of SOCS3 still retain sensitivity to leptin under obese conditions,Citation56 in mice with inactivation of SOCS3 in POMC neurons leptin sensitivity is only maintained in this neuron population; these animals are thus only mildly protected against diet-induced leptin resistance.Citation57 Evidently, these experiments demonstrate a critical role of SOCS3 as a negative regulator of LepRb-induced signaling in the CNS.
Gamber and colleagues demonstrated increased expression of the LepRb in the hypothalamus of obese mice, especially in POMC neurons. To investigate the relevance of this finding in the development of obesity, the authors generated mice overexpressing LepRb in POMC neurons.Citation51 Overexpression of LepRb specifically in POMC neurons had no effect on adiposity or SOCS3 expression under normal conditions; however, diet-induced obesity associated with elevated circulating leptin levels increased SOCS3 expression in POMC neurons and accelerated caloric intake and subsequently elevated body weight of these mice. These results indicate that an over-reactivity of LepR-induced signaling in POMC neurons—as present in obesity—increases SOCS3 expression, which in turn leads to the development of leptin resistance.Citation51 In line with these experiments, we could demonstrate that expression of a constitutively active variant of STAT3 specifically in POMC neurons increases SOCS3 expression independent of leptin action under normal conditions. This expression translated into increased food intake and adiposity as a result of the development of leptin resistance.Citation58 When exposed to diet-induced obesity, these mutant mice developed obesity as severe as that of control mice; both cohorts showed comparable leptin-resistant states and similarly activated STAT3 and elevated SOCS3 levels.Citation58 Taken together, these data suggest that high leptin in obesity increases basal LepRb-induced STAT3-mediated downstream signaling, particularly in POMC neurons, which eventually increases SOCS3 expression, thereby impairing acute leptin-activated signaling, namely by developing a resistance to leptin. However, while the role of leptin resistance in POMC neurons seems to meet the current model, the function of elevated leptin-induced STAT3 activation specifically in AgRP neurons resulted in a different outcome. Mice with overactivated STAT3 in AgRP neurons show a relative resistance to diet-induced obesity owing to increased locomotor activity.Citation59 Though STAT3 activation was supposed to alter AgRP and SOCS3 expression in this neuronal subpopulation, expression of those genes was unaltered in these mice. Thus, these experiments demonstrate that mechanisms of leptin resistance in AgRP neurons and POMC neurons might follow a different path. Nevertheless, signals other than leptin might well increase central SOCS3 expression under obese conditions that lead to leptin resistance.
Noteworthy, while numerous studies report the CNS as major site for leptin-induced STAT3 activity, also peripheral actions of leptin have been described. Of note, an unanticipated role for leptin-induced JAK-STAT3 in cardioprotection has been discovered.Citation60 Here, STAT3 activation by leptin reduced infarct size, a function that has been previously demonstrated for TNFα-induced STAT3 action.Citation61 Collectively, these studies reveal that leptin not only exerts central actions but can also have beneficial effects in peripheral organs. However, though such peripheral functions of leptin exist, chronic JAK-STAT3-SOCS3 action in obesity is mainly derived from other signals that vice versa not only act in the periphery but can also have substantial role in the CNS. Among these signals, not only inflammatory cytokines, such as IL-6 and tumor necrosis factor α, but also free fatty acids and other lipids.
Chronic JAK-STAT3-SOCS3 in Peripheral Organs Causes Insulin Resistance
As recently shown in obesity elevated circulating levels of cytokines impair insulin signaling in peripheral organs and even in the brain.Citation62,Citation63 Several studies have demonstrated a link between insulin resistance and obesity as a chronic inflammatory state. In obese mice and humans numerous inflammatory cytokines are released in excess by the white adipose tissue derived from adipocytes as well as from recruited inflammatory cells such as macrophages.Citation64-Citation66 Among the most prominent cytokines that are over-represented in the bloodstream of obese individuals with obesity are TNFα and IL-6; however only IL-6-induced signaling is mediated via JAK and STAT3.Citation42 In detail, IL-6 binds to its receptor, comprising the IL-6Rα chain (which confers specificity) and the GP130 signaling chain (which is common to other IL-6 type cytokine receptors).Citation42 The IL-6-bound receptor complex activates intracellular JAK2 subsequently leading to STAT3 activation, which in turn modulates gene expression, for example elevating SOCS3 expression. Of note, although certainly the IL-6Rα chain is expressed mainly by hepatocytes and immune cells, other cell types can also respond to IL-6 through a mechanism called IL-6 trans-signaling. In this process, the α chain is cleaved from the cell surface and in this soluble form jointly with IL-6 can bind to ubiquitously expressed GP130 to induce JAK-STAT3-mediated downstream signaling events.Citation67,Citation68
IL-6 derived from the muscle during intense exercise has been shown to exert beneficial effects on glucose homeostasis; however, increased basal IL-6 level in obese individuals impair insulin signaling presumably owing to its chronic nature. Clinical studies link elevated IL-6 serum levels with obesity, severity of insulin resistance and the risk of developing type 2 diabetes mellitus.Citation69-Citation72 Accordingly, obese women exhibited a reduction in IL-6 serum levels and increased insulin sensitivity after weight loss.Citation73
However, inactivation of IL-6 in mice has produced conflicting results and the expected increased insulin-sensitive phenotype could not be shown by two independent studies using the same IL-6 knockout mice.Citation74,Citation75 However, Wallenius et al. demonstrated mature-onset obesity and decreased glucose tolerance in IL-6-deficient mice,Citation74 whereas by contrast, Di Gregorio and colleagues could not detect any obvious phenotype related to maturity-onset obesity and diabetes.Citation75
The mechanism through which chronically raised IL-6 levels in obesity may cause insulin resistance is not yet defined, but may involve basal increases of SOCS protein levels in peripheral organs, similarly to the findings described for basal leptin-derived signal transduction in the CNS. Elevated IL-6 signaling in obesity leads to increased SOCS1 and SOCS3 protein levels in the three major insulin sensitive peripheral tissues, white adipose tissue, liver and muscle.Citation76 On the molecular level, SOCS1 and SOCS3 impair insulin action by binding to insulin receptor substrates (IRS)-1 and IRS-2, which leads to IRS-1 and IRS-2 ubiquitination and degradation.Citation77-Citation79 Interestingly, SOCS1 whole body knockout mice demonstrate hyperglycemia but die before reaching 3 weeks of age due to enhanced interferon γ signaling.Citation80 As mentioned previously, SOCS3 whole-body knockout mice are embryonically lethal due to inappropriate leukemia inhibitory factor signaling.Citation54,Citation55 SOCS3 heterozygous knockout mice are, however, protected against the development of obesity-associated insulin resistance.Citation53 On the other hand, overexpression of SOCS1 and SOCS3 leads to the development of insulin resistance by reducing tyrosine phosphorylation of IRS-1 and IRS-2, which is required for further insulin signal transduction.Citation77 These experiments point to a peripheral role of chronic IL-6-evoked JAK-STAT3 induced SOCS3 expression in the development of insulin resistance under obese conditions. As follows, we summarize the effects of chronic JAK-STAT3-SOCS3 signaling on the development of obesity-induced insulin resistance in the main peripheral metabolic organs.
White adipose tissue
Under basal conditions, up to 35% of systemic IL-6 is produced by the adipose tissue, predominantly by visceral fat depots.Citation81 The secretion from the visceral fat via the portal vein specifically affects the liver, given that almost 80% of total liver blood is derived from there.Citation81 Interestingly, increased diet-induced IL-6 release from the white adipose tissue was shown to crosstalk to the liver which displayed accelerated hepatic SOCS3 expression in turn impairing insulin action.Citation82 Nevertheless, locally, IL-6 induces the expression of SOCS3 in 3T3-L1 adipocytesCitation83,Citation84 whereas overexpression of SOCS3 in adipocytes reduces IRS-1 protein levels as well as insulin-stimulated IRS-1 and IRS-2 phosphorylation and binding of p85 to IRS-1.Citation79,Citation85 This action impairs insulin signaling and reduces glucose uptake in adipocytes and lipogenesis. However, mice which overexpress SOCS3 in adipocytes failed to develop diet-induced obesity owing to the fact that the insulin resistance occurred only locally in the white adipose tissue rather than systemically; this finding highlights the limited contribution of adipose tissue in whole-body glucose disposal.Citation85 Palanivel and colleagues investigated mice with white adipose tissue-specific SOCS3 deficiency, which showed only a mild protection against the development of obesity-associated insulin resistance.Citation86 Thus, although chronic JAK-STAT3-induced SOCS3 expression in white adipose tissue in obesity impairs local insulin sensitivity, systemic insulin action is unaltered. This phenomenon points to an important role of elevated SOCS3 levels in organs other than white adipose tissue to mitigate whole-body insulin sensitivity. However, in obesity, white adipose tissue is the predominant organ to release IL-6 into the circulation, which in turn inhibits insulin action in other organs.
Liver
The liver plays a key part in the regulation of whole-body energy homeostasis by controlling blood glucose levels during fasting and by storing excessive glucose as glycogen in the fed state. Studies in murine primary hepatocytes and human hepatocarcinoma cells revealed that IL-6 causes insulin resistance by suppressing tyrosine phosphorylation of IRS-1 through induction of SOCS3.Citation87 Moreover, SOCS1 and SOCS3 are elevated in the liver of obese miceCitation76 and the adenoviral-mediated gene transfer of SOCS1 or SOCS3 to the liver leads to glucose intolerance and insulin resistance.Citation77,Citation78 In contrast treatment of genetically obese db/db mice with antisense oligonucleotides against SOCS1 and SOCS3 improves the development of insulin resistance and hepatic steatosis in these mice.Citation78 Accordingly, the hepatocyte-specific disruption of SOCS3 first improves insulin sensitivity in chow-fed animals by ameliorating IRS-1 tyrosine phosphorylation,Citation88,Citation89 but with age these hepatocyte-specific SOCS3 knockout mice developed obesity and systemic insulin resistance owing to the hyperactivity of STAT3, which increases acute-phase proteins and inflammation.Citation88 The investigators conclude, that SOCS3 is a mediator of insulin resistance in the liver, but the lack of SOCS3 in the liver promotes systemic insulin resistance by mimicking chronic inflammation. In accordance with this study, Sachithanandan et al. demonstrate that diet-induced obesity in hepatocyte-specific SOCS3-deficient mice accelerates hepatic inflammation which leads to more pronounced development of systemic insulin resistance, hepatic steatosis and lipogenesis.Citation89 These data are also in line with our experiments which reveal a critical role of hepatic IL-6Rα-induced downstream signaling in the prevention of inflammatory cytokine expression derived from liver-resident Kupffer cells that impairs insulin action in the whole body.Citation90 Nevertheless, IL-6-induced STAT3 activation in hepatocytes is required for the proper suppression of hepatic glucose production during fasting, which is impaired under obesity conditions.Citation91 Taken together, hepatic IL-6-induced JAK-STAT3-SOCS3 signaling exerts a dual role on whole-body glucose homeostasis by preventing accelerated inflammation and inhibiting local insulin action.
Skeletal muscle
Skeletal muscle, which uses glucose and fatty acids as fuel, is a primary target tissue for the development of insulin resistance. Forty percent of the body mass in non-obese subjects consists of skeletal muscle and 30% of the resting metabolic rate can be traced back to the skeletal muscle. Numerous studies have revealed, that skeletal muscle-specific defects in insulin signaling contribute to systemic metabolic phenotypes.Citation92 However, IL-6 released from the muscle during intense exercise exerts beneficial effects on whole-body glucose metabolism, whereby the chronically IL-6 levels in the obese state impair insulin action.Citation93 Nevertheless, IL-6 increases SOCS3 as well a PTP1B mRNA in skeletal muscle, thus inhibiting local and systemic insulin sensitivityCitation94 and SOCS3 mRNA is increased in murine muscle in diet-induced and genetic obesity.Citation76,Citation95 A recent study from Yang et al. demonstrates that overexpressing SOCS3 in the murine muscle leads to the development of both skeletal muscle-specific and systemic insulin resistance by antagonizing IRS-1 phosphorylation.Citation95 However, in a similar approach, Lebrun et al. demonstrate that muscle-specific SOCS3 overexpression animals are overweight as a result of decreased locomotor activity rather than through the development of skeletal muscle specific insulin resistance.Citation96 Nevertheless, muscle-specific SOCS3 knockout mice exposed to diet-induced obesity are protected against hyperinsulinemia and insulin resistance as these animals exhibit enhanced skeletal muscle IRS-1 and AKT phosphorylation resulting in increased glucose uptake in the muscle.Citation97 Collectively, these studies reveal that chronic JAK-STAT3-SOCS3 in skeletal muscle impairs whole-body insulin sensitivity.
Conclusion
Chronic JAK-STAT3-SOCS3 during the course of obesity impairs proper leptin and insulin action and generates a futile cycle that further promotes weight gain. Recent advances in genetic mouse models combined with dietary composition have shed light on the underlying molecular mechanisms. Here, we report the mechanisms of chronic JAK-STAT3-SOCS3 signaling in obesity induced in the CNS via excessive leptin and in the periphery via elevations of IL-6 () although vice versa functions for both signals have been reported. We are aware of the existence of other signal transducers and crosstalk of the aforementioned mediators that regulate chronicity of JAK-STAT3-SOCS3 in the development of obesity, but are unable to address those in this framework. Moreover, numerous other high profile manuscripts exist that support our discussion, but have not been mentioned in this review. More effort has to be put forth to clarify the cell type-specific mechanisms of the JAK-STAT3-SOCS3 axis on the development of obesity-associated disorders such as leptin resistance and insulin resistance to approach suitable strategies in the therapeutic combat of this epidemiological disease. The discrepancy of acute vs. chronic effects of JAK-STAT3-SOCS3 complicates this task, the investigation of which has clear potential for further research.
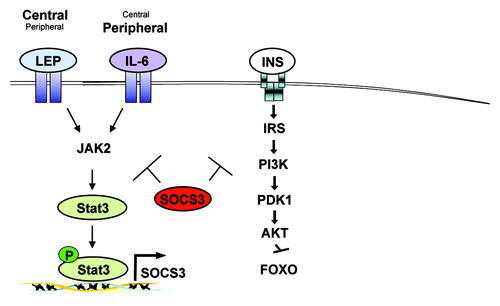
Figure 1. Chronic JAK-STAT3-SOCS3 signaling in obesity. Obesity increases circulating levels of leptin and IL-6 that in turn chronically activate intracellular JAK-STAT3 signaling. While Leptin (Lep) acts predominantly in the central nervous system, IL-6 has been reported to mainly function in peripheral organs, though both factors can also act vice versa. Chronic JAK-STAT3 signaling induced by leptin and IL-6 lead to the increased expression of the negative regulator SOCS3. SOCS3 in turn not only negatively regulates leptin and IL-6 signaling but also impairs insulin (INS) action eventually leading to obesity and insulin resistance.
Acknowledgments
We are grateful for intensive discussions with members of the Wunderlich and Brüning labs and for manuscript proofreading by Linda Koch. C.M.W. was funded from grants of CECAD and CMMC attributed to F.T.W. who received additional support from the DFG (SFB832 A15 and Z3, CRU286 RP5). N.H. obtained funding from the DFG (grant HO 4440/1-1)
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Ghoreschi K, Laurence A, O’Shea JJ. Janus kinases in immune cell signaling. Immunol Rev 2009; 228:273 - 87; http://dx.doi.org/10.1111/j.1600-065X.2008.00754.x; PMID: 19290934
- Kim JH, Bachmann RA, Chen J. Interleukin-6 and insulin resistance. Vitam Horm 2009; 80:613 - 33; http://dx.doi.org/10.1016/S0083-6729(08)00621-3; PMID: 19251052
- Kiu H, Nicholson SE. Biology and significance of the JAK/STAT signalling pathways. Growth Factors 2012; 30:88 - 106; http://dx.doi.org/10.3109/08977194.2012.660936; PMID: 22339650
- Sun B, Karin M. Obesity, inflammation, and liver cancer. J Hepatol 2012; 56:704 - 13; http://dx.doi.org/10.1016/j.jhep.2011.09.020; PMID: 22120206
- Cance WG, Liu ET. Protein kinases in human breast cancer. Breast Cancer Res Treat 1995; 35:105 - 14; http://dx.doi.org/10.1007/BF00694751; PMID: 7612897
- Firmbach-Kraft I, Byers M, Shows T, Dalla-Favera R, Krolewski JJ. tyk2, prototype of a novel class of non-receptor tyrosine kinase genes. Oncogene 1990; 5:1329 - 36; PMID: 2216457
- Harpur AG, Andres AC, Ziemiecki A, Aston RR, Wilks AF. JAK2, a third member of the JAK family of protein tyrosine kinases. Oncogene 1992; 7:1347 - 53; PMID: 1620548
- Rane SG, Reddy EP. JAK3: a novel JAK kinase associated with terminal differentiation of hematopoietic cells. Oncogene 1994; 9:2415 - 23; PMID: 7518579
- Wilks AF. Two putative protein-tyrosine kinases identified by application of the polymerase chain reaction. Proc Natl Acad Sci U S A 1989; 86:1603 - 7; http://dx.doi.org/10.1073/pnas.86.5.1603; PMID: 2466296
- Wilks AF, Harpur AG, Kurban RR, Ralph SJ, Zürcher G, Ziemiecki A. Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase. Mol Cell Biol 1991; 11:2057 - 65; PMID: 1848670
- Witthuhn BA, Silvennoinen O, Miura O, Lai KS, Cwik C, Liu ET, et al. Involvement of the Jak-3 Janus kinase in signalling by interleukins 2 and 4 in lymphoid and myeloid cells. Nature 1994; 370:153 - 7; http://dx.doi.org/10.1038/370153a0; PMID: 8022486
- Fu XY, Schindler C, Improta T, Aebersold R, Darnell JE Jr.. The proteins of ISGF-3, the interferon alpha-induced transcriptional activator, define a gene family involved in signal transduction. Proc Natl Acad Sci U S A 1992; 89:7840 - 3; http://dx.doi.org/10.1073/pnas.89.16.7840; PMID: 1502204
- Hou J, Schindler U, Henzel WJ, Ho TC, Brasseur M, McKnight SL. An interleukin-4-induced transcription factor: IL-4 Stat. Science 1994; 265:1701 - 6; http://dx.doi.org/10.1126/science.8085155; PMID: 8085155
- Mui AL, Wakao H, Harada N, O’Farrell AM, Miyajima A. Interleukin-3, granulocyte-macrophage colony-stimulating factor, and interleukin-5 transduce signals through two forms of STAT5. J Leukoc Biol 1995; 57:799 - 803; PMID: 7539031
- Schindler C, Fu XY, Improta T, Aebersold R, Darnell JE Jr.. Proteins of transcription factor ISGF-3: one gene encodes the 91-and 84-kDa ISGF-3 proteins that are activated by interferon alpha. Proc Natl Acad Sci U S A 1992; 89:7836 - 9; http://dx.doi.org/10.1073/pnas.89.16.7836; PMID: 1502203
- Wakao H, Harada N, Kitamura T, Mui AL, Miyajima A. Interleukin 2 and erythropoietin activate STAT5/MGF via distinct pathways. EMBO J 1995; 14:2527 - 35; PMID: 7781605
- Yamamoto K, Quelle FW, Thierfelder WE, Kreider BL, Gilbert DJ, Jenkins NA, et al. Stat4, a novel gamma interferon activation site-binding protein expressed in early myeloid differentiation. Mol Cell Biol 1994; 14:4342 - 9; PMID: 8007943
- Zhong Z, Wen Z, Darnell JE Jr.. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994; 264:95 - 8; http://dx.doi.org/10.1126/science.8140422; PMID: 8140422
- Fagerlund R, Mélen K, Kinnunen L, Julkunen I. Arginine/lysine-rich nuclear localization signals mediate interactions between dimeric STATs and importin alpha 5. J Biol Chem 2002; 277:30072 - 8; http://dx.doi.org/10.1074/jbc.M202943200; PMID: 12048190
- Ihle JN. STATs: signal transducers and activators of transcription. Cell 1996; 84:331 - 4; http://dx.doi.org/10.1016/S0092-8674(00)81277-5; PMID: 8608586
- Kawashima T, Bao YC, Minoshima Y, Nomura Y, Hatori T, Hori T, et al. A Rac GTPase-activating protein, MgcRacGAP, is a nuclear localizing signal-containing nuclear chaperone in the activation of STAT transcription factors. Mol Cell Biol 2009; 29:1796 - 813; http://dx.doi.org/10.1128/MCB.01423-08; PMID: 19158271
- Kawashima T, Bao YC, Nomura Y, Moon Y, Tonozuka Y, Minoshima Y, et al. Rac1 and a GTPase-activating protein, MgcRacGAP, are required for nuclear translocation of STAT transcription factors. J Cell Biol 2006; 175:937 - 46; http://dx.doi.org/10.1083/jcb.200604073; PMID: 17178910
- Kessler DS, Veals SA, Fu XY, Levy DE. Interferon-alpha regulates nuclear translocation and DNA-binding affinity of ISGF3, a multimeric transcriptional activator. Genes Dev 1990; 4:1753 - 65; http://dx.doi.org/10.1101/gad.4.10.1753; PMID: 2249773
- Sekimoto T, Imamoto N, Nakajima K, Hirano T, Yoneda Y. Extracellular signal-dependent nuclear import of Stat1 is mediated by nuclear pore-targeting complex formation with NPI-1, but not Rch1. EMBO J 1997; 16:7067 - 77; http://dx.doi.org/10.1093/emboj/16.23.7067; PMID: 9384585
- Shuai K, Stark GR, Kerr IM, Darnell JE Jr.. A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science 1993; 261:1744 - 6; http://dx.doi.org/10.1126/science.7690989; PMID: 7690989
- Kile BT, Alexander WS. The suppressors of cytokine signalling (SOCS). Cell Mol Life Sci 2001; 58:1627 - 35; http://dx.doi.org/10.1007/PL00000801; PMID: 11706989
- Krebs DL, Hilton DJ. SOCS proteins: negative regulators of cytokine signaling. Stem Cells 2001; 19:378 - 87; http://dx.doi.org/10.1634/stemcells.19-5-378; PMID: 11553846
- Hilton DJ, Richardson RT, Alexander WS, Viney EM, Willson TA, Sprigg NS, et al. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc Natl Acad Sci U S A 1998; 95:114 - 9; http://dx.doi.org/10.1073/pnas.95.1.114; PMID: 9419338
- Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, et al. A family of cytokine-inducible inhibitors of signalling. Nature 1997; 387:917 - 21; http://dx.doi.org/10.1038/43206; PMID: 9202125
- ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, Zhu W, Tremblay M, David M, et al. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol 2002; 22:5662 - 8; http://dx.doi.org/10.1128/MCB.22.16.5662-5668.2002; PMID: 12138178
- Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, et al. PTP1B regulates leptin signal transduction in vivo. Dev Cell 2002; 2:489 - 95; http://dx.doi.org/10.1016/S1534-5807(02)00148-X; PMID: 11970898
- Shuai K. Regulation of cytokine signaling pathways by PIAS proteins. Cell Res 2006; 16:196 - 202; http://dx.doi.org/10.1038/sj.cr.7310027; PMID: 16474434
- Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature 1998; 395:763 - 70; http://dx.doi.org/10.1038/27376; PMID: 9796811
- Könner AC, Brüning JC. Selective insulin and leptin resistance in metabolic disorders. Cell Metab 2012; 16:144 - 52; http://dx.doi.org/10.1016/j.cmet.2012.07.004; PMID: 22883229
- Belgardt BF, Brüning JC. CNS leptin and insulin action in the control of energy homeostasis. Ann N Y Acad Sci 2010; 1212:97 - 113; http://dx.doi.org/10.1111/j.1749-6632.2010.05799.x; PMID: 21070248
- Chua SC Jr., Koutras IK, Han L, Liu SM, Kay J, Young SJ, et al. Fine structure of the murine leptin receptor gene: splice site suppression is required to form two alternatively spliced transcripts. Genomics 1997; 45:264 - 70; http://dx.doi.org/10.1006/geno.1997.4962; PMID: 9344648
- White DW, Kuropatwinski KK, Devos R, Baumann H, Tartaglia LA. Leptin receptor (OB-R) signaling. Cytoplasmic domain mutational analysis and evidence for receptor homo-oligomerization. J Biol Chem 1997; 272:4065 - 71; http://dx.doi.org/10.1074/jbc.272.7.4065; PMID: 9020115
- Kloek C, Haq AK, Dunn SL, Lavery HJ, Banks AS, Myers MG Jr.. Regulation of Jak kinases by intracellular leptin receptor sequences. J Biol Chem 2002; 277:41547 - 55; http://dx.doi.org/10.1074/jbc.M205148200; PMID: 12196522
- Ihle JN, Kerr IM. Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet 1995; 11:69 - 74; http://dx.doi.org/10.1016/S0168-9525(00)89000-9; PMID: 7716810
- Taniguchi T. Cytokine signaling through nonreceptor protein tyrosine kinases. Science 1995; 268:251 - 5; http://dx.doi.org/10.1126/science.7716517; PMID: 7716517
- Banks AS, Davis SM, Bates SH, Myers MG Jr.. Activation of downstream signals by the long form of the leptin receptor. J Biol Chem 2000; 275:14563 - 72; http://dx.doi.org/10.1074/jbc.275.19.14563; PMID: 10799542
- Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J 2003; 374:1 - 20; http://dx.doi.org/10.1042/BJ20030407; PMID: 12773095
- Fuente-Martín E, García-Cáceres C, Granado M, de Ceballos ML, Sánchez-Garrido MA, Sarman B, et al. Leptin regulates glutamate and glucose transporters in hypothalamic astrocytes. J Clin Invest 2012; 122:3900 - 13; http://dx.doi.org/10.1172/JCI64102; PMID: 23064363
- Wang Y, Kuropatwinski KK, White DW, Hawley TS, Hawley RG, Tartaglia LA, et al. Leptin receptor action in hepatic cells. J Biol Chem 1997; 272:16216 - 23; http://dx.doi.org/10.1074/jbc.272.26.16216; PMID: 9195922
- Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 2008; 135:61 - 73; http://dx.doi.org/10.1016/j.cell.2008.07.043; PMID: 18854155
- Zhang X, Hecker M, Park JW, Tompsett AR, Jones PD, Newsted J, et al. Time-dependent transcriptional profiles of genes of the hypothalamic-pituitary-gonadal axis in medaka (Oryzias latipes) exposed to fadrozole and 17beta-trenbolone. Environ Toxicol Chem 2008; 27:2504 - 11; http://dx.doi.org/10.1897/08-082.1; PMID: 18693774
- Zhang X, Hecker M, Park JW, Tompsett AR, Newsted J, Nakayama K, et al. Real-time PCR array to study effects of chemicals on the Hypothalamic-Pituitary-Gonadal axis of the Japanese medaka. Aquat Toxicol 2008; 88:173 - 82; http://dx.doi.org/10.1016/j.aquatox.2008.04.009; PMID: 18534694
- Bailly-Maitre B, Belgardt BF, Jordan SD, Coornaert B, von Freyend MJ, Kleinridders A, et al. Hepatic Bax inhibitor-1 inhibits IRE1alpha and protects from obesity-associated insulin resistance and glucose intolerance. J Biol Chem 2010; 285:6198 - 207; http://dx.doi.org/10.1074/jbc.M109.056648; PMID: 19996103
- Butler AA, Cone RD. The melanocortin receptors: lessons from knockout models. Neuropeptides 2002; 36:77 - 84; http://dx.doi.org/10.1054/npep.2002.0890; PMID: 12359499
- Tartaglia LA. The leptin receptor. J Biol Chem 1997; 272:6093 - 6; PMID: 9102398
- Munzberg H. Leptin-signaling pathways and leptin resistance. Forum Nutr 2010; 63:123 - 32; http://dx.doi.org/10.1159/000264400; PMID: 19955780
- Bjørbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell 1998; 1:619 - 25; http://dx.doi.org/10.1016/S1097-2765(00)80062-3; PMID: 9660946
- Howard JK, Cave BJ, Oksanen LJ, Tzameli I, Bjørbaek C, Flier JS. Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat Med 2004; 10:734 - 8; http://dx.doi.org/10.1038/nm1072; PMID: 15220914
- Marine JC, McKay C, Wang D, Topham DJ, Parganas E, Nakajima H, et al. SOCS3 is essential in the regulation of fetal liver erythropoiesis. Cell 1999; 98:617 - 27; http://dx.doi.org/10.1016/S0092-8674(00)80049-5; PMID: 10490101
- Roberts AW, Robb L, Rakar S, Hartley L, Cluse L, Nicola NA, et al. Placental defects and embryonic lethality in mice lacking suppressor of cytokine signaling 3. Proc Natl Acad Sci U S A 2001; 98:9324 - 9; http://dx.doi.org/10.1073/pnas.161271798; PMID: 11481489
- Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron 2004; 42:983 - 91; http://dx.doi.org/10.1016/j.neuron.2004.06.004; PMID: 15207242
- Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gao XB, et al. Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron 2006; 51:801 - 10; http://dx.doi.org/10.1016/j.neuron.2006.08.023; PMID: 16982424
- Ernst MB, Wunderlich CM, Hess S, Paehler M, Mesaros A, Koralov SB, et al. Enhanced Stat3 activation in POMC neurons provokes negative feedback inhibition of leptin and insulin signaling in obesity. J Neurosci 2009; 29:11582 - 93; http://dx.doi.org/10.1523/JNEUROSCI.5712-08.2009; PMID: 19759305
- Mesaros A, Koralov SB, Rother E, Wunderlich FT, Ernst MB, Barsh GS, et al. Activation of Stat3 signaling in AgRP neurons promotes locomotor activity. Cell Metab 2008; 7:236 - 48; http://dx.doi.org/10.1016/j.cmet.2008.01.007; PMID: 18316029
- Smith CC, Dixon RA, Wynne AM, Theodorou L, Ong SG, Subrayan S, et al. Leptin-induced cardioprotection involves JAK/STAT signaling that may be linked to the mitochondrial permeability transition pore. Am J Physiol Heart Circ Physiol 2010; 299:H1265 - 70; http://dx.doi.org/10.1152/ajpheart.00092.2010; PMID: 20656889
- Lecour S, Suleman N, Deuchar GA, Somers S, Lacerda L, Huisamen B, et al. Pharmacological preconditioning with tumor necrosis factor-alpha activates signal transducer and activator of transcription-3 at reperfusion without involving classic prosurvival kinases (Akt and extracellular signal-regulated kinase). Circulation 2005; 112:3911 - 8; http://dx.doi.org/10.1161/CIRCULATIONAHA.105.581058; PMID: 16344382
- Hotamisligil GS. Inflammatory pathways and insulin action. Int J Obes Relat Metab Disord 2003; 27:Suppl 3 S53 - 5; http://dx.doi.org/10.1038/sj.ijo.0802502; PMID: 14704746
- Kleinridders A, Schenten D, Könner AC, Belgardt BF, Mauer J, Okamura T, et al. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab 2009; 10:249 - 59; http://dx.doi.org/10.1016/j.cmet.2009.08.013; PMID: 19808018
- Mohamed-Ali V, Pinkney JH, Coppack SW. Adipose tissue as an endocrine and paracrine organ. Int J Obes Relat Metab Disord 1998; 22:1145 - 58; http://dx.doi.org/10.1038/sj.ijo.0800770; PMID: 9877249
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr.. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003; 112:1796 - 808; PMID: 14679176
- Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest 2005; 115:1111 - 9; PMID: 15864338
- Wunderlich CM, Delić D, Behnke K, Meryk A, Ströhle P, Chaurasia B, et al. Cutting edge: Inhibition of IL-6 trans-signaling protects from malaria-induced lethality in mice. J Immunol 2012; 188:4141 - 4; http://dx.doi.org/10.4049/jimmunol.1102137; PMID: 22467660
- Scheller J, Rose-John S. Interleukin-6 and its receptor: from bench to bedside. Med Microbiol Immunol 2006; 195:173 - 83; http://dx.doi.org/10.1007/s00430-006-0019-9; PMID: 16741736
- Roytblat L, Rachinsky M, Fisher A, Greemberg L, Shapira Y, Douvdevani A, et al. Raised interleukin-6 levels in obese patients. Obes Res 2000; 8:673 - 5; http://dx.doi.org/10.1038/oby.2000.86; PMID: 11225716
- Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 2001; 286:327 - 34; http://dx.doi.org/10.1001/jama.286.3.327; PMID: 11466099
- Vozarova B, Weyer C, Hanson K, Tataranni PA, Bogardus C, Pratley RE. Circulating interleukin-6 in relation to adiposity, insulin action, and insulin secretion. Obes Res 2001; 9:414 - 7; http://dx.doi.org/10.1038/oby.2001.54; PMID: 11445664
- Hu FB, Meigs JB, Li TY, Rifai N, Manson JE. Inflammatory markers and risk of developing type 2 diabetes in women. Diabetes 2004; 53:693 - 700; http://dx.doi.org/10.2337/diabetes.53.3.693; PMID: 14988254
- Bastard JP, Jardel C, Bruckert E, Blondy P, Capeau J, Laville M, et al. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J Clin Endocrinol Metab 2000; 85:3338 - 42; http://dx.doi.org/10.1210/jc.85.9.3338; PMID: 10999830
- Wallenius V, Wallenius K, Ahrén B, Rudling M, Carlsten H, Dickson SL, et al. Interleukin-6-deficient mice develop mature-onset obesity. Nat Med 2002; 8:75 - 9; http://dx.doi.org/10.1038/nm0102-75; PMID: 11786910
- Di Gregorio GB, Hensley L, Lu T, Ranganathan G, Kern PA. Lipid and carbohydrate metabolism in mice with a targeted mutation in the IL-6 gene: absence of development of age-related obesity. Am J Physiol Endocrinol Metab 2004; 287:E182 - 7; http://dx.doi.org/10.1152/ajpendo.00189.2003; PMID: 15191885
- Ueki K, Kondo T, Tseng YH, Kahn CR. Central role of suppressors of cytokine signaling proteins in hepatic steatosis, insulin resistance, and the metabolic syndrome in the mouse. Proc Natl Acad Sci U S A 2004; 101:10422 - 7; http://dx.doi.org/10.1073/pnas.0402511101; PMID: 15240880
- Ueki K, Kondo T, Kahn CR. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol Cell Biol 2004; 24:5434 - 46; http://dx.doi.org/10.1128/MCB.24.12.5434-5446.2004; PMID: 15169905
- Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem 2002; 277:42394 - 8; http://dx.doi.org/10.1074/jbc.C200444200; PMID: 12228220
- Shi H, Tzameli I, Bjørbaek C, Flier JS. Suppressor of cytokine signaling 3 is a physiological regulator of adipocyte insulin signaling. J Biol Chem 2004; 279:34733 - 40; http://dx.doi.org/10.1074/jbc.M403886200; PMID: 15181014
- Starr R, Metcalf D, Elefanty AG, Brysha M, Willson TA, Nicola NA, et al. Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc Natl Acad Sci U S A 1998; 95:14395 - 9; http://dx.doi.org/10.1073/pnas.95.24.14395; PMID: 9826711
- Fontana L, Eagon JC, Trujillo ME, Scherer PE, Klein S. Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes 2007; 56:1010 - 3; http://dx.doi.org/10.2337/db06-1656; PMID: 17287468
- Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, et al. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 2008; 322:1539 - 43; http://dx.doi.org/10.1126/science.1160794; PMID: 19056984
- Fasshauer M, Kralisch S, Klier M, Lossner U, Bluher M, Klein J, et al. Insulin resistance-inducing cytokines differentially regulate SOCS mRNA expression via growth factor- and Jak/Stat-signaling pathways in 3T3-L1 adipocytes. J Endocrinol 2004; 181:129 - 38; http://dx.doi.org/10.1677/joe.0.1810129; PMID: 15072573
- Lagathu C, Bastard JP, Auclair M, Maachi M, Capeau J, Caron M. Chronic interleukin-6 (IL-6) treatment increased IL-6 secretion and induced insulin resistance in adipocyte: prevention by rosiglitazone. Biochem Biophys Res Commun 2003; 311:372 - 9; http://dx.doi.org/10.1016/j.bbrc.2003.10.013; PMID: 14592424
- Shi H, Cave B, Inouye K, Bjørbaek C, Flier JS. Overexpression of suppressor of cytokine signaling 3 in adipose tissue causes local but not systemic insulin resistance. Diabetes 2006; 55:699 - 707; http://dx.doi.org/10.2337/diabetes.55.03.06.db05-0841; PMID: 16505233
- Palanivel R, Fullerton MD, Galic S, Honeyman J, Hewitt KA, Jorgensen SB, et al. Reduced Socs3 expression in adipose tissue protects female mice against obesity-induced insulin resistance. Diabetologia 2012; 55:3083 - 93; http://dx.doi.org/10.1007/s00125-012-2665-3; PMID: 22872213
- Senn JJ, Klover PJ, Nowak IA, Zimmers TA, Koniaris LG, Furlanetto RW, et al. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J Biol Chem 2003; 278:13740 - 6; http://dx.doi.org/10.1074/jbc.M210689200; PMID: 12560330
- Torisu T, Sato N, Yoshiga D, Kobayashi T, Yoshioka T, Mori H, et al. The dual function of hepatic SOCS3 in insulin resistance in vivo. Genes Cells 2007; 12:143 - 54; http://dx.doi.org/10.1111/j.1365-2443.2007.01044.x; PMID: 17295835
- Sachithanandan N, Fam BC, Fynch S, Dzamko N, Watt MJ, Wormald S, et al. Liver-specific suppressor of cytokine signaling-3 deletion in mice enhances hepatic insulin sensitivity and lipogenesis resulting in fatty liver and obesity. Hepatology 2010; 52:1632 - 42; http://dx.doi.org/10.1002/hep.23861; PMID: 20799351
- Wunderlich FT, Ströhle P, Könner AC, Gruber S, Tovar S, Brönneke HS, et al. Interleukin-6 signaling in liver-parenchymal cells suppresses hepatic inflammation and improves systemic insulin action. Cell Metab 2010; 12:237 - 49; http://dx.doi.org/10.1016/j.cmet.2010.06.011; PMID: 20816090
- Inoue H, Ogawa W, Asakawa A, Okamoto Y, Nishizawa A, Matsumoto M, et al. Role of hepatic STAT3 in brain-insulin action on hepatic glucose production. Cell Metab 2006; 3:267 - 75; http://dx.doi.org/10.1016/j.cmet.2006.02.009; PMID: 16581004
- Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest 2000; 106:473 - 81; http://dx.doi.org/10.1172/JCI10842; PMID: 10953022
- Pedersen BK, Febbraio MA. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol 2012; 8:457 - 65; http://dx.doi.org/10.1038/nrendo.2012.49; PMID: 22473333
- Schéle E, Fekete C, Egri P, Füzesi T, Palkovits M, Keller E, et al. Interleukin-6 receptor α is co-localised with melanin-concentrating hormone in human and mouse hypothalamus. J Neuroendocrinol 2012; 24:930 - 43; http://dx.doi.org/10.1111/j.1365-2826.2012.02286.x; PMID: 22295972
- Yang Z, Hulver M, McMillan RP, Cai L, Kershaw EE, Yu L, et al. Regulation of insulin and leptin signaling by muscle suppressor of cytokine signaling 3 (SOCS3). PLoS One 2012; 7:e47493; http://dx.doi.org/10.1371/journal.pone.0047493; PMID: 23115649
- Lebrun P, Cognard E, Bellon-Paul R, Gontard P, Filloux C, Jehl-Pietri C, et al. Constitutive expression of suppressor of cytokine signalling-3 in skeletal muscle leads to reduced mobility and overweight in mice. Diabetologia 2009; 52:2201 - 12; http://dx.doi.org/10.1007/s00125-009-1474-9; PMID: 19672574
- Jorgensen SB, O'Neill HM, Sylow L, Honeyman J, Hewitt KA, Palanivel R, et al. Deletion of Skeletal Muscle SOCS3 Prevents Insulin Resistance in Obesity. Diabetes 2013; 62:56 - 64; http://dx.doi.org/10.2337/db12-0443; PMID: 22961088