Abstract
Signal transducers and activators of transcription (STATs) transduce extracellular signals that regulate the initiation, duration and intensity of immune responses. However, unbridled activation of STATs by pro-inflammatory cytokines or growth factors contributes to pathogenic autoimmunity. In this review, we briefly discuss STAT pathways that promote the development and expansion of T cells that mediate two CNS inflammatory diseases, multiple sclerosis (MS) and uveitis. Particular focus is on animal models of MS and uveitis and new approaches to the treatment of CNS autoimmune diseases based on therapeutic targeting of Th17 cells and STAT pathways.
Introduction
The vertebrate immune system is comprised of the adaptive and innate immune systems that have distinct, as well as, overlapping functions in initiating and regulating host immunity. During Ag-presentation, dendritic cells (DC) of the innate immune system secrete cytokines that instruct naïve T cells to differentiate toward one of several alternative developmental pathways resulting in the generation of Th1, Th2, Th17 or regulatory (Treg) T cells ( and ). Cytokines secreted by the DCs (IL-12, IL-23 and IL-27) or by differentiated T cells (IFN-γ, IL-2, IL-4, IL-6, IL-10 and IL-21) influence the quality and nature of the immune response. These cytokines mediate their biological activities through receptors associated with Janus kinases (JAK1, JAK2, JAK3 and Tyk2).Citation1 Upon binding of the cytokine to its cognate receptor, JAKs are activated by transphosphorylation, providing docking sites for recruitment of specific members of the STAT family of transcription factors.Citation2 STATs recruited to the receptor complex are phosphorylated at a critical tyrosine residue, form homo- or hetero-dimers and translocate into the nucleus where they bind to specific DNA sequences and activate gene transcription. STATs (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B and STAT6) thus provide a rapid membrane to nucleus mechanism for regulating gene expression.Citation2 However, cytokine responses are typically transient and in the absence of appropriate regulation may lead to imbalance in the immune system; resulting in immunodeficiency or autoimmunity. Suppressor of cytokine signaling (SOCS) proteins are cytokine-inducible negative feedback regulators that control the initiation, intensity, duration, and quality of cytokine responses ().Citation3 The family is comprised of eight members characterized by the presence of an SH2 domain and a SOCS box domain.Citation3 Their inhibitory effects derive from direct interaction of the SOCS proteins with cytokine receptors and/or JAKs, thereby preventing recruitment of STATs to the signaling complex. SOCS proteins also target proteins for polyubiquitination and proteosome-mediated degradation.Citation3
Figure 1. The IL-12 cytokine family and its effects on T-helper differentiation. The family is comprised of IL-12 (p35/p40), IL-23 (p19/p40), IL-27 (IL27p28/Ebi3) and IL-35 (p35/Ebi3), and each member interacts with high-affinity heterodimeric receptors comprising of the pairing between IL-12Rβ1, IL12Rβ2, IL-27Rα (WSX-1) or gp130. They mediate their biological effects through the activation of STAT pathways. The outcome of the response can be proinflammatory or immune suppression depending on the predominant IL-12 family cytokines secreted by dendritic cells during Ag priming.
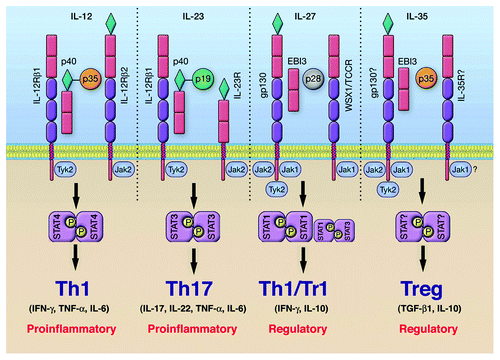
Figure 2. Cytokines and STAT pathways regulate T-helper cells that mediate uveitis and MS. Cytokine-binding facilitates recruitment and activation of requisite STAT proteins. Activated STATs induce the upregulation of lineage-specific master regulators such as T-bet, GATA3, ROR-γt or Foxp3 leading to the production of cytokines that promote the establishment and stability of the lineage. Cytokines produced by the T cell subset define the nature of the immune response. However, homeostatic balance of the various T-helper and regulatory T cells is orchestrated and exquisitely regulated by negative regulatory factors, including members of the SOCS family of latent cytoplasmic proteins.
Immune Privilege and Protection of the CNS (Central Nervous System) from Inflammation
The CNS is an immune privileged site, comprised of the brain, spinal cord and the ocular retina.Citation4 The intricate and highly vulnerable physiology of the CNS is shielded by the blood-brain barrier (BBB) and the blood-ocular barrier (BOB).Citation4 The barrier-forming constituents, comprised of endothelial and parenchymal basement membranes, pericytes and the perivascular space that inhibit paracellular diffusion, limit transcellular diffusion while allowing import of nutrients and export of toxic metabolites out of the CNS.Citation4 Epithelial cells of the choroid plexus and resident ocular parenchymal cells constitutively secrete immunosuppressive and anti-inflammatory cytokines that inactivate immunological effector cells.Citation5 However, during MS or uveitis encephalitogenic or uveitogenic lymphocytes bearing antigen-receptors specific to neuronal or retinal proteins breach the BOB or BBB, attack and destroy CNS tissues. Thus, extravasation of inflammatory cells from the blood into the retina, brain or spinal cord is required for the development of neuroinflammation ().
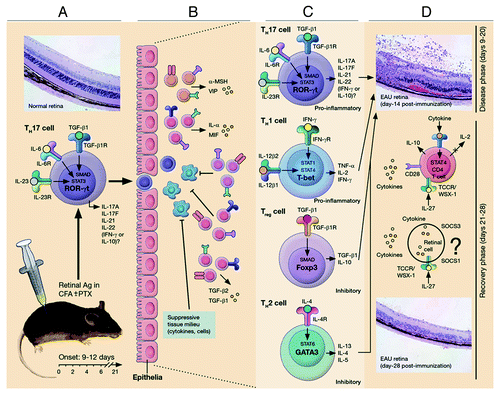
Figure 3. Schematic representation of early events leading to ocular inflammation, tissue destruction and induction of retinal protective mechanism in rodent model of uveitis. (A) Immunization of susceptible mouse strains (e.g., C57BL/6) in complete Freund’s adjuvant (CFA) triggers an immunological response characterized by abundance of Th17 and Th1 cells in lymph nodes, spleen and peripheral blood. (B) Activated Th17 cells expressing high levels of granzyme and activated α4β1 integrin facilitate breakdown of blood ocular barrier (BOB) retinal-blood extravasation into retina. The inflammatory cells entering the eye encounter hostile environment of the retina consisting of anti-inflammatory molecules (TGFβ, α-MSHa, VIP, MIF and IL-1rα) and resident retinal cells express inhibitory cell surface associated proteins (TGF-β, FAS/FAS ligand, CD46 and CD59). (C) Breakdown of BOB is accompanied by influx of other inflammatory cells and all major T-helper subsets are detectable in the reina during EAU. (D) Eventual elimination of cells from retina derives from endogenous adaptive mechanisms of ocular immune privilege. IFN-γ/STAT1-induced IL-27 production by resident ocular cells and cytokine-induced expression of SOCS1 and SOCS3 by retinal cells contribute to mitigation of uveitis.
Etiology and Control of Chronic Inflammation in the Central Nervous System (CNS)
Inflammation in the CNS presents unique challenges, as the need to avoid collateral damage that may compromise functional integrity of the retina or brain is as important as the need to eliminate the pathogen.Citation5 It is now widely accepted that unrestrained neuroinflammatory responses deriving from excessive secretion of cytokines by inflammatory cells contributes to neuronal or photoreceptor cell deficit that precede neurodegenerative changes in uveitis, multiple sclerosis, Alzheimer disease or age-related macular degeneration. Uveitis and multiple sclerosis are classical T cell mediated CNS autoimmune diseases that provide useful framework for understanding cross-talk between cytokines secreted by cells of the innate system (IL-12, IL-23 and IL-27) and cytokines produced by adaptive immune cells (IL-2, IL-17 and IFN-γ). The fact that these cytokines, as well as, neuronal-protective cytokines (CNTF, IGF-1, OSM and LIF) utilize STAT pathways, provides opportunity to study how aberrant regulation of STAT pathways contribute to pathogenic autoimmunity and other neurodegenerative diseases. Mice with targeted deletion of STAT proteins in T cells have therefore been used to characterize the role of each STAT member in CNS autoimmune diseases. In this review, we have focused on experimental findings in mouse models of multiple sclerosis and uveitis because most of what we know about the role of STATs in CNS autoimmune diseases derives from these models.
Multiple sclerosis (MS) is an autoimmune disease characterized by immunological response to myelin, a dielectric (electrically insulating) material that forms the myelin sheath surrounding the axon of neurons. In MS, encephalitogenic lymphocytes attack and destroy myelinated neurons, thereby interfering with synaptic transmission and communication between neurons. A characteristic feature of MS is its recurrent cycles of debilitating neurological disease.Citation6 Experimental autoimmune encephalomyelitis (EAE), induced in various rodent models by immunization with spinal cord or brain homogenate in CFA, has provided valuable insights into immunopathogenic mechanisms of MS.Citation6
Uveitis is a diverse group of intraocular inflammatory diseases that includes birdshot retinochoroidopathy, Behcet disease and ocular sarcoidosis, and is characterized by the destruction of uveal tissue by autoreactive uveitogenic T cells.Citation7 Experimental autoimmune uveitis (EAU) is an animal disease that shares essential pathological features with human uveitis.Citation8 EAU is the animal model of uveitis and is a predominantly T cell-mediated intraocular inflammatory disease induced in susceptible species by active immunization with retinal protein extracts in CFA.Citation8 Uveitis is also a relapsing-remitting CNS autoimmune disease.
Encephalitogenic or uveitogenic T cells that mediate EAE or EAU, respectively, preferentially enter the CNS across BBB or BOB as a result of the expression of P-selectins, LFA-1, chemokines (MIP-1 and RANTES) and chemokine receptors by the inflammatory cells. In fact, antibodies directed against ICAM-1 or LFA-1 have been used to inhibit EAUCitation9 and EAE,Citation10 underscoring the importance of adhesion molecules in pathogenesis of both diseases. Furthermore, integrins and osteopontin that promote chemotactic properties of leukocytes are important for homing of uveitogenic cells into neuroretina.Citation11,Citation12 Numerous studies have sought to identify STAT pathways that mediate recruitment of encephalitogenic or uveitogenic T cells into the brain or retina as they are considered to be potential therapeutic targets in MS and uveitis.
Role of STAT Proteins in CNS Autoimmune Diseases
All four major T cell subsets (Th1, Th2, Th17 and Tregs) are detected in the retina (), brain or spinal cord in active MS or uveitis. However, the presence of any T cell type in the CNS is undesirable because the cytokines they produce may have deleterious effects on neurons and photoreceptors. Although a major therapeutic goal is to prevent any type of T cell from entering the CNS, it is particularly important to limit the expansion of Th1 and Th17 cells that have been implicated as etiologic agents in MS, EAE, uveitis and EAU.Citation13,Citation14 STAT members essential to the differentiation of Th1 and Th17 subsets have now been identified by use of mice with targeted deletion of STATs in T cells.Citation15 We provide below a brief summary of our current understanding of the role of Th1 and Th17 cells in CNS autoimmune diseases.
The Th1 lineage
Secretion of IL-12 and/or IL-27 by DCs during Ag presentation activates STAT1 and STAT4 pathways that initiate epigenetic changes and reprogram the naïve T cell genetic program to differentiate into Th1 cells.Citation15 The differentiating Th1 cell upregulates expression of IL-12Rβ2 and T-bet.Citation16 Commitment to Th1 phenotype ultimately requires the remodeling of the IFN-γ chromosomal locus, acquisition of heritable competence for sustained expression of IFN-γ and IL-12Rβ2.Citation15 Detection of IL-12 and IFN-γ in MS lesions or vitreous of patients with active uveitis, together with reports of upregulated expression of pSTAT1, pSTAT4 and T-bet in PBMC of patients with relapsing-remitting multiple sclerosis, provided strong support for the role of Th1 cells in MS and uveitis.Citation17,Citation18 The capacity of the IFN-γ-producing Th1 cells to induce CNS autoimmune disease was attributed in part to IFN-γ-induced activation of CNS-resident microglia/macrophages, upregulation of MHC class II on CNS and recruitment of inflammatory cells into the CNS via induction of chemokine expression.Citation19 It is notable that changes in the profiles of IFN-γ-dependent chemokines have profound effects on cellular trafficking and inflammatory responses in EAE.Citation20 The role of Th1 cells in CNS inflammatory disease was subsequently supported by studies showing that CNS-infiltrating T cells in EAE or EAU secrete copious amounts of IFN-γ and mice lacking T-bet are resistant to EAE.Citation21 However, mice deficient in IFN-γ receptor, STAT1 or IL-12p35 develop EAE and EAU, undermining the notion that IFN-γ and Th1 cells are essential for the development of CNS inflammatory diseases.Citation21-Citation23
The Th17 lineage
The discovery that IL-23 shares the IL-12p40 subunit with IL-12 and drives expansion of Th17 cells eventually led to the discovery of the involvement of Th17 subset in the etiology of diseases previously attributed to Th1 cells. The Th17 subset is characterized by a unique transcriptional program induced by IL-6 and TGF-β1 and dependent on STAT3 and SMAD signal transduction pathways, respectively.Citation24,Citation25 Elevated secretion of IL-6 in inflamed peripheral tissues activates STAT3 pathways resulting in the induction of ROR-γt expression and activation of epigenetic changes that effect heritable competence for sustained expression of hallmark cytokines of the Th17 lineage (IL-17A, IL-17F and IL-22).Citation25 The Th17 master transcription factors ROR-γt and RORα also induce expression of IL-23 receptor through STAT3-dependent mechanisms, rendering the differentiating cells responsive to IL-23, an innate immune cell cytokine essential for stabilization of the Th17 phenotype.Citation25 There are strong links between Th17 cells and CNS autoimmune diseases.Citation26 In studies of MS patients, IL-17 mRNA is more abundant in the blood and cerebrospinal tissue.Citation26 In addition, IL-23p19 expression is elevated in brain tissue from patients with active and chronic lesions, indicating pivotal role of IL-23 in promoting the inflammatory responses in MS.Citation27 Similarly, Th17 levels are markedly elevated in the blood of uveitis patients.Citation13 Their levels increase significantly during active uveitis and decrease following treatment, suggesting a link between the expansion of Th17 cells and autoimmune uveitis.Citation13
Therapeutic Management of Patients with CNS Autoimmune Disease
There is currently no cure for MS. However, MS is currently managed by three main strategies: administration of interferon β (IFN-β), glatiramer acetate or mitoxantrone. More than 80% of patients are treated with IFN-β, making it the most common therapy for MS.Citation28 Treatment of relapsing-remitting MS with subcutaneous IFN-β injections reduces the frequency and severity of clinical relapses, progression to disability and development of new lesions.Citation29 While the mechanisms underlying the effects of IFN-β is not well understood, it is thought to function by inhibiting T cell activation and the expression of adhesion molecules necessary for leukocyte extravasation into CNS. Glatiramer acetate, which is used in about 20% of patients, is a polymer consisting of the four amino acids glutamic acid, alanine, lysine and tyrosine and it functions by inhibiting the production of inflammatory cytokines and preventing the acquisition of encephalitogenic effector functions by CD4+ T lymphocytes.Citation30 Mitoxantrone is another FDA approved immunosuppressive agents with proven efficacy against MS; however, toxicity severely limits its use.Citation28 Similar to MS, treatment of uveitis primarily focuses on immunosuppressive agents (e.g., cyclosporine, FK-506, daclizumab, rapamycin and infliximab) or antibodies or biologics that inhibit inflammatory cytokines (anti-IL-2, anti-IFN-γ, anti-TNF-α and anti-TNF receptor). Although these drugs and biologics are fairly effective in ameliorating disease symptoms, they are of limited value as long-term therapy because they can cause renal toxicity and other adverse effects that preclude prolonged use. These limitations have been the impetus to develop new safe and effective drugs for MS and uveitis.
Therapeutic Targeting of STAT Pathways: Potential Therapy for CNS Autoimmune Diseases
Targeting IL-4 and STAT6 pathway in Th2 cells
EAE and EAU studies in mice deficient in IL-4 or STAT6 were used to assess whether STAT6 is a potential therapeutic target for treatment of MS or uveitis. IL-4-deficient mice have similar incidence, EAE scores and fatality rate as wild-type mice.Citation31 Moreover, encephalitogenic T cells transduced with an IL-4-expressing vector reduced the severity of EAE in adoptive transfer studiesCitation20 and IL-4 was found to be required for induction of protective oral tolerance in EAU,Citation32 suggesting that IL-4 may confer protection from EAE and EAU. IL-4 activates STAT6 and STAT6-deficient mice exhibit a predominantly Th1 phenotype with markedly reduced Th2 cytokine production and experience more severe clinical course of EAE as compared with WT mice.Citation20 Taken together these observations suggest that targeting STAT6 or inhibiting the differentiation or expansion of Th2 cells would not be beneficial.
Targeting STAT1 and STAT4 pathways in Th1 cells
Prior to our current appreciation of the role of Th17 cells in auto-inflammatory diseases, Th1 cells were thought to mediate EAE and EAU. As IL-12 is required for the development of Th1 cells, it was therefore expected that IL-12 administration would have disease-enhancing effects. Thus, Curcumin, a naturally occurring polyphenolic phytochemical was tested and shown to inhibit EAE by blocking IL-12-induced activation of the STAT4 transcription factor.Citation33 Similarly, COX-2 inhibitors were found to ameliorate EAE by targeting IL-12 signaling and Th1 differentiation.Citation34 On the other hand, recent studies have shown that curcumin ameliorates EAE through inhibition of IL-17 productionCitation35 and prostaglandin E2 (PGE2), a direct target of COX-2 inhibitors, promotes differentiation and proinflammatory functions of human and murine Th17 cells.Citation36 Thus, how these compounds inhibit Th17 cells remains to be elucidated.
STAT4-deficient mice are defective in Th1 differentiation and are highly resistant to the induction of EAE,Citation20 suggesting that it may be beneficial to target STAT4 pathways in T cells as a treatment for EAE. Paradoxically, IL-12 treatment protects mice from EAU through mechanisms involving IFN-γ.Citation37 In fact, mice lacking IFN-γ receptor or STAT1 develop severe EAE or EAU, underscoring the protective role of IFN-γ.Citation21,Citation23 The use of STAT4-deficient mice to investigate whether targeting Th1 differentiation would be beneficial in treating EAE or EAU is complicated because STAT4 knockout mice display a predominantly Th2 phenotypeCitation20 and produce substantial amounts of IL-4 and IL-5. Thus, the protection from EAE observed in STAT4 knockout mice may derive from protective effects of Th2 cytokines.
Nonetheless, Th1 cytokines appear to have two-sided roles: They initiate disease by promoting Ag-priming while conferring protection by limiting the immune response through upregulation of IL-27 and induction of apoptosis.Citation37,Citation38 A major concern relating to therapeutic targeting of STAT4 in CNS disease is that the role of Th1 cytokines may depend on the target tissue.Citation19 For example, recent studies in EAE suggest that while both Th1 and Th17 cells mediate EAE, production of IFN-γ by Th1 cells can suppress inflammation in the brain but not in the spinal cord.Citation19 In EAU, Th1 cells can induce disease depending on whether CFA is used during disease induction. If CFA is used, the disease is mediated by Th17 cells while Th1 cells recruited into the retina during EAU contribute to the suppression of uveitis by inducing the expression of IL-27 through STAT1-dependent mechanism and IL-27-mediated expansion of IL-10-producing T cells.Citation13,Citation23 In view of these caveats, additional data are required to further evaluate whether therapeutic inhibition of STAT4 pathway and Th1 would be beneficial in uveitis or MS.
Targeting STAT3 pathway and Th17 cells
Genome-wide association studies (GWAS) have revealed some STAT3 variants are associated with MS and the STAT3 SNP rs744166 is thought to be a putative MS protective haplotype.Citation39 Consistent with the critical role of STAT3 in Th17 differentiation, mice with targeted deletion of STAT3 in CD4+ T-cells (STAT3KO) cannot generate Th17 and do not develop EAU or EAE.Citation11 A major reason why STAT3KO mice are resistant to developing EAU or EAE is that the pathogenic T cells are defective in the expression and activation of α4β1 or α4β7 integrins and cannot traffic into the CNS.Citation11,Citation12 They are also defective in the expression of osteopontin that promotes chemotactic properties of leukocytes.Citation6 In fact, osteopontin and α4β1 along with αB crystalline are implicated in the relapse and remission of multiple sclerosis.Citation6 Pertinent to the development of effective treatment for relapsing-remitting CNS autoimmune diseases is the age-old question of where autoreactive memory lymphocytes reside in between episodes of recurrent inflammation. In a recent study, a very sensitive assay was used to track autoreactive lymphocytes that mediate blinding uveitis. The autoreactive uveitogenic memory T cells that mediate chronic uveitis were found to preferentially reside in the bone marrow through STAT3-dependent mechanisms.Citation12 Taken together, these observations suggest that STAT3 pathways and Th17 cells are attractive targets for inhibiting CNS autoimmune disease.
STAT3 inhibitors
The requirement of STAT3 for the generation of Th17 and development of EAU or EAE, suggests that the STAT3 pathway is a potential therapeutic target that can be used to prevent or mitigate uveitis or MS. Several compounds have been developed and found to inhibit STAT3 pathway and Th17 cells in vitro. Although many of these compounds are commercially available their in vivo functions have not been assessed in animal models of CNS autoimmune diseases. ORLL-NIH001 is a synthetic 406-kDa small chemical compound developed by Orchid Research Laboratories in India. ORLL-NIH001 has been utilized to inhibit STAT3 pathways in preclinical models of oncology and has recently been used to treat uveitis.Citation40 ORLL-NIH001 substantially reduced levels of Th17 cells, as well as, the IFN-γ-expressing Th17 subset that correlates with development of EAU.Citation11,Citation13 The inhibitory effects of ORLL-NIH001 derived in part from the downregulation of α4β1, α4β7, CCR6 and CXCR3. ORLL-NIH001-mediated inhibition of proteins required for lymphocyte trafficking into the retina was validated using two commercially available selective inhibitors of STAT3: a cell-permeable phosphopeptide that inhibits STAT3 by binding to STAT3-SH2 domain (STAT3 peptide; Calbiochem) and an amidosalicylic acid compound that selectively inhibits STAT3 activation and STAT3-dependent transcription (S31-201; Calbiochem).Citation40 It is notable that ORLL-NIH001 suppressed EAU in mice treated with the drug before disease induction, as well as, mice that received the drug after establishment of EAU, suggesting that ORLL-NIH001 may be used in treating pre-existing uveitis.Citation40 However, a drawback to therapeutic use of ORLL-NIH001 is its bioavailablility as frequent administration of the drug is required. Although delivery of the drug by intravenous injection is effective, oral administration and subcutaneous injection are attractive alternatives.
SOCS1 and SOCS3 mimetic peptides
Some SOCS proteins possess a kinase inhibitory region (KIR) that binds to tyrosine-phosphorylated JAKs and suppress JAK activities.Citation41 SOCS1 and SOCS3 KIR mimetics have been shown to inhibit STAT pathwaysCitation41 and the small peptide mimetics of SOCS1 effectively inhibits IL-6 and IFN-γ signaling in vitro and in vivo by targeting JAK-STAT pathway.Citation42-Citation44 Several SOCS1 mimetic peptides that are attached to lipophilic palmitoyl-lysine and arginine groups (see ) are effective in penetrating cells and inhibiting JAK2 kinase activity.Citation42 SOCS1 mimetics have also been used to inhibit Th17 expansion in EAE.Citation42-Citation44 Orally administered SOCS1 mimetics are also effective in inhibiting the production of IL-17, IFN-γ, TNF-α or IL-23 and antagonizing STAT3 activation by inflammatory cells.Citation41 Because they readily cross the blood brain barrier, SOCS1-KIR are considered to be more clinical efficacious than therapeutic antibodies that have difficulty in crossing the BBB.
Table 1. SOCS mimetics
Synthetic chemical inhibitors of Th17 developmental pathway
In addition to STATs, other transcription factors that contribute to Th17 development and expansion are potential therapeutic targets in Th17-mediated disease. For example, the nuclear receptors, retinoic-acid-receptor-related orphan receptors α (RORα) and ROR-γt, play essential roles in the development of the Th17 phenotype and are therefore attractive targets for treating EAE or EAU. Two drugs that block the activities of RORα and ROR-γt have been shown to prevent Th17 differentiation and are therefore of therapeutic value.Citation45 One of these is digoxin, a cardiac glycoside used for the treatment of heart conditions. It is a highly specific ROR-γt antagonist with no effects on RORα.Citation45 A second ligand antagonist compound called SR1001 is a derivative of the benzenesulphonamide drug T0901317.Citation45 It is a specific antagonist of both RORα and ROR-γt and does not affect the activity of other nuclear receptors. Both digoxin and SR1001 compete for ROR-γt binding to 25-hydoxycholesterol, a molecule that binds to the ligand-binding domain of ROR-γt.Citation45 Treatment of naïve CD4+ T cells with digoxin or SR1001 inhibited Th17 differentiation and expression of Th17 signature genes including IL23R, IL-17A, IL-17F and IL-22. Most importantly, both digoxin and SR1001 delayed the onset and reduced the severity of EAE by inhibiting the number of Th17 cells recruited into the spinal cord.Citation45 Thus, ROR-γt antagonists are potential drugs for treating CNS autoimmune diseases. However, while SR1001 inhibited Th17 cells in the mouse and humans, it also induced in vitro, the expansion of human Th1 and Tregs, suggesting that its effects on EAE may not be directly applicable to human disease. Another concern relating to the therapeutic use of RORα and ROR-γt inhibitors is that their effect on other cell types that express these orphan receptors are unknown. For example, ROR-γt and RORα are known to regulate hepatic glucose metabolism and targeting these factors may have unforeseen effects that may promote type I diabetes.Citation45
Perspectives
Advances in cytokine research over the past two decades have provided remarkable information and insights into mechanisms utilized by cytokines to transmit extracellular signals to the nucleus and activate genes whose products influence the behavior of the cell. The JAK-STAT pathway is one of the most important pathways utilized by cells of the innate and adaptive immune systems to initiate and regulate immune responses. Despite the remarkable knowledge of the role STAT proteins in the differentiation and functions of inflammatory cells, translation of this knowledge to therapy has lagged behind. For example, many STAT inhibitors have been developed and widely used in research. However, few of these compounds have been tested or assessed as therapeutic agents in animal models of human diseases. It is therefore of note that current therapies for treating MS and uveitis primarily focus on immunosuppressive agents or biologics. Although these drugs are fairly effective in ameliorating disease symptoms, they are of limited value because of renal toxicity and other adverse effects that preclude prolonged use. The use of small molecule compounds that target STAT pathways or Th17 cells represent an important addition to the armamentarium of remedies for these paralyzing and blinding diseases of the CNS. These new treatment modalities can potentially be used in synergy with each other, or with existing therapeutics, as a means to reduce toxicities associated with established treatment strategies. Our major goal in this review is to highlight the therapeutic merits of these compounds in autoimmune disease.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Darnell JE Jr.. STATs and gene regulation. Science 1997; 277:1630 - 5; http://dx.doi.org/10.1126/science.277.5332.1630; PMID: 9287210
- Levy DE, Darnell JE Jr.. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 2002; 3:651 - 62; http://dx.doi.org/10.1038/nrm909; PMID: 12209125
- Alexander WS. Suppressors of cytokine signalling (SOCS) in the immune system. Nat Rev Immunol 2002; 2:410 - 6; PMID: 12093007
- Wraith DC, Nicholson LB. The adaptive immune system in diseases of the central nervous system. J Clin Invest 2012; 122:1172 - 9; http://dx.doi.org/10.1172/JCI58648; PMID: 22466659
- Streilein JW. Ocular immune privilege: therapeutic opportunities from an experiment of nature. Nat Rev Immunol 2003; 3:879 - 89; http://dx.doi.org/10.1038/nri1224; PMID: 14668804
- Steinman L. A molecular trio in relapse and remission in multiple sclerosis. Nat Rev Immunol 2009; 9:440 - 7; http://dx.doi.org/10.1038/nri2548; PMID: 19444308
- Nussenblatt RB. The natural history of uveitis. Int Ophthalmol 1990; 14:303 - 8; http://dx.doi.org/10.1007/BF00163549; PMID: 2249907
- Egwuagu CE, Charukamnoetkanok P, Gery I. Thymic expression of autoantigens correlates with resistance to autoimmune disease. J Immunol 1997; 159:3109 - 12; PMID: 9317106
- Whitcup SM, Kozhich AT, Lobanoff M, Wolitzky BA, Chan CC. Blocking both E-selectin and P-selectin inhibits endotoxin-induced leukocyte infiltration into the eye. Clin Immunol Immunopathol 1997; 83:45 - 52; http://dx.doi.org/10.1006/clin.1996.4324; PMID: 9073535
- Alvarez JI, Cayrol R, Prat A. Disruption of central nervous system barriers in multiple sclerosis. Biochim Biophys Acta 2011; 1812:252 - 64; http://dx.doi.org/10.1016/j.bbadis.2010.06.017; PMID: 20619340
- Liu X, Lee YS, Yu CR, Egwuagu CE. Loss of STAT3 in CD4+ T cells prevents development of experimental autoimmune diseases. J Immunol 2008; 180:6070 - 6; PMID: 18424728
- Oh HM, Yu CR, Lee Y, Chan CC, Maminishkis A, Egwuagu CE. Autoreactive memory CD4+ T lymphocytes that mediate chronic uveitis reside in the bone marrow through STAT3-dependent mechanisms. J Immunol 2011; 187:3338 - 46; http://dx.doi.org/10.4049/jimmunol.1004019; PMID: 21832158
- Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, et al. TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat Med 2007; 13:711 - 8; http://dx.doi.org/10.1038/nm1585; PMID: 17496900
- Laurence A, O’Shea JJT. T(H)-17 differentiation: of mice and men. Nat Immunol 2007; 8:903 - 5; http://dx.doi.org/10.1038/ni0907-903; PMID: 17712339
- Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol 2010; 28:445 - 89; http://dx.doi.org/10.1146/annurev-immunol-030409-101212; PMID: 20192806
- Murphy KM, Ouyang W, Farrar JD, Yang J, Ranganath S, Asnagli H, et al. Signaling and transcription in T helper development. Annu Rev Immunol 2000; 18:451 - 94; http://dx.doi.org/10.1146/annurev.immunol.18.1.451; PMID: 10837066
- Nicoletti F, Patti F, Cocuzza C, Zaccone P, Nicoletti A, Di Marco R, et al. Elevated serum levels of interleukin-12 in chronic progressive multiple sclerosis. J Neuroimmunol 1996; 70:87 - 90; http://dx.doi.org/10.1016/S0165-5728(96)00101-4; PMID: 8862139
- Meleth AD, Agrón E, Chan CC, Reed GF, Arora K, Byrnes G, et al. Serum inflammatory markers in diabetic retinopathy. Invest Ophthalmol Vis Sci 2005; 46:4295 - 301; http://dx.doi.org/10.1167/iovs.04-1057; PMID: 16249511
- Pierson E, Simmons SB, Castelli L, Goverman JM. Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol Rev 2012; 248:205 - 15; http://dx.doi.org/10.1111/j.1600-065X.2012.01126.x; PMID: 22725963
- Chitnis T, Khoury SJ. Cytokine shifts and tolerance in experimental autoimmune encephalomyelitis. Immunol Res 2003; 28:223 - 39; http://dx.doi.org/10.1385/IR:28:3:223; PMID: 14713716
- Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med 2004; 200:79 - 87; http://dx.doi.org/10.1084/jem.20031819; PMID: 15238607
- Nishibori T, Tanabe Y, Su L, David M. Impaired development of CD4+ CD25+ regulatory T cells in the absence of STAT1: increased susceptibility to autoimmune disease. J Exp Med 2004; 199:25 - 34; http://dx.doi.org/10.1084/jem.20020509; PMID: 14699080
- Lee YS, Amadi-Obi A, Yu CR, Egwuagu CE. Retinal cells suppress intraocular inflammation (uveitis) through production of interleukin-27 and interleukin-10. Immunology 2011; 132:492 - 502; http://dx.doi.org/10.1111/j.1365-2567.2010.03379.x; PMID: 21294722
- Egwuagu CE. STAT3 in CD4+ T helper cell differentiation and inflammatory diseases. Cytokine 2009; 47:149 - 56; http://dx.doi.org/10.1016/j.cyto.2009.07.003; PMID: 19648026
- Ivanov II, Zhou L, Littman DR. Transcriptional regulation of Th17 cell differentiation. Semin Immunol 2007; 19:409 - 17; http://dx.doi.org/10.1016/j.smim.2007.10.011; PMID: 18053739
- Matusevicius D, Kivisäkk P, He B, Kostulas N, Ozenci V, Fredrikson S, et al. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler 1999; 5:101 - 4; PMID: 10335518
- Li Y, Chu N, Hu A, Gran B, Rostami A, Zhang GX. Increased IL-23p19 expression in multiple sclerosis lesions and its induction in microglia. Brain 2007; 130:490 - 501; http://dx.doi.org/10.1093/brain/awl273; PMID: 17003070
- Javed A, Reder AT. Therapeutic role of beta-interferons in multiple sclerosis. Pharmacol Ther 2006; 110:35 - 56; http://dx.doi.org/10.1016/j.pharmthera.2005.08.011; PMID: 16229894
- Codarri L, Fontana A, Becher B. Cytokine networks in multiple sclerosis: lost in translation. Curr Opin Neurol 2010; 23:205 - 11; http://dx.doi.org/10.1097/WCO.0b013e3283391feb; PMID: 20442570
- Aharoni R. The mechanism of action of glatiramer acetate in multiple sclerosis and beyond. Autoimmun Rev 2012; In press PMID: 23051633
- Liblau R, Steinman L, Brocke S. Experimental autoimmune encephalomyelitis in IL-4-deficient mice. Int Immunol 1997; 9:799 - 803; http://dx.doi.org/10.1093/intimm/9.5.799; PMID: 9184926
- Rizzo LV, Morawetz RA, Miller-Rivero NE, Choi R, Wiggert B, Chan CC, et al. IL-4 and IL-10 are both required for the induction of oral tolerance. J Immunol 1999; 162:2613 - 22; PMID: 10072503
- Natarajan C, Bright JJ. Curcumin inhibits experimental allergic encephalomyelitis by blocking IL-12 signaling through Janus kinase-STAT pathway in T lymphocytes. J Immunol 2002; 168:6506 - 13; PMID: 12055272
- Muthian G, Raikwar HP, Johnson C, Rajasingh J, Kalgutkar A, Marnett LJ, et al. COX-2 inhibitors modulate IL-12 signaling through JAK-STAT pathway leading to Th1 response in experimental allergic encephalomyelitis. J Clin Immunol 2006; 26:73 - 85; http://dx.doi.org/10.1007/s10875-006-8787-y; PMID: 16418805
- Xie L, Li XK, Takahara S. Curcumin has bright prospects for the treatment of multiple sclerosis. Int Immunopharmacol 2011; 11:323 - 30; http://dx.doi.org/10.1016/j.intimp.2010.08.013; PMID: 20828641
- Boniface K, Bak-Jensen KS, Li Y, Blumenschein WM, McGeachy MJ, McClanahan TK, et al. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J Exp Med 2009; 206:535 - 48; http://dx.doi.org/10.1084/jem.20082293; PMID: 19273625
- Tarrant TK, Silver PB, Wahlsten JL, Rizzo LV, Chan CC, Wiggert B, et al. Interleukin 12 protects from a T helper type 1-mediated autoimmune disease, experimental autoimmune uveitis, through a mechanism involving interferon gamma, nitric oxide, and apoptosis. J Exp Med 1999; 189:219 - 30; http://dx.doi.org/10.1084/jem.189.2.219; PMID: 9892605
- Fitzgerald DC, Zhang GX, El-Behi M, Fonseca-Kelly Z, Li H, Yu S, et al. Suppression of autoimmune inflammation of the central nervous system by interleukin 10 secreted by interleukin 27-stimulated T cells. Nat Immunol 2007; 8:1372 - 9; http://dx.doi.org/10.1038/ni1540; PMID: 17994023
- Jakkula E, Leppä V, Sulonen AM, Varilo T, Kallio S, Kemppinen A, et al. Genome-wide association study in a high-risk isolate for multiple sclerosis reveals associated variants in STAT3 gene. Am J Hum Genet 2010; 86:285 - 91; http://dx.doi.org/10.1016/j.ajhg.2010.01.017; PMID: 20159113
- Yu CR, Lee YS, Mahdi RM, Surendran N, Egwuagu CE. Therapeutic targeting of STAT3 (signal transducers and activators of transcription 3) pathway inhibits experimental autoimmune uveitis. PLoS One 2012; 7:e29742; http://dx.doi.org/10.1371/journal.pone.0029742; PMID: 22238646
- Jager LD, Dabelic R, Waiboci LW, Lau K, Haider MS, Ahmed CM, et al. The kinase inhibitory region of SOCS-1 is sufficient to inhibit T-helper 17 and other immune functions in experimental allergic encephalomyelitis. J Neuroimmunol 2011; 232:108 - 18; http://dx.doi.org/10.1016/j.jneuroim.2010.10.018; PMID: 21131060
- Ahmed CM, Dabelic R, Martin JP, Jager LD, Haider SM, Johnson HM. Enhancement of antiviral immunity by small molecule antagonist of suppressor of cytokine signaling. J Immunol 2010; 185:1103 - 13; http://dx.doi.org/10.4049/jimmunol.0902895; PMID: 20543109
- Flowers LO, Johnson HM, Mujtaba MG, Ellis MR, Haider SM, Subramaniam PS. Characterization of a peptide inhibitor of Janus kinase 2 that mimics suppressor of cytokine signaling 1 function. J Immunol 2004; 172:7510 - 8; PMID: 15187130
- Waiboci LW, Ahmed CM, Mujtaba MG, Flowers LO, Martin JP, Haider MI, et al. Both the suppressor of cytokine signaling 1 (SOCS-1) kinase inhibitory region and SOCS-1 mimetic bind to JAK2 autophosphorylation site: implications for the development of a SOCS-1 antagonist. J Immunol 2007; 178:5058 - 68; PMID: 17404288
- Jetten AM. Immunology: A helping hand against autoimmunity. Nature 2011; 472:421 - 2; http://dx.doi.org/10.1038/472421a; PMID: 21525918