Abstract
The redox regulation of Janus kinases (JAKs) is a complex subject. Due to other redox-sensitive kinases in the kinome, redox-sensitive phosphatases, and cellular antioxidant systems and reactive oxygen species (ROS) production systems, the net biological outcomes of oxidative stress on JAK-dependent signal transduction vary according to the specific biological system examined. This review begins with a discussion of the biochemical evidence for a cysteine-based redox switch in the catalytic domain of JAKs, proceeds to consider direct and indirect regulatory mechanisms involved in biological experiments, and ends with a discussion of the role(s) of redox regulation of JAKs in various diseases.
As recounted in John Godfrey Saxe’s 19th century poem, “The Blind Men and the Elephant”, blind men attempted to describe an elephant, each comparing it to a wall, a spear, a snake, a tree, a fan, or a rope, depending on which part of the elephant he touched. “And so these men of Indostan / Disputed loud and long, / Each in his own opinion / Exceeding stiff and strong, / Though each was partly in the right, / And all were in the wrong!” This unresolved quarrel applies to the modern elephant sometimes known as “oxidative stress”, sometimes as “redox regulation”, and often described in terms of reactive oxygen species (ROS), reactive nitrogen species (RNS), hypoxia, ischemia, inflammation, lipid peroxidation, or the cornucopia of molecules with pro-oxidant or antioxidant properties. Unsurprisingly, when one appraises the literature surrounding the relationship between the elephant and Janus kinases (JAKs), one encounters a spirited disagreement over basic observations. To wit, some have argued that JAK activity was enhanced by oxidative stress while others argued that oxidative stress inhibited JAK activity, “though each was partly in the right”. In this review I will first discuss in vitro biochemical evidence to provide a plausible molecular mechanism for the direct redox regulation of JAK’s catalytic activity. Second, I will review in situ and in vivo experiments demonstrating that redox regulation and oxidative stress can affect JAK-dependent cellular outcomes, and discuss these results in terms of direct and indirect regulatory mechanisms. Thereafter I will speculate on a number of pathological conditions in which the redox regulation of JAKs may be significant.
Part I: The Beauty of Biochemical Simplicity
The term redox simply reminds us that whenever one molecule (or reaction center within a macromolecule) gains electrons to become reduced to a lower oxidation state, another molecule must yield those electrons to become oxidized to a higher oxidation state. From the author's perspective, the original clue that JAK2 might be susceptible to redox regulation was simply that it lost considerable activity once a few hours intervened between the isolation of the recombinant enzyme and the initiation of the radiolabelling autokinase assay, indicative of thermodynamically-favorable, spontaneous oxidation upon equilibration with atmospheric oxygen. By using two well-established thiol modification reagents (the reductant dithiothreitol (DTT)Citation1 and the oxidant ortho-iodosobenzoate (oIBZ)Citation2,Citation3), it was relatively simple to demonstrate that JAK2's activity could be inhibited by pre-treatment with oIBZ, maximized by pre-treatment with DTT, and that the effect of each reagent could be completely reversed by re-treatment with the reciprocal reagent.Citation4 Note that the pre-treated enzyme was removed from the redox reagents prior to initiating the autokinase assay; some commercial assay cocktails contain DTT or other thiol-reducing agents which mask the inhibitory effect of any oxidant and lead to erroneous conclusions. The dramatic response of partially-purified JAK2 to these reagents implied that cysteine residues within the enzyme were intimately involved in the process, and allowed one to predict that other thiol-reactive oxidants would evoke a similar response as oIBZ pre-treatment. Nitric oxide is known to react with free thiols to form S-nitrosothiolsCitation5 and is capable of oxidizing vicinal thiols into disulfidesCitation6; direct treatment of immunoprecipitated JAK2 with nitric oxide also resulted in DTT-reversible inhibition of radiolabelling autokinase activity.Citation4 These preliminary in vitro experiments involving partially-purified enzymes and thiol-selective reagents inspired the search for cysteine residues within JAK2 responsible for this phenomenon. Recombinant rat JAK2 contained 27 cysteine residues, but fortuitously, 18 of those residues in the N-terminal domains were eliminated from consideration by observing that a truncated, hyperactive form of the enzyme exhibited the same redox-sensitivity as the full-length form of the enzyme.Citation7 Assuming that serine residues would be the most conservative substitution for cysteine residues, a series of Cys-to-Ser mutant forms of the truncated, hyperactive JAK2 were generated in individual and combinatorial fashion, leading to the identification of four cysteine residues within the catalytic domain (Cys866, Cys917, Cys1094, and Cys1105) which cooperatively maintained catalytic competency. Further scrutiny of these four residues via Cys-to-Ala mutations, again in individual and pairwise fashion, revealed a functional role(s) of the Cys866,Cys917 pair in the N-lobe of the catalytic domain which was clearly distinct from that of the Cys1094,Cys1105 pair in the C-lobe of the catalytic domain.Citation8 Simultaneous mutation of both N-lobe cysteine residues to alanine residues provided evidence that these two served as a redox switch in the catalytic domain. This mutant possessed the requisite high level of in vitro radiolabelling autokinase activity to clearly demonstrate that the activity was completely unaffected by redox reagents oIBZ and DTT, unlike all other active JAK2 mutants examined to date.
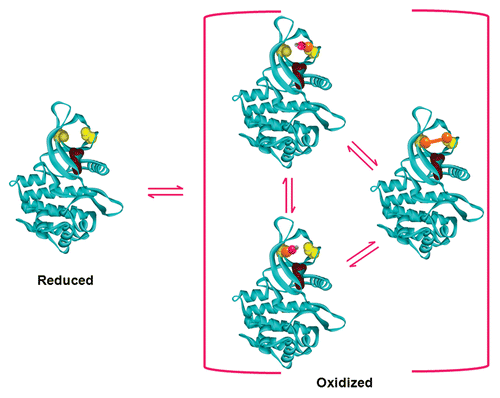
Other experimental insights into the nature of the oxidized state of the enzyme included: (1) serine substitutions at positions 866 and/or 917 consistently exhibited lower activities than their alanine counterparts, which was unexpected due to the assumption that a polar substitution would be more conservative than an apolar substitution; (2) individual mutations at either position 866 or 917 did not completely abolish the redox sensitivity of the enzyme, which was unexpected due to the assumption that the pair of proximal cysteine residues formed a disulfide bond upon oxidation. Thus, the reversible oxidation of these cysteine residues to sulfenic acids provided the most satisfactory explanation for the aforementioned biochemical observations. This does not exclude the possibility of disulfide formation under physiological conditions, because the two sulfur centers of Cys866 and Cys917 are approximately 9 Å apart, residing in the β2 and β4 sheets. It is perfectly reasonable to envision that these sulfur centers can flex closer to each other to reach the permissive distance for disulfide bond formation. The current best model for the direct redox regulation of Janus kinases is depicted in , in which the two N-lobe cysteines can exist in dynamic equilibrium between thiol, sulfenic acid, and disulfide states.Citation9 Substitution of serine residues in the N-lobe appeared to mimic a state in which the two cysteine residues have been oxidized to sulfenic acids, and these mutant enzymes had extremely low autokinase activity, as evidenced by the near lack of observable activity in the radiolabelling autokinase assay and grossly impaired in situ autophosphorylation activity. However, such mutants were not completely inactive kinases. As of this writing, the exact mechanism(s) to explain (i.e., altered nucleotide binding properties, destabilized coordination of the divalent cation bound to ATP, increased futile cycle hydrolysis of ATP, etc.) why the oxidized form of JAK2 has such dramatically-altered activity is as yet undefined. The site of the redox switch is in close proximity to the ATP binding site of JAK2, and it is also close to other N-lobe amino acid residues which increase JAK2’s enzymatic activity when mutated (T875N,Citation10 Y931C,Citation11 D873N, and P933RCitation12). Thus, the exact mechanism whereby the redox switch modulates JAK2’s activity is most likely to involve one or more of the biochemical steps involved in the catalysis of tyrosine phosphorylation.
Figure 1. A simple equilibrium model for the direct redox regulation of Janus kinases. This model, based on the 3D structural coordinates of the JAK2 catalytic domain,Citation125 shows the reduced (thiol) state of the redox switch in yellow, with Cys866 on the left and Cys917 on the right, and with the essential Lys882 residue shown in purple. Upon oxidation, these two residues can become oxidized to form a disulfide bond, shown in orange, or either one can become independently oxidized to a sulfenic acid, where the S-OH moiety is shown as bonded orange-pink-gray spheres. The dynamic equilibrium between these states shifts in response to the redox state of the environment, which can be naturally or artificially manipulated by an excess of reductants (shifting left) or oxidants (shifting right). Note: the disulfide form does not need to transition through a sulfenic acid state to become the fully reduced state, and vice versa.
Cysteine-based redox switches have been identified in a broad range of other enzyme systems and are currently appreciated as an important post-translational regulatory mechanism which allows these systems to respond to changes in the redox environment.Citation13 In addition to extra-catalytic redox switches, enzymes such protein tyrosine phosphatase 1B (PTP1B) contain active-site cysteine residues which are absolutely essential to the catalytic mechanism of action.Citation14 Unlike an essential active-site cysteine, the JAK2 redox switch dramatically modulates the enzyme's kinase activity rather than switching it between absolute “on/off” states. When one aligns the three dimensional structures of JAK2 and JAK1, the two cysteine residues of the redox switch are absolutely conserved,Citation8 consistent with evidence that JAK1 exhibits comparable redox-sensitivity.Citation15,Citation16 JAK3 has also been shown to be redox-sensitive,Citation4 yet only one of the two redox switch cysteine residues (Cys839 in JAK3, corresponding to Cys866 in JAK2) are aligned in 3D space; a serine residue (Ser890) is located in the space occupied by Cys917 in JAK2. This is consistent with mutational evidence showing that only one of the two cysteines is required for redox-sensitivity in JAK2, and is consistent with the notion that sulfenic acid formation is sufficient for a functional switch response. Evidence of the redox-sensitivity of TYK2 also existsCitation16 and is consistent with the comparable spatial arrangement of the redox switch cysteine residues: Cys866 of JAK2 and Cys915 of TYK2 are adjacent, rather than overlapping, and while Cys917 of JAK2 and Cys966 of TYK2 overlap, TYK2 contains an additional adjacent Cys965. A representative sample of 22 JAK amino acid sequences from diverse metazoan species were aligned using the CLUSTAL OMEGA multiple sequence alignment algorithmCitation17,Citation18; sections of the alignment including rat JAK2 cysteine residues 866, 917, 1094, and 1105 are shown in . The redox sensor switch is found in JAK1 and JAK2, whether from mammals, birds, reptiles or fish, but is found in neither HOP expressed in insects nor in JAK expressed in tunicates and crustaceans. Conservation of the redox sensor switch cysteine residues in vertebrate JAK3 and TYK2 is variable ().
Figure 2. Conservation of four critical cysteine residues within 22 Janus kinases. Twenty-two JAK amino acid sequences from diverse metazoan species were aligned using the CLUSTAL OMEGA multiple sequence alignment algorithm.Citation17,Citation18 Sections of the alignment including rat JAK2 cysteine residues 866, 917, 1094, and 1105 are shown, with conserved cysteine residues highlighted in yellow.

It is unclear whether this particular redox switch motif, which requires further experimental characterization, regulates the function of other protein-tyrosine kinases. Of the 90 mammalian protein-tyrosine kinase catalytic domains examined, only the TRK family members TRKA, TRKB, and TRKC contain such a spatially-conserved two-cysteine motif. Although no direct evidence could be found to show that this spatially-conserved two-cysteine motif functions as a redox switch in TRK family members, Cys866 of JAK2 closely approximates the space occupied by Cys529, Cys573, and Cys557 of TRKA, TRKB, and TRKC, respectively, and Cys917 of JAK2 overlaps the space occupied by Cys579, Cys623, and Cys607 of those respective kinases. Yet 51 of the mammalian protein-tyrosine kinases examined contain a cysteine residue in the space occupied by Cys917 of JAK2. In contrast, EPH family members lack such a cysteine, as do most of the SRC family members. Src is directly redox-regulated, although the precise mechanism remains a matter of debate.Citation19 Some have proposed that hydrogen peroxide activates Src activity via disulfide linkage between Cys245 in the SH2 domain and Cys487 in the kinase domain.Citation20 Others have proposed that oxidation of Cys277 creates an inactive Src dimer via intermolecular disulfide bond formation, a model which is proposed to extend to FGFR family members.Citation21 In addition to the aforementioned TRK family members, mammalian protein-tyrosine kinase families containing an equivalent of JAK2's Cys917 residue include the FGFR, VEGFR, PDGFR, TEC, LMR, and EGFR families.
Before ending this discussion of the in vitro biochemical aspects of the redox regulation of Janus kinases, the author wishes to mention another curiosity which may be either significant or merely happenstance. Two of the four cysteine residues (Cys1094 and Cys1105) which cooperatively maintain catalytic competency flank the opposite ends of the H-helix within the C-lobe.Citation7 The kinase-deficient B-form splice variant of human JAK3 contains the equivalent of Cys1094 but lacks both the Cys1105 equivalent and the I-helix.Citation22 No specific mechanistic role(s), other than maintaining structural and conformational integrity, has been proposed for these cysteine residues, yet this CPX6MX2CW motif appears in 54 protein-tyrosine kinases, with the less-constrained CX9C motif appearing in 70 mammalian protein-tyrosine kinases. While the equivalent of rat JAK2 Cys1094 did not appear in 2 of the 22 Janus kinases examined, the equivalent of Cys1105 was completely conserved in this sample, including in the insect HOPs. Should these cysteine residues serve a specific functional role, it should be of broad significance; otherwise, it may simply be a cherished evolutionary heirloom.
Part II: The Complexity of Biological Permutations
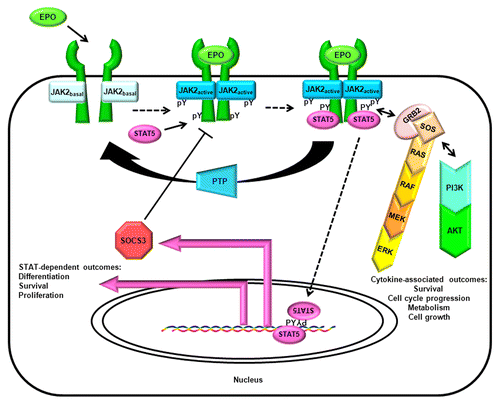
The literal biochemical purity required for the elucidation of structures via X-ray crystallography cannot be found in the messy realm of biology. The elegantly simple canonical cytokine-receptor-JAK-STAT-SOCS pathway provides a clear path from extracellular signal to intracellular response, but such pristine orderliness only exists in review figures (). JAK-dependent cytokines and cytokine-like hormones such as prolactinCitation23 and erythropoietinCitation24 are notoriously pleiotropic, which is unsurprising in light of the myriad other signaling molecules which can cross-talk with their associated receptors, receptor variants, JAKs, STATs, and SOCS within any given cell type. In these canonical cytokine receptor pathways, JAK transitions from a basal to an active state upon cytokine-stimulated autophosphorylation of tyrosine residues within its activation loop.Citation25,Citation26 JAKs also participate in a number of non-canonical pathways, most notably certain G-protein-coupled receptor (GPCR) pathways.Citation27,Citation28 JAKs are but one part of a dynamically-interactive kinome which involves interconnected cell-dependent patterns of synergies, trans-phosphorylations, and/or trans-activations involving various protein kinases.Citation29 This kinome network has various regulatory counterweights in protein tyrosine phosphatases, dual-specificity phosphatases and assorted post-translational modifiers. Were this not enough complexity to affect the net biological outcome of JAK-mediated signaling, one should consider that JAK (most notably JAK2) can occasionally be found in the cell nucleus.Citation30 While JAK has low catalytic turnover rates, it promiscuously recognizes substrates,Citation31-Citation33 so there may be a host of substrates phosphorylated by nuclear JAK awaiting characterization.Citation34 Finally, Janus kinases remain full of enzymatic surprises, ranging from unconventional responses to enzyme inhibitorsCitation35,Citation36 to novel catalytic properties outside of the well-known JH1 catalytic domainCitation37,Citation38.
Figure 3. The canonical cytokine/JAK/STAT/SOCS pathway. The canonical cytokine/JAK/STAT/SOCS pathway is illustrated with the example of erythropoieitin/JAK2/STAT5/SOCS3. Canonical pathways stimulate cytokine and cytokine-like hormone receptor-dependent activation of tyrosine kinase activity in JAKs, resulting in an elevation of JAK activity state, phosphorylation of docking sites on the receptor, and the recruitment and tyrosine phosphorylation of STATs. Tyrosine-phosphorylated STATs oligomerize, translocate into the nucleus, and stimulate gene transcription. One of the transcribed genes encodes SOCS, which primarily provides negative feedback via inhibition of JAK's action and initiating proteasomal degradation of the activated receptor complex; upon tyrosine phosphorylation it also promotes survival via the Ras pathway.Citation126 Other tyrosine phosphorylation-dependent interactions initially catalyzed by JAK, such as coupling the EPO receptor (EPOR) to the RAS/RAF/MEK/ERK pathway via GRB2/SOS, or coupling EPOR to the PI3K/AKT pathway, may not be considered by all authors to be part of the canonical pathway per se, yet they must be considered to understand the cytokine-proximal events of cell biology.
As stated above, the cellular redox state may regulate JAK directly via cysteines in the redox switch. Several of the PTKs capable of transphosphorylating and/or synergizing with JAK, such as Src,Citation20,Citation21 EGFR,Citation39 or FynCitation40 are directly affected by oxidants acting on regulatory cysteine residues. In some PTKs, most notably EGFR,Citation39 a biphasic response to increasing amounts of oxidants has been observed, which might be explained by the presence of stimulatory and inhibitory cysteine residues within the same kinase. At any given cellular redox state, the regulatory cysteine residues of some proteins will be more prone to oxidation than others. However, the fundamental kinetic and thermodynamic parameters (such as pKa values for regulatory cysteine residues and standard half-cell potentials for such cysteine/disulfide couples) which dictate this hierarchy are largely undefined for most PTKs. Tremendous progress made in computational modeling of the biochemistry of cysteine residuesCitation41 does not yet allow ranking of PTKs according to their individual reduced/oxidized equilibrium distributions at any given cellular redox state. Without further advances in both of these areas, one cannot predict which redox switches will be the first to be activated by mild oxidative stress, nor can one predict how mild oxidative stress will alter biological outcomes due to an ensemble of PTKs.
This argument also applies to the redox regulation of protein tyrosine phosphatases (PTPs). PTP1B,Citation42 CD45,Citation43 SHP-2,Citation44 and other mammalian PTPs and dual-function phosphatases contain a conserved catalytic-site cysteine, and uniformly become inactivated when it becomes oxidized. Other post-translational modifications of this cysteine, such as S-nitrosylation or glutathionylation, potentially protect the phosphatase from oxidative inhibition. If the composition and abundance of phosphatases within a cell are such that dephosphorylation dominates the balance of phosphorylated-to-nonphosphorylated STATs, and those phosphatases possess a higher oxidation sensitivity than do the resident JAKs, then one would expect oxidative stress to result in a net increase in STAT phosphorylation. If the situation is reversed in a different cell type, then one would expect oxidative stress to result in a net decrease in STAT phosphorylation. Indeed, the Halvorsen lab has shown that oxidative stress caused by hydrogen peroxide treatment inhibited JAK-coupled STAT phosphorylation in neuronal cells, but had no effect or enhanced such STAT phosphorylation in non-neuronal cells examined in the same report.Citation45 Hydrogen peroxide significantly impaired the ability of either growth hormone or prolactin to stimulate STAT5 phosphorylation in pancreatic β-islet cells.Citation8 In general, oxidative inhibition of JAK-mediated signaling has been observed when investigations focused on the ability of cytokines to evoke canonical JAK/STAT signals under oxidizing conditions, whereas the oxidative stimulation of JAK-mediated signaling was observed under more complex circumstances. Cell types in which the net canonical JAK/STAT outcome was inhibited by oxidative or nitrosative stress includes leptin- or CNTF-stimulated neuronal cells,Citation45-Citation48 IL-2- or IL-7-stimulated T cells,Citation49-Citation51 LIF-stimulated cardiomyocytes,Citation15,Citation52 IFN-α-stimulated hepatocellular carcinoma cells,Citation16 IL-3-stimulated pro-B cellsCitation4 and GH- or PRL-stimulated pancreatic β-islet cells.Citation8 In contrast, cell types in which resting, growth-arrested, or non-canonical JAK-associated outcomes were stimulated by oxidative stress include unsynchronized or growth-arrested fibroblasts,Citation40,Citation53 quiescent vascular smooth muscle cellsCitation54 or aortic endothelial cells,Citation55 and Ang II-stimulated aortic smooth muscle cells.Citation56
Unlike the canonical JAK-STAT pathways, the Ang II/JAK2 system found in vascular smooth muscle cells and glomerular mesangial cells has been associated consistently with enhanced JAK2 activity under oxidative stress. This non-canonical GPCR system stimulates JAK2,Citation27 stimulates the intracellular production of ROS,Citation57 is enhanced by hyperglycemia,Citation58 and requires ancillary signaling components such as SHP-2Citation59 to couple extracellular Ang II stimulation to JAK2 activation. Unlike most canonical systems in which JAK2 activity transduced pro-survival, mitogenic, differentiation, or growth arrest outcomes, in the Ang II and/or hydrogen peroxide systems, JAK2 activity has been reported to induce apoptosis in endothelial cells,Citation55,Citation60 vascular smooth muscle cells,Citation61 and cardiomyocytes.Citation62
Because no group has yet identified a second regulatory switch in JAK2 which could account for an increase in activity upon oxidation, how can one explain the observed stimulatory effects of hydrogen peroxide and ROS on JAK2 activity in these non-canonical systems? The loss of low molecular weight–protein tyrosine phosphatase (LMW-PTP) activity resulting from NADPH oxidase-catalyzed ROS generation appears to contribute to in the sustained JAK2 activity observed in pancreatic cancer cells.Citation63 The loss of phosphatase counteraction partially explains the net increase in STAT phosphorylation in non-stimulated B cells treated with hydrogen peroxide.Citation64,Citation65 Yet phosphatase inhibition cannot logically account for the increased in vitro radiolabelling autokinase activity in immunoprecipitated JAK2 isolated from fibroblasts following H2O2 exposure.Citation53 Trans-phosphorylating kinases, such as Fyn, might supplement the explanation, as evidenced by the stimulation of JAK2 by hydrogen peroxide in a Src-deficient cell, but not in a Fyn-deficient cell.Citation40 Yet Fyn activation was dependent upon JAK2 activity in the Ang II systemCitation66 in transfected COS-7 cells and vascular smooth muscle cells, while in the thymus of rodents Fyn transduced the biological effects of leptin in a JAK2-independent manner,Citation67 despite leptin's dependence upon JAK2 in most other cells. It has been proposedCitation68 that in vascular smooth muscle cells the ROS-mediated activation of JAK2 begins with the ROS activation of PKC-δ, which in turn activates PYK2 to transphosphorylate JAK2. Of course, one must be careful about assumptions surrounding in vivo and in situ PTK experiments, as illustrated by the demonstration that AG490 (originally developed as an EGFR kinase inhibitor,Citation69 yet erroneously typecast as a “specific” JAK2 inhibitor) had unexpected antioxidant properties that were independent of JAK2; this was critical to the recognition that the induction of ROS and the activation of STAT1 were separate H2O2-stimulated events occurring in glial cells.Citation70 One of the most important caveats about the interpretation of JAK2 inhibitor studies was the discovery that JAK2 inhibitors can lead to an increase of activation loop phosphorylation in a manner that is binding mode dependent.Citation35 In the context of myeloproliferative disorders, chronic treatment of JAK2-dependent cells with JAK2 inhibitors, rather than eradicate the cells, can result in the formation of heterodimeric complexes with JAK1 or TYK2 which transphosphorylate JAK2 and allow these cells to persist in the presence of type 1 JAK2 inhibitors.Citation36 It appears that JAK2 and hyperactive JAK2(V617F) induce ROS production in some cells,Citation71 indicating the existence of a feedback model to explain the dynamic interactions of JAK2 and intracellular ROS. However, one must exercise caution in interpreting biological studies involving JAK2, where there is often more to the picture than originally meets the eye.
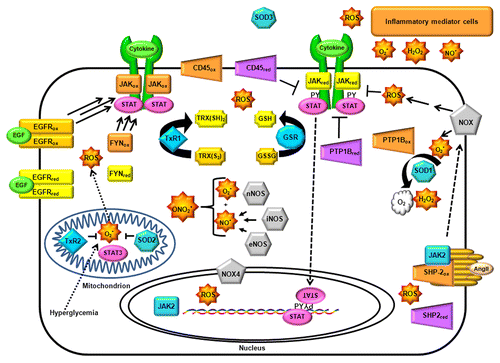
The complex portrait involving the many molecular partners known to interact with canonical and non-canonical JAK-STAT pathways makes it difficult to generate an accurate systems biology model of JAK-STAT signal transduction. In order to predict the outcome of oxidative stress on JAK-mediated signal transduction in a given cell type, one must consider the intrinsic redox sensitivity of all JAK-associated macromolecules, as well as the effect of these macromolecules on the cellular generation of oxidants and antioxidants, along with the capacity and responsiveness of the cell's autonomous antioxidant defense system (). In the absence of such an idealized computational model, researchers must continue to pay extremely close attention to specific experimental conditions to understand how ostensibly similar experiments can give rise to apparently conflicting results.
Figure 4. Permutations of JAK signal transduction outcomes due to the complexity of redox-associated pathways within the cell. To illustrate the importance of cell-specific context in determining the net biological outcomes of redox-regulation of JAK-dependent signaling, this cartoon depicts a few of the redox-related pathways, biomolecules, and processes capable of interacting with JAKs. Each of these components, such as protein tyrosine phosphatases (PTP, purple tetragons) and protein tyrosine kinases (yellow and orange rectangles) will be present in variable abundances from cell type to cell type, and their expression dynamically fluctuates in response to redox and non-redox regulation. Moreover, ROS is not generated uniformly throughout the cell, but is compartmentalized, such that their concentrations are gradients which can be transient or sustained according to the intensity and duration of their production. NADPH oxidases (NOX, gray pentagons) have specific subcellular localizations, as do thioredoxin reductases (TxR) and superoxide dismutases (SOD); cell-specific nitric oxide synthetases (NOS), glutathione reductase (GSR), and neighboring cells also affect the cellular redox state. Several of these redox regulators are in turn regulated by JAKs and other key signal transduction enzymes.
Part III: The Joy of Idle Speculation
Given the multitude of molecular determinants which complicate the role of redox regulation of JAK in cellular biology, modesty may be the best policy when making claims about the role(s) of redox regulation of JAKs in disease and in age-related health problems. Oxidants are key contributors to the free radical theory of aging,Citation72,Citation73 and oxidative stress is a pathogenic factor in a myriad of diseases, including cardiovascular diseases, type II diabetes, cancers, immunosuppressive diseases, asthma and neurodegenerative diseases. It would be disingenuous to claim that the redox regulation of JAKs is central to the pathogenic processes of all oxidant-associated diseases, yet naive to think it is relevant to none. Indeed, because the redox regulation of Janus kinases may be involved in so many disease pathologies, this sub-topic is more appropriate for a thorough review, and most diseases will be only briefly mentioned. For instance, the role of oxidative stress in cardiovascular diseaseCitation74 is a topic of vast importance, and the medical community continues to seek successful antioxidant therapies which remain elusive.Citation75-Citation77 To give justice to the redox regulation of JAK/STAT in cardiovascular medicine, the reader is referred to a more specialized review.Citation78 Oxidative stress is also a major contributing factor to the cardiovascular and microvascular complications of type II diabetes mellitus (TTDM).Citation79 Many of the prominent pathogenic processes of TTDM are intimately associated with both oxidative stress and JAK-dependent signal transduction, ranging from the failure of the pancreatic β islet cellCitation80-Citation85 to the development of diabetic nephropathy.Citation86-Citation88
Aging
Consistent with the free radical theory of aging, one might expect that the cumulative effect of oxidants on the JAK redox switch would impair the canonical response of cells to JAK-associated cytokines, such as growth hormone and interleukins, and that this would contribute to loss of muscle mass, immunosenescence, and other aging-associated characteristics. For example, age-related decline in growth hormone receptor signal transduction has been demonstrated in mice,Citation89,Citation90 and this would be a predictable consequence of the oxidative inhibition of JAK2. The age-associated decline of the immune system is well-documented but poorly understood,Citation91 and one should note that canonical JAK-STAT pathways are essential to the survival, expansion and function of many hematopoietic and immune cell types affected in age-associated immunosuppression.
Immunosuppressive diseases
JAKs, especially JAK3, are vital to the immune system,Citation92 where they are predominantly involved as intracellular mediators of signals from type I cytokines. Human inborn errors of the genes encoding several JAKs and STATs are often manifested in dysfunctional hematological and immunological phenotypes.Citation93 JAKs are now pharmacological targets of interest for allograft rejection prophylaxis, psoriasis, rheumatoid arthritis, and other disorders where immunosuppression is currently indicated.Citation94 The immune system has an intimate and complex dependency upon redox biology, in which ROS and RNS serve important dual roles as intra-system signal mediators involved in tissue repair and as cytotoxic defense molecules against pathogens.Citation95 While the immune system requires oxidative stress for functionality, uncontrolled and sustained oxidative stress is deleterious and leads to immunosuppression. This immunosuppression is manifested through the loss of functional T-cell subsets which show differential susceptibilities to oxidative stress, as illustrated by the ability of CD4−CD25bright Tregs to tolerate hydrogen peroxide levels lethal to CD4−CD25−/low T cells.Citation96 The impairment of normal JAK-coupled canonical signal transduction would be expected to occur under sustained and uncontrolled oxidative stress, and such molecular impairments would contribute to the cellular impairments observed in immunosuppression.
It is possible that the role of redox regulation of JAKs has been completely overlooked in AIDS, perhaps the most devastating immunosuppressive disease. The HIV-1-associated Tat enhances activation-induced cell death (AICD) in human T cellsCitation97 by increasing oxidative stressCitation98 via increased H2O2 production,Citation99 which may arise from Tat's ability to repress manganese superoxide dismutase.Citation98,Citation100 Interestingly, Tat does not typically repress superoxide dismutase, nor does it induce oxidative stress, nor does it induce AICD in chimpanzee T cells.Citation101 Recently it has been shown that IL-7 responsiveness in T cells from HIV-infected individuals, including the IL-7-stimulated phosphorylation of STAT, is impaired due to increased oxidative stress.Citation51 This observation would be consistent with the oxidative inhibition of JAK, essential to IL-7 signal transduction; this relationship between CD127 expression and IL-7 responsiveness in CD8+ T cells may be instructive given the fact that T-cell subsets have such variable abilities to tolerate oxidative stress.
It is also important to bear in mind that most laboratory studies of lymphocytes were performed on cells grown under non-physiologic, atmospheric oxygen concentrations which resulted in dramatic increases of the intracellular oxidative state as compared with lower physiological oxygen levels.Citation102,Citation103 The oxygen tension can dramatically affect IL-2-induced T-cell responses that are sometimes consistent with a simple oxidative inhibition of JAKCitation104 and sometimes quite surprising.Citation105 Given such evidence, existing assumptions about redox regulation and oxidative stress in lymphocytes, if not all mammalian cells, should be critically reassessed under more physiologically-relevant laboratory conditions.
Cancer
The importance of constitutively-active STATs, especially STAT3,Citation106,Citation107 has been demonstrated in a remarkably broad range of cancers. Due to the prevalence of the JAK2(V617F) mutation in polycythemia vera, essential thrombocythemia, and primary myelofibrosis, systematic efforts quickly led to the development of JAK-targeted drugs,Citation108,Citation109 and JAK1/JAK2 are now targets of ruxolitinib, an FDA-approved drug for myelofibrosis.Citation110 JAK-targeted drugs are now being tested in clinical trials against a variety of cancers, including various hematological malignancies caused by JAK2 chimeric proteins arising from chromosomal rearrangements.Citation111 From a medicinal chemistry perspective, the reactive cysteines of the redox switch may provide novel targets for drug designers now grappling with the problem of resistance to inhibitors which bind to the enzyme's ATP binding site.Citation11,Citation112 From a biological perspective, the relevance of redox regulation of JAKs to their roles in oncogenesis, cancer progression and the development of therapeutic resistance will have a significant influence on whether these drugs will succeed in clinical applications. Redox modulators play multiple roles in cancer, just as they play both positive and negative roles in the immune system. Oxidative stress is a major factor in cancer biology.Citation113 Many cancer chemotherapeutic agents generate ROS as a part of their mechanisms of action,Citation114 and similarly, the oxygen effect of radiation therapy depends upon the generation of ROS which increase the efficacy of DNA strand breakage.Citation115 Thus it is not a complete surprise that many cancer cells become resistant to chemotherapy and radiotherapy by upregulating antioxidant defense molecules, and resistance has been linked to increased thioredoxin/thioredoxin reductase in multiple cancers.Citation116,Citation117 JAKs have also been associated with therapeutic resistance,Citation118 and the JAK-associated STAT1 gene expression signature has emerged as a hallmark of many radioresistant tumors.Citation119,Citation120 One of the major hypotheses of cancer biology contends that chemoresistant and radioresistant cancers emerge from “stem-like” sub-populations of a tumor. Indeed, the aggressive and stem-like “triple-negative” breast cancer phenotype has an IL-6/JAK2/STAT3 gene expression profile.Citation121 If it is true that the behavior of JAKs in cancer are related to their roles in maintaining “stemness”,Citation122,Citation123 then the questions surrounding the relevance of redox regulation of JAK suddenly become quite pressing, because the importance of low oxygen tension in the stem cell niche is of undisputed importance.Citation124
Closing Comments
The preceding speculation concerning the role of redox regulation of JAKs in cancer stem cells brings this review to an appropriate end. Are there fundamental differences in the biological outcomes of oxidative stress upon JAK-associated signals, be they canonical or non-canonical, in the context of stem-like vs. fully differentiated cells and tissues? If so, then considerably more research will be required to fully understand the redox regulation of Janus kinases. Some cell lines more closely resemble stem-like cells than do others, but the vast majority of them are cultured under conditions that resemble neither stem cell niches nor the typical physiological oxygen environment. And while stem cells have a prominent, if not controversial, role in normal biology and pathobiology, most in vivo tissue biology experiments focus on bulk cellular responses of fully-differentiated cells unless experiments are intentionally designed otherwise. This review has answered only a few questions with reasonable certainty. Is there a molecular basis for the direct redox regulation of JAK2? This answer is “yes”. One hopes that this review will inspire more JAK-STAT researchers to carefully reconsider their experimental designs in the context of the redox environment and the many parameters which affect it. There is an elephant in the room, and it is raising many tantalizing questions just waiting to be answered.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
I wish to thank Drs Naila M Mamoon, John K Smith, Kiranam Chatti, Sheeyong Lee, Kanakadurga Kundrapu, Chetan N Patil, and others for their contributions to this research and their time in my laboratory. I especially appreciate the insights of Dr George W Booz and the opportunity to collaborate with him and members of his laboratory. Finally, I apologize that I was unable to cite all of the important primary literature summarized in this review, and I hope the readers will be guided to those manuscripts by reading the reviews I occasionally cited.
References
- Cleland WW. Dithiothreitol, a New Protective Reagent for Sh Groups. Biochemistry 1964; 3:480 - 2; http://dx.doi.org/10.1021/bi00892a002; PMID: 14192894
- Vallejos RH, Ravizzini RA, Andreo CS. Sulphydryl groups in photosynthetic energy conservation. IV. Inhibition of the ATPase of chloroplast coupling factor 1 by sulphydryl reagents. Biochim Biophys Acta 1977; 459:20 - 6; http://dx.doi.org/10.1016/0005-2728(77)90004-4; PMID: 137745
- Kim DS, Churchich JE. The reversible oxidation of vicinal SH groups in 4-aminobutyrate aminotransferase. Probes of conformational changes. J Biol Chem 1987; 262:14250 - 4; PMID: 3654661
- Duhé RJ, Evans GA, Erwin RA, Kirken RA, Cox GW, Farrar WL. Nitric oxide and thiol redox regulation of Janus kinase activity. Proc Natl Acad Sci U S A 1998; 95:126 - 31; http://dx.doi.org/10.1073/pnas.95.1.126; PMID: 9419340
- Seth D, Stamler JS. The SNO-proteome: causation and classifications. Curr Opin Chem Biol 2011; 15:129 - 36; http://dx.doi.org/10.1016/j.cbpa.2010.10.012; PMID: 21087893
- Pryor WA, Church DF, Govindan CK, Crank G. Oxidation of thiols by nitric oxide and nitrogen dioxide: synthetic utility and toxicological implications. J Org Chem 1982; 47:156 - 9; http://dx.doi.org/10.1021/jo00340a038
- Mamoon NM, Smith JK, Chatti K, Lee S, Kundrapu K, Duhé RJ. Multiple cysteine residues are implicated in Janus kinase 2-mediated catalysis. Biochemistry 2007; 46:14810 - 8; http://dx.doi.org/10.1021/bi701118u; PMID: 18052197
- Smith JK, Patil CN, Patlolla S, Gunter BW, Booz GW, Duhé RJ. Identification of a redox-sensitive switch within the JAK2 catalytic domain. Free Radic Biol Med 2012; 52:1101 - 10; http://dx.doi.org/10.1016/j.freeradbiomed.2011.12.025; PMID: 22281400
- Rehder DS, Borges CR. Cysteine sulfenic acid as an intermediate in disulfide bond formation and nonenzymatic protein folding. Biochemistry 2010; 49:7748 - 55; http://dx.doi.org/10.1021/bi1008694; PMID: 20712299
- Mercher T, Wernig G, Moore SA, Levine RL, Gu TL, Fröhling S, Cullen D, Polakiewicz RD, Bernard OA, Boggon TJ, et al. JAK2T875N is a novel activating mutation that results in myeloproliferative disease with features of megakaryoblastic leukemia in a murine bone marrow transplantation model. Blood 2006; 108:2770 - 9; http://dx.doi.org/10.1182/blood-2006-04-014712; PMID: 16804112
- Hornakova T, Springuel L, Devreux J, Dusa A, Constantinescu SN, Knoops L, Renauld JC. Oncogenic JAK1 and JAK2-activating mutations resistant to ATP-competitive inhibitors. Haematologica 2011; 96:845 - 53; http://dx.doi.org/10.3324/haematol.2010.036350; PMID: 21393331
- Mullighan CG, Zhang J, Harvey RC, Collins-Underwood JR, Schulman BA, Phillips LA, Tasian SK, Loh ML, Su X, Liu W, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 2009; 106:9414 - 8; http://dx.doi.org/10.1073/pnas.0811761106; PMID: 19470474
- Brandes N, Schmitt S, Jakob U. Thiol-based redox switches in eukaryotic proteins. Antioxid Redox Signal 2009; 11:997 - 1014; http://dx.doi.org/10.1089/ars.2008.2285; PMID: 18999917
- van Montfort RL, Congreve M, Tisi D, Carr R, Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature 2003; 423:773 - 7; http://dx.doi.org/10.1038/nature01681; PMID: 12802339
- Kurdi M, Booz GW. Evidence that IL-6-type cytokine signaling in cardiomyocytes is inhibited by oxidative stress: parthenolide targets JAK1 activation by generating ROS. J Cell Physiol 2007; 212:424 - 31; http://dx.doi.org/10.1002/jcp.21033; PMID: 17385713
- Di Bona D, Cippitelli M, Fionda C, Cammà C, Licata A, Santoni A, Craxì A. Oxidative stress inhibits IFN-alpha-induced antiviral gene expression by blocking the JAK-STAT pathway. J Hepatol 2006; 45:271 - 9; http://dx.doi.org/10.1016/j.jhep.2006.01.037; PMID: 16595158
- Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res 2010; 38:Web Server issue W695-9; http://dx.doi.org/10.1093/nar/gkq313; PMID: 20439314
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 2011; 7:539; http://dx.doi.org/10.1038/msb.2011.75; PMID: 21988835
- Corcoran A, Cotter TG. Redox regulation of protein kinases. FEBS J 2013; 280:1944 - 65; http://dx.doi.org/10.1111/febs.12224; PMID: 23461806
- Giannoni E, Buricchi F, Raugei G, Ramponi G, Chiarugi P. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol Cell Biol 2005; 25:6391 - 403; http://dx.doi.org/10.1128/MCB.25.15.6391-6403.2005; PMID: 16024778
- Kemble DJ, Sun G. Direct and specific inactivation of protein tyrosine kinases in the Src and FGFR families by reversible cysteine oxidation. Proc Natl Acad Sci U S A 2009; 106:5070 - 5; http://dx.doi.org/10.1073/pnas.0806117106; PMID: 19273857
- Lai KS, Jin Y, Graham DK, Witthuhn BA, Ihle JN, Liu ET. A kinase-deficient splice variant of the human JAK3 is expressed in hematopoietic and epithelial cancer cells. J Biol Chem 1995; 270:25028 - 36; http://dx.doi.org/10.1074/jbc.270.42.25028; PMID: 7559633
- Grattan DR, Kokay IC. Prolactin: a pleiotropic neuroendocrine hormone. J Neuroendocrinol 2008; 20:752 - 63; http://dx.doi.org/10.1111/j.1365-2826.2008.01736.x; PMID: 18601698
- Nairz M, Sonnweber T, Schroll A, Theurl I, Weiss G. The pleiotropic effects of erythropoietin in infection and inflammation. Microbes Infect 2012; 14:238 - 46; http://dx.doi.org/10.1016/j.micinf.2011.10.005; PMID: 22094132
- Feng J, Witthuhn BA, Matsuda T, Kohlhuber F, Kerr IM, Ihle JN. Activation of Jak2 catalytic activity requires phosphorylation of Y1007 in the kinase activation loop. Mol Cell Biol 1997; 17:2497 - 501; PMID: 9111318
- Chatti K, Farrar WL, Duhé RJ. Tyrosine phosphorylation of the Janus kinase 2 activation loop is essential for a high-activity catalytic state but dispensable for a basal catalytic state. Biochemistry 2004; 43:4272 - 83; http://dx.doi.org/10.1021/bi036109b; PMID: 15065871
- Marrero MB, Schieffer B, Paxton WG, Heerdt L, Berk BC, Delafontaine P, Bernstein KE. Direct stimulation of Jak/STAT pathway by the angiotensin II AT1 receptor. Nature 1995; 375:247 - 50; http://dx.doi.org/10.1038/375247a0; PMID: 7746328
- Lukashova V, Chen Z, Duhé RJ, Rola-Pleszczynski M, Stanková J. Janus kinase 2 activation by the platelet-activating factor receptor (PAFR): roles of Tyk2 and PAFR C terminus. J Immunol 2003; 171:3794 - 800; PMID: 14500680
- Kilpinen S, Ojala K, Kallioniemi O. Analysis of kinase gene expression patterns across 5681 human tissue samples reveals functional genomic taxonomy of the kinome. PLoS One 2010; 5:e15068; http://dx.doi.org/10.1371/journal.pone.0015068; PMID: 21151926
- Zouein FA, Duhé RJ, Booz GW. JAKs go nuclear: emerging role of nuclear JAK1 and JAK2 in gene expression and cell growth. Growth Factors 2011; 29:245 - 52; http://dx.doi.org/10.3109/08977194.2011.614949; PMID: 21892841
- Sanz A, Ungureanu D, Pekkala T, Ruijtenbeek R, Touw IP, Hilhorst R, Silvennoinen O. Analysis of Jak2 catalytic function by peptide microarrays: the role of the JH2 domain and V617F mutation. PLoS One 2011; 6:e18522; http://dx.doi.org/10.1371/journal.pone.0018522; PMID: 21533163
- Jäkel H, Weinl C, Hengst L. Phosphorylation of p27Kip1 by JAK2 directly links cytokine receptor signaling to cell cycle control. Oncogene 2011; 30:3502 - 12; http://dx.doi.org/10.1038/onc.2011.68; PMID: 21423214
- Duhé RJ, Clark EA, Farrar WL. Characterization of the in vitro kinase activity of a partially purified soluble GST/JAK2 fusion protein. Mol Cell Biochem 2002; 236:23 - 35; http://dx.doi.org/10.1023/A:1016186907376; PMID: 12190118
- Dawson MA, Bannister AJ, Göttgens B, Foster SD, Bartke T, Green AR, Kouzarides T. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature 2009; 461:819 - 22; http://dx.doi.org/10.1038/nature08448; PMID: 19783980
- Andraos R, Qian Z, Bonenfant D, Rubert J, Vangrevelinghe E, Scheufler C, Marque F, Régnier CH, De Pover A, Ryckelynck H, et al. Modulation of activation-loop phosphorylation by JAK inhibitors is binding mode dependent. Cancer Discov 2012; 2:512 - 23; http://dx.doi.org/10.1158/2159-8290.CD-11-0324; PMID: 22684457
- Koppikar P, Bhagwat N, Kilpivaara O, Manshouri T, Adli M, Hricik T, Liu F, Saunders LM, Mullally A, Abdel-Wahab O, et al. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature 2012; 489:155 - 9; http://dx.doi.org/10.1038/nature11303; PMID: 22820254
- Ungureanu D, Wu J, Pekkala T, Niranjan Y, Young C, Jensen ON, Xu CF, Neubert TA, Skoda RC, Hubbard SR, et al. The pseudokinase domain of JAK2 is a dual-specificity protein kinase that negatively regulates cytokine signaling. Nat Struct Mol Biol 2011; 18:971 - 6; http://dx.doi.org/10.1038/nsmb.2099; PMID: 21841788
- Bandaranayake RM, Ungureanu D, Shan Y, Shaw DE, Silvennoinen O, Hubbard SR. Crystal structures of the JAK2 pseudokinase domain and the pathogenic mutant V617F. Nat Struct Mol Biol 2012; 19:754 - 9; http://dx.doi.org/10.1038/nsmb.2348; PMID: 22820988
- Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat Chem Biol 2011; 8:57 - 64; http://dx.doi.org/10.1038/nchembio.736; PMID: 22158416
- Abe J, Berk BC. Fyn and JAK2 mediate Ras activation by reactive oxygen species. J Biol Chem 1999; 274:21003 - 10; http://dx.doi.org/10.1074/jbc.274.30.21003; PMID: 10409649
- Marino SM, Gladyshev VN. Redox biology: computational approaches to the investigation of functional cysteine residues. Antioxid Redox Signal 2011; 15:135 - 46; http://dx.doi.org/10.1089/ars.2010.3561; PMID: 20812876
- Guan KL, Dixon JE. Evidence for protein-tyrosine-phosphatase catalysis proceeding via a cysteine-phosphate intermediate. J Biol Chem 1991; 266:17026 - 30; PMID: 1654322
- Streuli M, Krueger NX, Thai T, Tang M, Saito H. Distinct functional roles of the two intracellular phosphatase like domains of the receptor-linked protein tyrosine phosphatases LCA and LAR. EMBO J 1990; 9:2399 - 407; PMID: 1695146
- Weibrecht I, Böhmer SA, Dagnell M, Kappert K, Ostman A, Böhmer FD. Oxidation sensitivity of the catalytic cysteine of the protein-tyrosine phosphatases SHP-1 and SHP-2. Free Radic Biol Med 2007; 43:100 - 10; http://dx.doi.org/10.1016/j.freeradbiomed.2007.03.021; PMID: 17561098
- Kaur N, Lu B, Monroe RK, Ward SM, Halvorsen SW. Inducers of oxidative stress block ciliary neurotrophic factor activation of Jak/STAT signaling in neurons. J Neurochem 2005; 92:1521 - 30; http://dx.doi.org/10.1111/j.1471-4159.2004.02990.x; PMID: 15748169
- Monroe RK, Halvorsen SW. Cadmium blocks receptor-mediated Jak/STAT signaling in neurons by oxidative stress. Free Radic Biol Med 2006; 41:493 - 502; http://dx.doi.org/10.1016/j.freeradbiomed.2006.04.023; PMID: 16843830
- Monroe RK, Halvorsen SW. Mercury abolishes neurotrophic factor-stimulated Jak-STAT signaling in nerve cells by oxidative stress. Toxicol Sci 2006; 94:129 - 38; http://dx.doi.org/10.1093/toxsci/kfl073; PMID: 16896058
- Jang EH, Park CS, Lee SK, Pie JE, Kang JH. Excessive nitric oxide attenuates leptin-mediated signal transducer and activator of transcription 3 activation. Life Sci 2007; 80:609 - 17; http://dx.doi.org/10.1016/j.lfs.2006.10.007; PMID: 17097687
- Bingisser RM, Tilbrook PA, Holt PG, Kees UR. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J Immunol 1998; 160:5729 - 34; PMID: 9637481
- Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, Zanovello P, Segal DM. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol 2002; 168:689 - 95; PMID: 11777962
- Kalinowska M, Bazdar DA, Lederman MM, Funderburg N, Sieg SF. Decreased IL-7 responsiveness is related to oxidative stress in HIV disease. PLoS One 2013; 8:e58764; http://dx.doi.org/10.1371/journal.pone.0058764; PMID: 23505558
- Kurdi M, Sivakumaran V, Duhé RJ, Aon MA, Paolocci N, Booz GW. Depletion of cellular glutathione modulates LIF-induced JAK1-STAT3 signaling in cardiac myocytes. Int J Biochem Cell Biol 2012; 44:2106 - 15; http://dx.doi.org/10.1016/j.biocel.2012.08.016; PMID: 22939972
- Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. Am J Physiol 1998; 275:C1640 - 52; PMID: 9843726
- Madamanchi NR, Li S, Patterson C, Runge MS. Reactive oxygen species regulate heat-shock protein 70 via the JAK/STAT pathway. Arterioscler Thromb Vasc Biol 2001; 21:321 - 6; http://dx.doi.org/10.1161/01.ATV.21.3.321; PMID: 11231909
- Tawfik A, Jin L, Banes-Berceli AK, Caldwell RB, Ogbi S, Shirley A, Barber D, Catravas JD, Stern DM, Fulton D, et al. Hyperglycemia and reactive oxygen species mediate apoptosis in aortic endothelial cells through Janus kinase 2. Vascul Pharmacol 2005; 43:320 - 6; http://dx.doi.org/10.1016/j.vph.2005.08.018; PMID: 16257269
- Schieffer B, Luchtefeld M, Braun S, Hilfiker A, Hilfiker-Kleiner D, Drexler H. Role of NAD(P)H oxidase in angiotensin II-induced JAK/STAT signaling and cytokine induction. Circ Res 2000; 87:1195 - 201; http://dx.doi.org/10.1161/01.RES.87.12.1195; PMID: 11110778
- Kirabo A, Kearns PN, Jarajapu YP, Sasser JM, Oh SP, Grant MB, Kasahara H, Cardounel AJ, Baylis C, Wagner KU, et al. Vascular smooth muscle Jak2 mediates angiotensin II-induced hypertension via increased levels of reactive oxygen species. Cardiovasc Res 2011; 91:171 - 9; http://dx.doi.org/10.1093/cvr/cvr059; PMID: 21354995
- Modesti A, Bertolozzi I, Gamberi T, Marchetta M, Lumachi C, Coppo M, Moroni F, Toscano T, Lucchese G, Gensini GF, et al. Hyperglycemia activates JAK2 signaling pathway in human failing myocytes via angiotensin II-mediated oxidative stress. Diabetes 2005; 54:394 - 401; http://dx.doi.org/10.2337/diabetes.54.2.394; PMID: 15677497
- Godeny MD, Sayyah J, VonDerLinden D, Johns M, Ostrov DA, Caldwell-Busby J, Sayeski PP. The N-terminal SH2 domain of the tyrosine phosphatase, SHP-2, is essential for Jak2-dependent signaling via the angiotensin II type AT1 receptor. Cell Signal 2007; 19:600 - 9; http://dx.doi.org/10.1016/j.cellsig.2006.08.010; PMID: 17027227
- Duan W, Yang Y, Yi W, Yan J, Liang Z, Wang N, Li Y, Chen W, Yu S, Jin Z, et al. New role of JAK2/STAT3 signaling in endothelial cell oxidative stress injury and protective effect of melatonin. PLoS One 2013; 8:e57941; http://dx.doi.org/10.1371/journal.pone.0057941; PMID: 23483946
- Sandberg EM, Sayeski PP. Jak2 tyrosine kinase mediates oxidative stress-induced apoptosis in vascular smooth muscle cells. J Biol Chem 2004; 279:34547 - 52; http://dx.doi.org/10.1074/jbc.M405045200; PMID: 15159394
- Mascareno E, Beckles DL, Siddiqui MA. Janus kinase-2 signaling mediates apoptosis in rat cardiomyocytes. Vascul Pharmacol 2005; 43:327 - 35; http://dx.doi.org/10.1016/j.vph.2005.08.023; PMID: 16269269
- Lee JK, Edderkaoui M, Truong P, Ohno I, Jang KT, Berti A, Pandol SJ, Gukovskaya AS. NADPH oxidase promotes pancreatic cancer cell survival via inhibiting JAK2 dephosphorylation by tyrosine phosphatases. Gastroenterology 2007; 133:1637 - 48; http://dx.doi.org/10.1053/j.gastro.2007.08.022; PMID: 17983808
- Carballo M, Conde M, El Bekay R, Martín-Nieto J, Camacho MJ, Monteseirín J, Conde J, Bedoya FJ, Sobrino F. Oxidative stress triggers STAT3 tyrosine phosphorylation and nuclear translocation in human lymphocytes. J Biol Chem 1999; 274:17580 - 6; http://dx.doi.org/10.1074/jbc.274.25.17580; PMID: 10364193
- Crump KE, Juneau DG, Poole LB, Haas KM, Grayson JM. The reversible formation of cysteine sulfenic acid promotes B-cell activation and proliferation. Eur J Immunol 2012; 42:2152 - 64; http://dx.doi.org/10.1002/eji.201142289; PMID: 22674013
- Sayeski PP, Ali MS, Safavi A, Lyles M, Kim SO, Frank SJ, Bernstein KE. A catalytically active Jak2 is required for the angiotensin II-dependent activation of Fyn. J Biol Chem 1999; 274:33131 - 42; http://dx.doi.org/10.1074/jbc.274.46.33131; PMID: 10551884
- Girasol A, Albuquerque GG, Mansour E, Araújo EP, Degasperi G, Denis RG, Carvalheira JB, Saad MJ, Velloso LA. Fyn mediates leptin actions in the thymus of rodents. PLoS One 2009; 4:e7707; http://dx.doi.org/10.1371/journal.pone.0007707; PMID: 19888448
- Frank GD, Eguchi S. Activation of tyrosine kinases by reactive oxygen species in vascular smooth muscle cells: significance and involvement of EGF receptor transactivation by angiotensin II. Antioxid Redox Signal 2003; 5:771 - 80; http://dx.doi.org/10.1089/152308603770380070; PMID: 14588150
- Osherov N, Gazit A, Gilon C, Levitzki A. Selective inhibition of the epidermal growth factor and HER2/neu receptors by tyrphostins. J Biol Chem 1993; 268:11134 - 42; PMID: 8098709
- Gorina R, Sanfeliu C, Galitó A, Messeguer A, Planas AM. Exposure of glia to pro-oxidant agents revealed selective Stat1 activation by H2O2 and Jak2-independent antioxidant features of the Jak2 inhibitor AG490. Glia 2007; 55:1313 - 24; http://dx.doi.org/10.1002/glia.20542; PMID: 17607690
- Marty C, Lacout C, Droin N, Le Couédic JP, Ribrag V, Solary E, Vainchenker W, Villeval JL, Plo I. A role for reactive oxygen species in JAK2V617F myeloproliferative neoplasm progression. Leukemia 2013; In press http://dx.doi.org/10.1038/leu.2013.102; PMID: 23558526
- Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev 1998; 78:547 - 81; PMID: 9562038
- Sohal RS, Orr WC. The redox stress hypothesis of aging. Free Radic Biol Med 2012; 52:539 - 55; http://dx.doi.org/10.1016/j.freeradbiomed.2011.10.445; PMID: 22080087
- Lee R, Margaritis M, Channon KM, Antoniades C. Evaluating oxidative stress in human cardiovascular disease: methodological aspects and considerations. Curr Med Chem 2012; 19:2504 - 20; http://dx.doi.org/10.2174/092986712800493057; PMID: 22489713
- Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury: Part II: animal and human studies. Circulation 2003; 108:2034 - 40; http://dx.doi.org/10.1161/01.CIR.0000093661.90582.c4; PMID: 14581381
- Myung SK, Ju W, Cho B, Oh SW, Park SM, Koo BK, Park BJ, Korean Meta-Analysis Study Group. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: systematic review and meta-analysis of randomised controlled trials. BMJ 2013; 346:f10; http://dx.doi.org/10.1136/bmj.f10; PMID: 23335472
- Sugamura K, Keaney JF Jr.. Reactive oxygen species in cardiovascular disease. Free Radic Biol Med 2011; 51:978 - 92; http://dx.doi.org/10.1016/j.freeradbiomed.2011.05.004; PMID: 21627987
- Kurdi M, Booz GW. JAK redux: a second look at the regulation and role of JAKs in the heart. Am J Physiol Heart Circ Physiol 2009; 297:H1545 - 56; http://dx.doi.org/10.1152/ajpheart.00032.2009; PMID: 19717737
- Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res 2010; 107:1058 - 70; http://dx.doi.org/10.1161/CIRCRESAHA.110.223545; PMID: 21030723
- Fujinaka Y, Takane K, Yamashita H, Vasavada RC. Lactogens promote beta cell survival through JAK2/STAT5 activation and Bcl-XL upregulation. J Biol Chem 2007; 282:30707 - 17; http://dx.doi.org/10.1074/jbc.M702607200; PMID: 17728251
- Robertson RP, Harmon J, Tran PO, Poitout V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 2004; 53:Suppl 1 S119 - 24; http://dx.doi.org/10.2337/diabetes.53.2007.S119; PMID: 14749276
- Ma ZA, Zhao Z, Turk J. Mitochondrial dysfunction and β-cell failure in type 2 diabetes mellitus. Exp Diabetes Res 2012; 2012:703538; http://dx.doi.org/10.1155/2012/703538; PMID: 22110477
- Choi D, Schroer SA, Lu SY, Wang L, Wu X, Liu Y, Zhang Y, Gaisano HY, Wagner KU, Wu H, et al. Erythropoietin protects against diabetes through direct effects on pancreatic beta cells. J Exp Med 2010; 207:2831 - 42; http://dx.doi.org/10.1084/jem.20100665; PMID: 21149549
- Liu JL, Coschigano KT, Robertson K, Lipsett M, Guo Y, Kopchick JJ, Kumar U, Liu YL. Disruption of growth hormone receptor gene causes diminished pancreatic islet size and increased insulin sensitivity in mice. Am J Physiol Endocrinol Metab 2004; 287:E405 - 13; http://dx.doi.org/10.1152/ajpendo.00423.2003; PMID: 15138153
- Freemark M, Avril I, Fleenor D, Driscoll P, Petro A, Opara E, Kendall W, Oden J, Bridges S, Binart N, et al. Targeted deletion of the PRL receptor: effects on islet development, insulin production, and glucose tolerance. Endocrinology 2002; 143:1378 - 85; http://dx.doi.org/10.1210/en.143.4.1378; PMID: 11897695
- Matsui F, Meldrum KK. The role of the Janus kinase family/signal transducer and activator of transcription signaling pathway in fibrotic renal disease. J Surg Res 2012; 178:339 - 45; http://dx.doi.org/10.1016/j.jss.2012.06.050; PMID: 22883438
- Marrero MB, Banes-Berceli AK, Stern DM, Eaton DC. Role of the JAK/STAT signaling pathway in diabetic nephropathy. Am J Physiol Renal Physiol 2006; 290:F762 - 8; http://dx.doi.org/10.1152/ajprenal.00181.2005; PMID: 16527921
- Berthier CC, Zhang H, Schin M, Henger A, Nelson RG, Yee B, Boucherot A, Neusser MA, Cohen CD, Carter-Su C, et al. Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes 2009; 58:469 - 77; http://dx.doi.org/10.2337/db08-1328; PMID: 19017763
- Xu X, Sonntag WE. Growth hormone-induced nuclear translocation of Stat-3 decreases with age: modulation by caloric restriction. Am J Physiol 1996; 271:E903 - 9; PMID: 8944679
- Xu X, Sonntag WE. Moderate caloric restriction prevents the age-related decline in growth hormone receptor signal transduction. J Gerontol A Biol Sci Med Sci 1996; 51:B167 - 74; http://dx.doi.org/10.1093/gerona/51A.2.B167; PMID: 8612101
- Gruver AL, Hudson LL, Sempowski GD. Immunosenescence of ageing. J Pathol 2007; 211:144 - 56; http://dx.doi.org/10.1002/path.2104; PMID: 17200946
- O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med 2013; 368:161 - 70; http://dx.doi.org/10.1056/NEJMra1202117; PMID: 23301733
- Casanova JL, Holland SM, Notarangelo LD. Inborn errors of human JAKs and STATs. Immunity 2012; 36:515 - 28; http://dx.doi.org/10.1016/j.immuni.2012.03.016; PMID: 22520845
- O’Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012; 36:542 - 50; http://dx.doi.org/10.1016/j.immuni.2012.03.014; PMID: 22520847
- Wink DA, Hines HB, Cheng RY, Switzer CH, Flores-Santana W, Vitek MP, Ridnour LA, Colton CA. Nitric oxide and redox mechanisms in the immune response. J Leukoc Biol 2011; 89:873 - 91; http://dx.doi.org/10.1189/jlb.1010550; PMID: 21233414
- Mougiakakos D, Johansson CC, Kiessling R. Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood 2009; 113:3542 - 5; http://dx.doi.org/10.1182/blood-2008-09-181040; PMID: 19050306
- Westendorp MO, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, Debatin KM, Krammer PH. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature 1995; 375:497 - 500; http://dx.doi.org/10.1038/375497a0; PMID: 7539892
- Westendorp MO, Shatrov VA, Schulze-Osthoff K, Frank R, Kraft M, Los M, Krammer PH, Dröge W, Lehmann V. HIV-1 Tat potentiates TNF-induced NF-kappa B activation and cytotoxicity by altering the cellular redox state. EMBO J 1995; 14:546 - 54; PMID: 7859743
- Gülow K, Kaminski M, Darvas K, Süss D, Li-Weber M, Krammer PH. HIV-1 trans-activator of transcription substitutes for oxidative signaling in activation-induced T cell death. J Immunol 2005; 174:5249 - 60; PMID: 15843521
- Flores SC, Marecki JC, Harper KP, Bose SK, Nelson SK, McCord JM. Tat protein of human immunodeficiency virus type 1 represses expression of manganese superoxide dismutase in HeLa cells. Proc Natl Acad Sci U S A 1993; 90:7632 - 6; http://dx.doi.org/10.1073/pnas.90.16.7632; PMID: 8395050
- Ehret A, Westendorp MO, Herr I, Debatin KM, Heeney JL, Frank R, Krammer PH. Resistance of chimpanzee T cells to human immunodeficiency virus type 1 Tat-enhanced oxidative stress and apoptosis. J Virol 1996; 70:6502 - 7; PMID: 8709290
- Atkuri KR, Herzenberg LA, Herzenberg LA. Culturing at atmospheric oxygen levels impacts lymphocyte function. Proc Natl Acad Sci U S A 2005; 102:3756 - 9; http://dx.doi.org/10.1073/pnas.0409910102; PMID: 15738407
- Atkuri KR, Herzenberg LA, Niemi AK, Cowan T, Herzenberg LA. Importance of culturing primary lymphocytes at physiological oxygen levels. Proc Natl Acad Sci U S A 2007; 104:4547 - 52; http://dx.doi.org/10.1073/pnas.0611732104; PMID: 17360561
- Carswell KS, Weiss JW, Papoutsakis ET. Low oxygen tension enhances the stimulation and proliferation of human T lymphocytes in the presence of IL-2. Cytotherapy 2000; 2:25 - 37; http://dx.doi.org/10.1080/146532400539026; PMID: 12042052
- Sahaf B, Atkuri K, Heydari K, Malipatlolla M, Rappaport J, Regulier E, Herzenberg LA, Herzenberg LA. Culturing of human peripheral blood cells reveals unsuspected lymphocyte responses relevant to HIV disease. Proc Natl Acad Sci U S A 2008; 105:5111 - 6; http://dx.doi.org/10.1073/pnas.0712363105; PMID: 18364393
- Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene 2000; 19:2474 - 88; http://dx.doi.org/10.1038/sj.onc.1203527; PMID: 10851046
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 2009; 9:798 - 809; http://dx.doi.org/10.1038/nrc2734; PMID: 19851315
- Sayyah J, Sayeski PP. Jak2 inhibitors: rationale and role as therapeutic agents in hematologic malignancies. Curr Oncol Rep 2009; 11:117 - 24; http://dx.doi.org/10.1007/s11912-009-0018-2; PMID: 19216843
- Baskin R, Majumder A, Sayeski PP. The recent medicinal chemistry development of Jak2 tyrosine kinase small molecule inhibitors. Curr Med Chem 2010; 17:4551 - 8; http://dx.doi.org/10.2174/092986710794182953; PMID: 21062251
- Deisseroth A, Kaminskas E, Grillo J, Chen W, Saber H, Lu HL, Rothmann MD, Brar S, Wang J, Garnett C, et al. U.S. Food and Drug Administration approval: ruxolitinib for the treatment of patients with intermediate and high-risk myelofibrosis. Clin Cancer Res 2012; 18:3212 - 7; http://dx.doi.org/10.1158/1078-0432.CCR-12-0653; PMID: 22544377
- Chase A, Bryant C, Score J, Haferlach C, Grossmann V, Schwaab J, Hofmann WK, Reiter A, Cross NC. Ruxolitinib as potential targeted therapy for patients with JAK2 rearrangements. Haematologica 2013; 98:404 - 8; http://dx.doi.org/10.3324/haematol.2012.067959; PMID: 22875628
- Marit MR, Chohan M, Matthew N, Huang K, Kuntz DA, Rose DR, Barber DL. Random mutagenesis reveals residues of JAK2 critical in evading inhibition by a tyrosine kinase inhibitor. PLoS One 2012; 7:e43437; http://dx.doi.org/10.1371/journal.pone.0043437; PMID: 22916261
- Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked?. Free Radic Biol Med 2010; 49:1603 - 16; http://dx.doi.org/10.1016/j.freeradbiomed.2010.09.006; PMID: 20840865
- Chandra J. Oxidative stress by targeted agents promotes cytotoxicity in hematologic malignancies. Antioxid Redox Signal 2009; 11:1123 - 37; http://dx.doi.org/10.1089/ars.2008.2302; PMID: 19018667
- Wardman P. The importance of radiation chemistry to radiation and free radical biology (The 2008 Silvanus Thompson Memorial Lecture). Br J Radiol 2009; 82:89 - 104; http://dx.doi.org/10.1259/bjr/60186130; PMID: 19168690
- Yokomizo A, Ono M, Nanri H, Makino Y, Ohga T, Wada M, Okamoto T, Yodoi J, Kuwano M, Kohno K. Cellular levels of thioredoxin associated with drug sensitivity to cisplatin, mitomycin C, doxorubicin, and etoposide. Cancer Res 1995; 55:4293 - 6; PMID: 7671238
- Arnold NB, Ketterer K, Kleeff J, Friess H, Büchler MW, Korc M. Thioredoxin is downstream of Smad7 in a pathway that promotes growth and suppresses cisplatin-induced apoptosis in pancreatic cancer. Cancer Res 2004; 64:3599 - 606; http://dx.doi.org/10.1158/0008-5472.CAN-03-2999; PMID: 15150118
- Carmo CR, Lyons-Lewis J, Seckl MJ, Costa-Pereira AP. A novel requirement for Janus kinases as mediators of drug resistance induced by fibroblast growth factor-2 in human cancer cells. PLoS One 2011; 6:e19861; http://dx.doi.org/10.1371/journal.pone.0019861; PMID: 21625473
- Khodarev NN, Beckett M, Labay E, Darga T, Roizman B, Weichselbaum RR. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc Natl Acad Sci U S A 2004; 101:1714 - 9; http://dx.doi.org/10.1073/pnas.0308102100; PMID: 14755057
- Pitroda SP, Wakim BT, Sood RF, Beveridge MG, Beckett MA, MacDermed DM, Weichselbaum RR, Khodarev NN. STAT1-dependent expression of energy metabolic pathways links tumour growth and radioresistance to the Warburg effect. BMC Med 2009; 7:68; http://dx.doi.org/10.1186/1741-7015-7-68; PMID: 19891767
- Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ, Choudhury SA, Maruyama R, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44⁺CD24⁻ stem cell-like breast cancer cells in human tumors. J Clin Invest 2011; 121:2723 - 35; http://dx.doi.org/10.1172/JCI44745; PMID: 21633165
- Hernandez-Vargas H, Ouzounova M, Le Calvez-Kelm F, Lambert MP, McKay-Chopin S, Tavtigian SV, Puisieux A, Matar C, Herceg Z. Methylome analysis reveals Jak-STAT pathway deregulation in putative breast cancer stem cells. Epigenetics 2011; 6:428 - 39; http://dx.doi.org/10.4161/epi.6.4.14515; PMID: 21266853
- Birnie R, Bryce SD, Roome C, Dussupt V, Droop A, Lang SH, Berry PA, Hyde CF, Lewis JL, Stower MJ, et al. Gene expression profiling of human prostate cancer stem cells reveals a pro-inflammatory phenotype and the importance of extracellular matrix interactions. Genome Biol 2008; 9:R83; http://dx.doi.org/10.1186/gb-2008-9-5-r83; PMID: 18492237
- Mohyeldin A, Garzón-Muvdi T, Quiñones-Hinojosa A. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell 2010; 7:150 - 61; http://dx.doi.org/10.1016/j.stem.2010.07.007; PMID: 20682444
- Lucet IS, Fantino E, Styles M, Bamert R, Patel O, Broughton SE, Walter M, Burns CJ, Treutlein H, Wilks AF, et al. The structural basis of Janus kinase 2 inhibition by a potent and specific pan-Janus kinase inhibitor. Blood 2006; 107:176 - 83; http://dx.doi.org/10.1182/blood-2005-06-2413; PMID: 16174768
- Cacalano NA, Sanden D, Johnston JA. Tyrosine-phosphorylated SOCS-3 inhibits STAT activation but binds to p120 RasGAP and activates Ras. Nat Cell Biol 2001; 3:460 - 5; http://dx.doi.org/10.1038/35074525; PMID: 11331873