Abstract
Bispecific antibodies (BsAbs) represent an emerging class of biologics that achieve dual targeting with a single agent. Recombinant DNA technologies have facilitated a variety of creative bispecific designs with many promising therapeutic applications; however, practical methods for producing high quality BsAbs that have good product stability, long serum half-life, straightforward purification, and scalable production have largely been limiting. Here we describe a protein-engineering approach for producing stable, scalable tetravalent IgG-like BsAbs. The stability-engineered IgG-like BsAb was envisioned to target and crosslink two TNF family member receptors, TRAIL-R2 (TNF-Related Apoptosis Inducing Ligand Receptor-2) and LTβR (Lymphotoxin-beta Receptor), expressed on the surface of epithelial tumor cells with the goal of triggering an enhanced anti-tumor effect. Our IgG-like BsAbs consists of a stability-engineered anti- LTβR single chain Fv (scFv) genetically fused to either the N- or C-terminus of the heavy chain of a full-length anti-TRAIL-R2 IgG1 monoclonal antibody. Both N- or C-terminal BsAbs were active in inhibiting tumor cell growth in vitro, and with some cell lines demonstrated enhanced activity relative to the combination of parental Abs. Pharmacokinetic studies in mice revealed long serum half-lives for the BsAbs. In murine tumor xenograft models, therapeutic treatment with the BsAbs resulted in reduction in tumor volume either comparable to or greater than the combination of parental antibodies, indicating that simultaneously targeting and cross-linking receptor pairs is an effective strategy for treating tumor cells. These studies support that stability-engineering is an enabling step for producing scalable IgG-like BsAbs with properties desirable for biopharmaceutical development.
Introduction
Recombinant bispecific antibodies (BsAbs) are designed to perform as single agent, dual antigen-binding molecules. BsAbs can bind to two different epitopes on the same antigen or to two entirely distinct antigens. For treating human disease, such as cancer, the biological consequence of engaging more than one epitope or target, for example cell membrane-associated receptors, with a BsAb may result in increased potency or efficacy due to: (1) enhanced avidity or specificity towards target or (2) simultaneous targeting of two distinct disease-related pathways resulting in an additive, synergistic or novel mechanism-of-action. Bispecific antibodies can also be employed for recruiting or redirecting a natural killer (NK) cell, T cell or monocyte/neutrophil cellular response to tumors by bringing these two cell types close together.Citation1 BsAbs have also been used in pretargeting strategies where first the BsAb is administered to a patient such that it localizes to its target, and unbound BsAb is then allowed to clear from the bloodstream. A second agent, such as a derivatized cytotoxic or radionuclide, is subsequently administered whereupon it is captured by the immobilized BsAb concentrating the cell-killing agent to the target tissue.Citation2
Over the last decade, a spectrum of innovative BsAb formats have been constructed, with single-chain Fv (scFv) and diabody fragments being extensively used as fundamental building blocks.Citation1,Citation3,Citation4 A subgroup of these, designed as full-length IgG-like BsAbs, have the added advantage of preserving an intact Fc domain, thereby readily allowing purification, providing active cellular immune effector functions and imparting greatly extended serum pharmacokinetic properties.Citation5 While many of these IgG-like BsAbs have shown promising activity in in vitro assays, low production yields, inadequate product quality and loss of activity or short serum half- life have largely remained significant impediments to conducting rigorous in vivo studies and advancing these compounds towards clinical development.
It is well documented that insufficient thermodynamic stability of Fv and scFv fragments can often limit scFv production, quality and yield.Citation6,Citation7 Many methods, including both rational design and molecular evolution techniques, have been described for improving the stability of scFvs.Citation6,Citation8–Citation10 Given the interest in using scFv fragments as “building blocks” for constructing IgG-like BsAbs, we surmised that a scFv engineered to have sufficient stability should, upon incorporation into an IgG-like BsAb, result in a BsAb that also has improved and acceptable stability properties.
Our hypothesis that stabilization of scFv domains would enable production of therapeutically useful BsAb molecules was tested by evaluating a series of tetravalent IgG-like BsAbs constructed using scFvs selected with varying levels of resistance to thermal challenge. Here we report on the biochemical, biophysical and biological properties of BsAbs constructed from stabilized scFvs.
The stability-engineered BsAbs were built from two agonist antibodies targeting TNF family member receptors, TRAIL-R2 (TNF-Related Apoptosis Inducing Ligand Receptor-2) and LTβR (Lymphotoxin-β Receptor) which are co-expressed on the surface of a variety of human tumor epithelial cell lines. The death domain containing TRAIL-R2 receptor represents an attractive new target in cancer, with early clinical trials of soluble TRAIL or agonist antibodies to TRAIL-R2 demonstrating acceptable safety and tolerability and promising signs of anti-tumor activity.Citation11 LTβR, one of several non-death domain containing TNF family receptors, has shown preclinical efficacy in tumor xenograft models when targeted with an agonist antibody.Citation12 We set out to construct a BsAb targeting TRAIL-R2 and LTβR to explore the possibility that a BsAb might trigger an enhanced, synergistic, or broader anti-tumor response than that achieved by treating with a mixture of the two antibodies.
The full-length anti-TRAIL-R2 antibody 14A2 was used as the IgG component of the BsAb, and the anti-LTβR antibody BHA10 served as a source for the stability-engineered scFv moiety. Once stabilized, the BHA10 scFv could be appended to either the amino- or carboxyl terminus of the 14A2 antibody heavy chain enabling scalable production of high quality tetravalent IgG-like BsAbs in a CHO cell expression system. The stability-engineered BsAbs demonstrated excellent serum half-lives in mice and showed anti-tumor activity in mouse tumor xenograft models. These results indicate that incorporating stability-engineered scFvs into IgG-like BsAb designs may greatly facilitate scalable evaluation of this novel class of biotherapeutics.
Results
Production of BsAb containing wild-type scFv.
The IgG-like BsAb design consisted of a scFv derived from the anti-LTβR humanized mAb, BHA10, genetically connected through a flexible linker to either the amino (N-BsAb) or carboxyl terminus (C-BsAb) of the full-length anti-TRAIL-R2 chimeric, IgG 14A2 (). The BHA10 scFv was built in the VH □ VL orientation and joined in-frame via a (Gly4Ser)n linker to either the amino-terminal VH domain or the carboxyl end of the 14A2 IgG in the bicistronic mammalian expression vector pN5KG1 as shown in . Plasmids were used to stably transfect CHO cells for protein production. Preliminary experiments with the C-BsAb containing wild-type BHA10 scFv revealed that a transfected pool of CHO cells secreted a moderate level of C-BsAb into the culture supernatant with an accumulated titer of approximately 40 mg per liter. However, nearly 40% of the Protein A purified BsAb was present as high MW aggregates (), and even isolated monomeric BsAb containing wild-type scFv was still prone to forming aggregates (Bailly V, unpublished observation).
In order to determine whether the intrinsic stability of the scFv moiety might be a contributing factor to the poor quality of the wild-type C-BsAb, we compared the relative thermal stability of purified wild-type BHA10 scFv produced in E. coli to BHA10 FAb using differential scanning calorimetry. All four domains of the BHA10 FAb (VH, VL, CH1 and CL) unfolded cooperatively with a Tm of 78°C (). Similar to other reported antibody fragments, the wild-type BHA10 scFv variable domains, lacking CH1 and CL, unfolded at much lower temperatures than the FAb.Citation13 The VL domain unfolded with a Tm = 68°C, while the VH domain unfolded at a Tm = 58°C, twenty degrees lower than the observed unfolding transition of the BHA10 FAb. As expected, the measured calorimetric enthalpy of unfolding (ΔHoU(TM)) was also significantly reduced in the scFv format;
ΔHoU(TM) = 260 kcal/mol for the FAb; ΔHoU(TM) = 44 kcal/mol for the VL domain; and
ΔHoU(TM) = 22 kcal/mol for the VH domain.
Design, expression and characterization of stabilized BHA10 scFvs.
Based on the results from the DSC analysis, we hypothesized that enhancing the thermal stability of the BHA10 scFv might, in turn, lead to a reduced tendency of the IgG-like BsAb to form aggregates. A molecular model of the BHA10 scFv was used to visually inspect the integrity of the 15 amino acid (Gly4Ser)3 linker used to connect the VH and VL domains on the flanking secondary structure elements as well as to determine the energetic contributions (). The (Gly4Ser)3 linker was found to be too short to adequately span the distance between the two variable domains possibly imposing structural constraints.Citation14 Accordingly, we built and tested BHA10 scFvs containing a longer linker, (Gly4Ser)4.
In addition, the model was used to evaluate pairs of cysteine residues substituted at framework positions VH44 and VL100, VH44 and VL105, and VH105 and VL43 that could participate in creating a stabilizing disulfide bond ().Citation10 The modeled conformation of the cysteines at positions VH44 and VL100 supported the geometry for forming a disulfide bond [δ(Sγ − Sγ) = 2.1 Å, δ(Cβ − Cβ) = 4.2Å, δ(Cα − Cα) = 5.5 Å]. However, the modeled cysteine at position VH105 was oriented away from the VL − VH interface [δ(Sγ − Sγ) = 5.4 Å and δ(Cβ − Cβ) = 5.8 Å, δ(Cα − Cα) = 5.7 Å] and was predicted to require reorientation of the VH104–106 backbone to form the VH105-VL43 disulfide. Modeled cysteines at positions VH44 and VL105 appeared to be incompatible with the length of a disulfide bond δ(Cα − Cα) = 17.6 Å, and would likely require realignment of VH FR4 region towards VL.
Four plasmids were constructed from the parent BHA10 scFv (). One scFv contained the longer (Gly4Ser)4 linker and the remaining three constructs each contained one of the three pairs of cysteine mutations described above. The variant BHA10 scFvs were expressed in the E. coli strain W3110 and culture supernatants containing secreted scFv proteins were analyzed by western blot. The scFv constructed with the (Gly4Ser)4 linker was produced by W3110 and the major protein product migrated according to its predicted molecular weight (∼30 kDa, data not shown). ScFvs constructed with the different pairs of cysteine substitutions, however, varied greatly in levels and quality of proteins with only the BHA10 scFv containing the cysteine pair at positions VL100 and VH44 produced and fully intact (data not shown). We also tested the effect of combining the longer (Gly4Ser)4 linker with the cysteine substitutions at VL100 and VH44 in the BHA10 scFv. Supernatants containing the various engineered BHA10 scFvs were first compared to wild-type BHA10 scFv by determining the temperature (T50) at which 50% of scFv molecules retained binding to LTβR antigen following thermal challenge. ScFvs were subjected to a range of temperatures spanning the thermal transition temperature of wild-type BHA10 scFv (previously determined to be T50 = 49°C). All of the engineered scFv molecules showed improved resistance to thermal challenge relative to the wild-type scFv (). The scFv with the longer linker (BHA10-GS4 scFv) showed a +4°C increase in thermal resistance relative to the wild-type BHA10 scFv in this assay. Introduction of the disulfide bond at positions VL100 and VH44 (BHA10-SS scFv) improved scFv thermal resistance by +10°C. Combining the engineered disulfide bond with the longer linker (BHA10-SS/GS4 scFv) increased the T50 by as much as 13°C, resulting in a scFv that was equally resistant to thermal challenge as the BHA10 Fab.
To further assess the stability and conformation of wild-type BHA10 and the engineered BHA10-GS4 and BHA10-SS/GS4 scFvs, we examined temperature-dependent binding of the hydrophobic dye 1-anilino-8-naphthalene sulfonate (ANS) to the purified scFvs (). The intrinsic fluorescence of ANS is quenched in aqueous solvent and increases when bound to exposed hydrophobic surfaces such as those that occur in partially unfolded proteins. Wild-type BHA10 scFv containing the original (Gly4Ser)3 linker had higher baseline ANS binding compared to the engineered BHA10 scFvs at all temperatures below 37°C. Increasing the temperature led to a steady increase of ANS binding to wild-type BHA10 scFv up to the transition temperature (∼60°C) indicative of additional exposure of hydrophobic sites during thermal denaturation. Increasing the length of the linker from (Gly4Ser)3 to (Gly4Ser)4 nearly halved the fluorescence signal at lower temperatures and had a sustained stabilizing effect at all temperatures approaching the Tm. Addition of the disulfide bond (BHA10-SS/GS4 scFv) had a stabilizing effect at temperatures below the 60°C transition temperature but also appeared to have some stabilizing effect at higher temperatures. BHA10-GS4 and BHA10-SS/GS4 demonstrated marked increases in ANS-binding only as the scFv approached its Tm, suggesting that increases in ANS-binding occur only as the native folded states of the stabilized scFvs begin to unfold.
Thermal unfolding of the purified wild-type BHA10 and engineered BHA10-GS4 and BHA10-SS/GS4 scFvs was monitored by differential scanning calorimetry (DSC) (). DSC data was analyzed using the non 2-state curve-fitting model. All three scFv VL domains demonstrated nearly identical thermal transition temperatures. Incremental increases in VH stability were observed with BHA10-GS4 and BHA10-SS/GS4 scFvs suggesting that most of the stability gain in these scFvs was associated with stabilizing the folded state of the VH domain. Lastly, binding affinities of the engineered BHA10 scFvs to LTβR were measured by surface plasmon resonance. BHA10 GS4 and BHA10-SS/GS4 binding properties were found to be essentially unchanged (±2-fold) compared to wild-type BHA10 scFv and FAb ().
Expression, production and characterization of stability- engineered BsAbs.
Wild-type and stability-engineered BHA10 scFvs were used to generate a panel of eight IgG-like BsAbs as listed in . Plasmids encoding the eight BsAbs were first used to transiently express protein in CHO cells. Supernatants containing BsAbs were collected and evaluated by western blot analysis. Both N- and C-heavy chain fusion proteins migrated according to their predicted molecular weights (calculated protein molecular weights ∼77 kDa) (). Light chain proteins also migrated according to their predicted molecular weights (∼25 kDa). Western blot analysis showed that the stability-engineered BsAbs containing either the BHA10-SS or BHA10-SS/GS4 scFvs expressed well independent of whether the scFv was fused to the amino- or carboxyl- termini (, lanes 4, 5, 8 and 9). Surprisingly, little to no secreted protein was detected for the non-stabilized N-BsAb or N-BsAb-GS4 constructs (, lanes 6 and 7 compared to lanes 8 and 9). While intact heavy chain fusion protein was detected in supernatants from all four C-terminal BsAb transfections, the presence of a ∼55–60 kDa molecular weight immunoreactive byproduct was substantially reduced in supernatants from both of the disulfide-bonded (SS) stabilized scFv-containing BsAbs (arrow, , lanes 2 and 3 versus lanes 4 and 5). While we did not further characterize the 55–60 kDa molecular weight byproduct, given that it was found to be immunoreactive with the anti-IgG detection reagent we speculate that the fragment contains heavy chain Fc sequences.
Based on these results, four stable CHO cell lines-N-BsAb-SS, N-BsAb-SS/GS4, C-BsAb-SS and C-BsAb-SS/GS4 were established and chosen for scale-up BsAb production. Stability-engineered BsAb titers ranged from 16–87 mg/liter. Protein A eluates of the stability-engineered BsAbs versus wild-type C-BsAb were first examined by analytical size-exclusion chromatography (). The chromatograms revealed that compared to the wild-type C-BsAb eluate, which previously had been shown to contain ∼40% aggregates, the C-BsAb-SS eluate contained a reduced level of aggregates at ∼20%, and the C-BsAb-SS/GS4 eluate contained the lowest level of aggregates at ∼10%. In comparison, control 14A2 IgG contained 6–8% aggregates (data not shown). The Protein A eluates of N- and C-BsAb-SS/GS4 BsAbs were further purified by anion exchange chromatography. Greater than 1 gram of highly purified material was readily obtained for each of the two BsAbs. Analytical size-exclusion chromatography and SDS-PAGE demonstrated >98% monomeric BsAb for both N- and C-BsAb-SS/GS4 BsAbs (Suppl. ).
We were interested in determining whether the engineered modifications impacted the long-term stability of the N-BsAb-SS/GS4 and C-BsAb-SS/GS4 BsAb molecules, a critical attribute for a biological therapeutic. Real-time stability studies of the BsAbs stored at 4°C for three months were conducted under buffer conditions and protein concentrations described in the Methods. Protein quality was assessed for (1) aggregation/precipitation (2) polypeptide cleavage or proteolysis, and (3) post-translational modifications such as oxidation or changes in oligosaccharide distribution. SEC/light scattering analyses showed little evidence of aggregation or material loss that might occur through precipitation (Suppl. ). Both the N-BsAb-SS/GS4 and C-BsAb-SS/ GS4 BsAbs contained ≥96.7% monomer after three months storage at 4°C for both high and low protein concentrations as compared to 97.4% monomer for the BHA10 IgG control. Increases of 1–2% oligomeric material were detected at the end of the study for both BsAbs and were predominately composed of BsAb dimers. Neither of the BsAb samples accumulated detectable levels of high-order aggregates, even though this type of aggregate would be readily observed by light scattering detection. In comparison, the BHA10 IgG control antibody accumulated less than 1% dimers over the three month study. The integrity of the BsAbs was monitored by SDS-PAGE and LC/MS analyses. Non-reducing SDS-PAGE showed that the C-BsAb-SS/GS4 remained intact with no evidence of proteolysis observed during the three month study at 4°C (Suppl. ). Analysis revealed extremely low and comparable levels of fragmentation by the end of the three month study for both the low and high concentration N-BsAb-SS/GS4 samples. LC/MS analyses of N-BsAb-SS/GS4 and C-BsAb-SS/GS4 were performed to detect any changes in protein molecular masses, an indication of potential degradation. Deconvoluted N-BsAb-SS/GS4 and C-BsAb-SS/GS4 light and heavy chain mass spectra showed no discernable modifications arising during the three month study indicating absence of chemical modifications that could adversely affect bispecific antibody function or stability (). Lastly, the two BsAbs were subjected to three cycles of freeze-thaw and agitation stress over 12 hours at room temperature. SEC/LS results showed a slight reduction of 0.2–0.6% in monomeric peak area for all samples with exception of the BHA10 antibody control (Suppl. and Suppl. ). Aggregates were mostly dimers as determined by SEC retention time as well as static light scattering detector (data not shown). The results of the agitation studies yielded no visible precipitates. SEC/LS analyses showed a minor decrease of 1–1.5% in monomeric peak area for the two low concentration N-BsAb-SS/GS4 and C-BsAb-SS/GS4 samples (Suppl. ). Reductions in monomeric peak areas were found to be inversely proportional with increases in BsAb dimers (data not shown).
In vitro activity of stability-engineered BsAbs.
The N- and C-BsAb-SS/GS4 BsAbs were tested for dual binding activity to the soluble receptor targets, TRAIL-R2 and LTβR, by surface plasmon resonance (SPR). N- and C-BsAb-SS/GS4 BsAb kinetic association/dissociation curves were generated to immobilized TRAIL-R2 followed by a secondary association/dissociation phase with soluble LTβR (). These results confirmed that both the N- and C-BsAb-SS/GS4 BsAbs simultaneously bound TRAIL-R2 and LTβR with nearly identical association and dissociation kinetics suggesting equivalent binding activity for both the N- and C-terminal BsAbs in this assay format. The control antibody BHA10 IgG showed no detectable binding to immobilized TRAIL-R2 while the control antibody 14A2 showed binding activity similar to both of the BsAbs towards TRAIL-R2 (data not shown).
To assay for in vitro cellular activity, tumor cells that stain positive for expression of both TRAIL-R2 and LTβR by fluorescence cytometry (unpublished data) were treated with the stability-engineered BsAbs or with control mAbs (BHA10 and 14A2) alone or in combination, and cell viability was measured after 3–4 days in culture. In the human WiDr colon carcinoma cell line, the individual mAbs were effective in reducing cell number, while the combination of mAbs showed somewhat enhanced activity. Both the N- and C-terminal BsAbs showed activity comparable to the mAb combination (), achieving similar total reduction in cell number as well as statistically similar IC50 values, that is the concentration of antibodies resulting in 50% growth inhibition (IC50 = 18–36 pM). Preincubation of the BsAbs with soluble receptor, either LTβR-Fc or TRAIL-R2-Fc, resulted in an expected reduction in activity in accordance with the contribution of the individual target (data not shown). In the MDA-MB231 breast tumor cell line, while treatment with the individual mAbs or the combination of mAbs revealed negligible activity, the C-BsAb-SS/GS4 BsAb had a pronounced effect (IC50 = 46 pM) in reducing tumor cell number (). The N-BsAb-SS/GS4 BsAb, in contrast, exhibited no activity in the MDA-MB231 cell line. In the Me180 tumor cell line, both BsAbs showed stronger anti-tumor cell activity relative to the combination of mAbs, with the C-terminal version exhibiting greater activity (p = 0.03) relative to the N-terminal BsAb (). Importantly, the BsAbs lacked detectable cytotoxic activity on a normal cell line (HUVEC) in this assay (), despite the presence of both receptors on the cell surface as measured by fluorescence cytometry (data not shown), suggesting tumor specific activity of the BsAbs.
Stability-engineered BsAbs exhibit good pharmacokinetic properties and inhibit tumor cell growth in vivo.
We next assessed the pharmacokinetic parameters of the stability-engineered N- and C- BsAbs. A single 10 mg/kg bolus of BsAb was administered by intraperitoneal injection into CB17-scid mice. The stability-engineered BsAbs exhibited in vivo half-lives comparable to that of standard monoclonal antibodies (N-BsAb-SS/GS4: T1/2 = 10.3 days; C-BsAb-SS/GS4: T1/2 = 15.1 days) (), as measured in an ELISA detecting IgG in the serum samples. Cmax values were 85.7 µg/ml and 105.7 µg/ml for N-BsAb-SS/GS4 and C-BsAb-SS/GS4 BsAbs, respectively. To confirm that the bispecific antibody remained intact in the serum, we employed an antigen-specific ELISA, using LTβR-Fc coated on the plate and detection with TRAIL-R2-Fc-His6. Serum concentration-time curves of the BsAbs detected by the antigen-specific ELISA were comparable to those in the standard IgG detection ELISA (Suppl. ), indicating that the BsAbs remained functionally intact in the serum throughout the course of the study. Both BsAb designs bound mouse () and human () neonatal Fc receptor, FcRn, with affinities equivalent to that of native IgG antibodies. The observed affinities are consistent with the demonstrated IgG-like PK properties of the BsAbs. Next we proceeded to test for efficacy in a tumor xenograft model. Immunodeficient mice with established WiDr tumors were treated by intraperitoneal administration with stability-engineered BsAbs (1×/week at 2 mg/kg), or with control mAbs alone (2×/week at 2 mg/kg), or in combination (2×/week each at 1 mg/kg). Both N- and C- BsAbs were efficacious in achieving greater than 60% tumor growth inhibition in the mice (). The in vivo results closely reflected those observed in WiDr cells in vitro, with the BsAbs showing comparable tumor inhibition to the combination of the individual antibodies, and enhanced activity (p < 0.05) relative to the individual antibodies.
We then tested the C-BsAb-SS/GS4 BsAb in the MDA-MB231 xenograft model because in vitro data suggested improved potency of the BsAb relative to the combination of mAbs in this cell line. The C-BsAb-SS/GS4 BsAb indeed demonstrated significant antitumor activity (p < 0.001) achieving up to 50% tumor inhibition in the MDA-MB231 model (), whereas the individual mAbs, and even the combination of mAbs, showed no efficacy. As was observed in vitro, the N-terminal BsAb did not inhibit tumor growth in the MDA-MB-231 model (data not shown).
Discussion
Here we show that enhancing the thermal stability of a scFv domain, a commonly used building block for constructing IgG-like BsAbs, greatly improves BsAb production and quality. Once sufficiently stabilized, scFv gene sequences can be appended to either the amino- or carboxyl-terminus of an IgG for producing biologically active, physically stable tetravalent IgG-like BsAbs. We found that stability-engineered BsAbs could be produced at reasonably high levels (16–87 mg/L) in a mammalian host cell line without gene amplification. The BsAbs could be purified to a high level of purity (>98%) using standard and scalable purification strategies and also demonstrated excellent biophysical stability. Only minor (<2%) changes in protein quality, such as aggregation or precipitation, were detected over a three month storage period at 4°C in standard saline buffer. No chemical modifications, such as oxidized or deamidated amino acid residues, were detected. The oligosaccharide distribution pattern was indistinguishable from that of normal IgGs. The biophysical and biochemical behavior of the stability-engineered BsAbs under these conditions compares favorably to that of normal IgGs as no attempt was made to optimize formulation of the BsAbs. Pharmacokinetic studies showed that both the N- and C- BsAbs had long serum half-lives and remained functionally intact. The lengthy serum half-lives of the stability-engineered BsAbs is consistent with our finding that the BsAbs bind to the neonatal Fc receptor FcRn and therefore likely do not interfere with the IgG salvage pathway.Citation15 Finally, both the in vitro and in vivo studies demonstrated that the BsAbs are biologically active with inhibition of tumor growth occurring at levels comparable to or greater than the combination of parental antibodies.
Numerous methods have previously been described for preparing recombinant IgG-like BsAbs with many showing excellent activity in vitro.Citation16–Citation20 However, preparation of IgG-like BsAbs that are suitable for preclinical evaluation in vivo has been particularly challenging, and few study results have been reported. Recently, a di-diabody IgG-like BsAb (assembled from two cross-over scFv chains) was described that was shown to simultaneously target and block activity of EGF and IGF-1 growth factor receptors expressed on the surface of cultured tumor cells.Citation17 While the di-diabody BsAb was shown to be effective at inhibiting tumor growth in vivo in a mouse xenograft model, ≥60% activity to both receptors was reportedly lost within a 24 h period following administration, suggesting that the di-diabody construct may have lacked sufficient stability. More recently, a report by Wu and colleagues described a promising technology for producing dual-targeting N-terminal Ig-like BsAbs referred to as dual-variable domain (DVD) IgGs that were shown to exhibit favorable PK properties and in vivo activity comparable to that of the combined mAbs.Citation21 The DVD technology links the VL and VH domains (Fv) specific to one target in series to the corresponding domains of an IgG specific to a second target using the naturally occurring conserved “elbow” sequences located at the amino terminus of CL and CH1. It was proposed that the flexibility of the elbow sequences and absence of strong secondary structure allows for stable assembly of the added N-terminal Fvs.
In our study we provide proof-of-concept evidence that a sufficiently stable scFv can be a useful building block for producing high quality IgG-like BsAbs. Increasing the BHA10 scFv linker length from 15 to 20 amino acids resulted in a moderate increase in scFv thermal stability. Consistent with the computational model, the longer (Gly4Ser)3 linker was likely relieving strain on the VH domain, favoring a more stable scFv folded-state. Also consistent with scFv model, only the cysteine residues introduced at positions VH44 and VL100 between the BHA10 scFv VH/VL interface led to a stable, functional BHA10 scFv. However, given the likelihood that introducing disulfides may not be a general solution for stabilizing scFvs, we anticipate that linker length optimization combined with rational and molecular evolution strategies will make it possible to routinely improve scFv stability for producing quality IgG-like bispecific antibodies.Citation6,Citation9,Citation14,Citation22–Citation25 While ongoing studies are addressing whether a threshold thermal transition value can be defined for selecting a suitably stabilized scFv for constructing a stability-engineered IgG-like BsAbs, we surmise that a T50 value ∼65°C or a Tm value ≥70°C may be desirable. Presumably, related approaches could be applied for stabilizing other immunoglobulin variable regions to be used for building IgG-like BsAbs such as domain antibodies, diabodies and tandem diabodies.
Antibody and antibody-derived therapeutics must have sufficient physical and chemical stability to progress towards clinical application. Preliminary stability studies of the N-BsAb-SS/GS4 and C-BsAb-SS/GS4 BsAbs showed good biophysical stability under conditions similar to those encountered during storage and handling of biotherapeutics. To fully validate the biophysical properties of our IgG-like BsAb molecules more comprehensive studies should be carried out. Such studies might include long-term stability studies at various temperatures in different formulation buffers. In addition, studies should be pursued to quantitatively investigate the effect of long-term storage on the binding activity of the BsAbs towards both target antigens.
The IgG-like BsAbs in this study were designed to target two TNF receptor family members, TRAIL-R2 and LTβR, to explore the possibility that a BsAb might trigger an enhanced or broader anti-tumor response than that achieved by treating with a mixture of the two antibodies. Significantly, the BsAbs exhibited enhanced anti-tumor activity relative to the combination of parental antibodies in a subset of cell lines in vitro. In vivo the enhanced activity of the C-BsAb was recapitulated in the MDA-MB231 model. Notably, normal HUVEC cells, despite being comparable to MDA-MB231 and Me180 in expression of both LTβR and TRAIL-R2 receptors on the cell surface and in their insensitivity to the mAbs, were uniquely insensitive to induction of cell death by the BsAbs, suggesting that tumor cells may be preferentially sensitive to the killing activity.
These studies suggest that the pronounced anti-tumor activity of the BsAbs may be due to crosslinking of LTβR and TRAIL-R2 receptors, an event which does not occur by simply combining the individual antibodies. We speculate that the anti-LTβR × anti-TRAIL-R2 BsAbs might be inducing the formation of non-physiological receptor complexes that promote novel or enhanced signaling events such as was described with Fas activation by varying the oligomer states of Fas ligand.Citation26 Alternatively, the BsAb could alter signal intensity or quality by forcing receptor relocalization into a new environment such as a lipid raft or by altering the kinetics of receptor internalization.Citation27 Further studies are warranted to gain a better understanding of the BsAb-mediated anti-tumor response. While the mechanism of cooperation is not fully understood, this is the first example of a BsAb with enhanced activity over the individual antibody combination, highlighting a potential feature of BsAb-based therapeutics using agonist antibodies.
While both the N- and C-BsAbs exhibited similar and equivalent dual binding activity to soluble TRAIL-R2 and LTβR receptors, the C-BsAb showed enhanced biological activity relative to the N-BsAb in some cell lines. It is possible that the differing activities of the N- and C-BsAbs may depend on the varying densities of LTβR and TRAIL-R2 receptors on the different tumor cell lines. The enhanced activity of the C-BsAb may also be explained by the possibility that the N-terminal configuration cannot fully engage and cross-link LTβR and TRAIL-R2 receptors due to close physical proximity and steric interference between the BHA10 scFv and the 14A2 Fab arm. We estimated that the 25 amino acid (G4S)5 linker used to append the BHA10 scFv to the amino terminus of 14A2 heavy chain generates about 50 angstroms distance between the complementarity determining regions of the 14A2 and BHA10 antibody binding domains and is expected to allow sufficient separation of the two sites. This suggests that the BsAb format (N- versus C-terminal BsAb) and spatial orientation of the antibody binding domains may be critical for effective engagement of receptors on a cellular surface, although this may not be the case for targeting soluble antigens. We believe that the flexibility in design offered by this stability engineering approach is an important feature allowing assessment of various IgG-like BsAb formats to select a biologic with optimal therapeutic activity.
Materials and Methods
Cloning, design and stability engineering of BHA10 scFv.
BHA10 scFv was cloned into the pBAD bacterial expression vector (Invitrogen, Carlsbad, CA) using the Polymerase Chain Reaction with primers 5′-CAG TAG CAT GCA GGT CCA ACT GGT GCA G-3′ and 5′-GTT CTA GAA AGC TTT TGT CGT CGT CGT CTT TGA TCT CCA CCT TGG TAC CCT G-3′. The PCR product was purified using the Millipore Ultrafree-DNA extraction kit (Millipore; Bedford, MA), digested with Sph I, made blunt-end by digesting with DNA Polymerase I in the presence of dNTPs, and then digested with Hind III. The blunt-ended/Hind III digested PCR product was ligated to Sca I/Hind III digested pBAD vector and used to transform E. coli strain XL1-Blue. The final BHA10 scFv construct contained an N-terminal Gene III signal peptide to direct secretion to the periplasmic space of E. coli and a C-terminal His6 tag for detection and protein purification.
Homology models of BHA10 single chain Fvs comprising (Gly4Ser)n linkers of varying lengths were built with MODELER using a structure of an in-house anti-VLA4 FAb fragment as a template.Citation28 The conformations of the VH-VL connecting (Gly4Ser)n loops were predicted using a hierarchical technique implemented in Prime.Citation29 The conformations of the side chains were predicted using SCWRL3 algorithm.Citation30 The models were subsequently refined in CHARMM and analyzed using Pymol visualization software (DeLano Scientific LLC, San Carlos, CA, http://www.pymol.org).Citation31
Amino acid substitutions were introduced using a commercially available site-directed mutagenesis kit (Stratagene; La Jolla, CA). Mutagenesis reactions were transformed into E. coli strain XL10-GOLD® (Stratagene). Paired cysteine substitutions were introduced at VH and VL positions 44 and 100, 44 and 105, and 105 and 43, respectively, numbering according to Kabat.Citation32 Variant (Gly4Ser)n linker lengths were introduced by PCR. DNA sequence analysis confirmed the correct sequence of the final constructs.
ScFv expression and purification.
E. coli strain W3110 (ATCC No. 27325, Manassas, VA) was used for expressing BHA10 scFv proteins. Transformants were grown in 4 × 250 ml SB media (Teknova, Half Moon Bay, CA) containing 50 µg/ml carbenicillin in 1 liter baffled flasks to OD600 □ 0.8, induced by adding to 0.02% arabinose, and cultured overnight. Bacteria were collected by centrifugation. The pellets were solubilized and lysed using 40 ml B-PER protein extraction reagent (Pierce Biotechnology, Thermo Fisher Scientific Inc., Rockford, IL). Solubilized scFv was applied to a 5 ml Ni-NTA-Superflow column (Qiagen, Valencia, CA). Bound scFv was washed with 60 mM imidazole, pH 8.0 and eluted with 300 mM imidazole, pH 8.0. Eluted scFv was loaded onto a 6 ml Protein L agarose column (Pierce). Bound protein was washed with phosphate buffered saline (PBS) and eluted with 0.1 M glycine, pH 3.0. Purified scFvs were dialyzed against PBS and stored at −20°C. Protein concentrations were determined using an ε280 nm = 2.1 ml mg−1 cm−1. BHA10 FAb fragment was prepared essentially as described with the exception that no alkylating agents were used.Citation33 LTβR-Fc was prepared as described.Citation34
Thermal challenge assays.
Plasmid DNAs encoding wild-type and engineered BHA10 scFvs were used to transform W3110. Colonies were grown overnight in SB expression media supplemented with 1% glycine, 1% Triton X-100, 0.02% arabinose, and 50 µg/ml carbenicillin at either 37°C or 32°C.Citation35 Bacterial supernatants were collected and heat-treated in either PCR strip tubes or 96-well plates (Applied Biosystems, Foster City, CA) for 60–90 min in a thermal cycler (iCycler, Bio-Rad, Gaithersburg, MD). Cooled samples were transferred to 96-well v-bottomed plates (Corning, Corning, NY) and centrifuged at 2,000 rpm, 4°C for 30 min (IEC Centra 8R, Thermo Electron, Waltham, MA). Supernatants were then assayed for binding to LTβR-Fc antigen by DELFIA. MaxiSorp 96-well plates (Nalge Nunc, Rochester, NY) were coated with 1 µg/ml LTβR-Fc in 0.1 M sodium carbonate buffer, pH 9.5 overnight at 4°C, and then blocked with DELFIA assay buffer (DAB, 10 mM Tris HCl pH 7.4, 150 mM NaCl, 20 µM EDTA, 0.5% BSA, 0.02% Tween 20, 0.01% NaN3) for 1 h with shaking at room temperature and washed 3 times with DAB without BSA (Wash buffer). Test samples diluted in DAB were added to plates in a final volume of 100 µl, incubated for 1 h with shaking at room temperature, and then rinsed with Wash buffer. Bound BHA10 scFv was detected by incubating with 40 ng/ml of Eu-labeled anti-His6 antibody followed by a wash step and addition of DELFIA enhancement solution (Perkin Elmer, Boston, MA). Plates were read using the Europium method on a Victor 2 (Perkin Elmer). For thermal gradients, the data was analyzed with Prism 4 software (GraphPad Software, San Diego, CA) using a sigmoidal dose response with variable slope as the model. The values obtained for the mid-point of the thermal denaturation curves are referred to as T50 values and are not construed as being equivalent to biophysically derived Tm values.
Differential scanning calorimetry analyses.
The thermal unfolding profiles of BHA10 FAb and scFv antibody fragments were measured using an automated capillary differential scanning calorimeter (capDSC; MicroCal LLC, Northampton, MA). Scans were performed at 1°C/min using the medium feedback mode for enhanced peak resolution. Protein concentrations ranged between 0.4–1.0 mg/ml. The scan range was 20–95°C. All 96-well plates containing protein were stored within the instrument at 6°C. Transition midpoints (Tm's) were determined by fitting the curves using the non-two-state model with the Origin 7 software provided by the manufacturer.
ANS assay.
The hydrophobic dye 1-anilino-8-naphthalene sulfonate was used to probe BHA10 scFv protein conformation. Twenty micrograms per milliliter scFv protein solutions in PBS were treated with 10 µM final concentration of dye and fluorescence measured on a J-810 circular dichroism spectropolarimeter (Jasco, Easton, MD) equipped with a thermoelectric peltier device and a fluorescence detector perpendicular to the light path. Excitation and emission monochromators were set to 370 nm (10 nm bandwidth) and 480 nm, respectively. Thermal denaturation was performed with a data pitch of 1.0°C and a temperature slope of 120°C/hour. Photomultiplier voltage was set to 600 V.
Affinity measurements.
Isothermal titration calorimetry (ITC) was used to measure the affinity of BHA10 FAb to LTβR-Fc. ITC was performed on a VP-ITC unit (MicroCal LLC) set to 30°C. Approximately 500 µl of a 7 µM LTβR-Fc solution was placed into the sample cell, and PBS dialysate was placed in the reference cell. A total of 234 µl of 70 µM BHA10 FAb was titrated into the sample cell in 7 × 10 µl, 12 × 7 µl, followed by 8 × 10 µl injections. The reaction stoichiometry was 1:1. ITC curves were analyzed using the Origin Software supplied by the manufacturer.
KD measurements of BHA10 scFv preparations were performed using surface plasmon resonance (SPR) on a Biacore 3000 instrument (Biacore Inc., Piscataway, NJ). All experiments were performed in HBS-EP buffer, pH 7.4. Twenty µg/ml biotinylated PENTA-His antibody (Qiagen) was immobilized onto a streptavidin-coated CM5 chip at a flow rate of 10 µl/min for approximately 1 min. 0.1 µM solutions of wild-type and engineered BHA10 scFvs were injected over the chip at a flow rate of 5 µl/min for 10 min. A series of LTβR-Fc solutions ranging from 1–200 nM were subsequently injected at a flow rate of 30 µl/min onto the scFv-coated surface. Background was subtracted using buffer only samples. Sensorgram curves were analyzed using the BiaEval 3.0 manufacturer's software. KD values were calculated by fitting kinetic association and dissociation curves to a 1:1 Langmuir binding model.
BsAb expression and purification.
Wild-type and engineered BHA10 scFv gene sequences were amplified in a two-step PCR reaction and used to construct a panel of BsAbs as N- and C-terminal fusions to the chimeric anti-TRAIL-R2 IgG (14A2) for expression in CHO cells using the mammalian expression vector pN5KG1.Citation36 For the N-terminal bispecific constructs (N- BsAbs), a (Gly4Ser)5 linker was used to connect the BHA10 scFvs to the mature amino terminus of 14A2 heavy chain. For the C-terminal bispecific constructs (C- BsAbs), a Ser(Gly4Ser)3 linker was used to connect the BHA10 scFvs to the carboxyl terminus of 14A2 heavy chain.
Plasmid DNAs were used to transform DHFR-deficient CHO DG44 cells for transient and stable production of antibody protein.Citation37 For transient transfection 20 µg of plasmid DNA was transfected into 4 × 106 cells by electroporation and the cells cultured in CHO-SSFM II media containing 100 µM hypoxanthine and 16 µM thymidine (Invitrogen) for four days at 37°C. Transient protein expression was evaluated by western Blot. Briefly, supernatant samples were fractionated on 4–20% Tris-glycine SDS-PAGE, transferred to nitrocellulose membranes and probed with anti-human IgG antibody-HRP conjugate. Membranes were processed using the ECL western blotting analysis system (Amersham ECL, GE Healthcare Life Sciences, Piscataway, NJ).
For stable protein production, transfected DHFR positive cell lines were adapted to serum-free conditions and scaled for antibody production. Supernatants from 11-day bioreactor runs were harvested and precleared by ultrafiltration. The BsAbs were affinity purified by Protein A Sepharose FF (GE Healthcare) chromatography. Bound antibodies were eluted with 0.1 M glycine, pH 3.0 and neutralized to approximately pH 8 by addition of Tris base. Eluants were diluted 10-fold into 50 mM Tris, pH 8.5 and proteins purified to homogeneity using a TMAE anion exchange column (Fractogel®; EMD Biosciences, San Diego, CA). BsAbs were dialyzed into PBS (Irvine Scientific, Santa Ana, CA). Endotoxin levels were assayed by Kinetic Quantitative Chromogenic LAL Analysis (Lonza, Allendale, NJ). Purity and percentage of monomer BsAb product was assessed by 4–20% Tris-glycine SDS-PAGE and analytical size-exclusion HPLC (SEC), respectively.
BsAb dual binding assay.
Surface Plasmon Resonance (SPR) was used to measure dual binding activity of the BsAbs. Penta-His Biotin Conjugate antibody immobilized sensorchips were prepared as described above. Twenty microliters of 50 nM TRAIL-R2-Fc-His6 (R&D Systems, Minneapolis, MN) was injected at 2 µl/min flow rate. Flow rates were then increased to 10 µl/min. Seventy microliters of (1) HBS-EP buffer, (2) 14A2 antibody, (3) BHA10 antibody, (4) 25 nM N-BsAb-SS/GS4 or (5) 25 nM C-BsAb-SS/GS4 were injected followed by 10 minutes of dissociation in buffer prior to subsequent 70 µl injections of 0–100 nM dilutions of LTβR-Fc. Complexes were allowed to dissociate for approximately 8 additional minutes. Sensorgrams were processed using BiaEval software provided by the manufacturer and plotted using Kaleida-Graph™ (Synergy Software, Reading, PA). KD values were calculated by fitting kinetic association and dissociation curves to a 1:1 Langmuir binding model.
FcRn binding assay.
mAb or BsAb were coated on Nunc MaxiSorp 96-well plates at 3 µg/ml in PBS, pH 6 overnight at 4°C. The plates were washed once with PBS, pH 6 and blocked with 300 µl/well of 0.1 M sodium phosphate, 0.1 M sodium chloride, 0.05% Tween 20 and 0.1% gelatin, pH 6 for 2 h at room temperature. The plates were washed again with PBS, pH 6 and incubated with 100 µL of the appropriate concentration of recombinantly produced biotinylated mouse or human FcRn-Fc. The FcRn-Fc constructs contain mutations within the Fc that abrogate self-association of the FcRn with the Fc domains.Citation38 After 1.5 h plates were washed with PBS, pH 6 and incubated with 100 µL of streptavidin HRP at room temperature for 1.5 h, washed with PBS, pH 6 and developed with TMB (3, 3′, 5, 5′-tetramethylbenzidine) and read at 450 nm. A four-parameter logistic fit was used to model the binding data.
Stability studies of BsAbs.
High and low concentration BsAb samples were formulated in PBS and used for 2–8°C stability studies. N-BsAb-SS/GS4 was used at 1.8 mg/ml and 10.3 mg/ml and C-BsAb-SS/GS4 at 5.0 mg/ml and 11.4 mg/ml. Initial (T = 0), intermediate (T = 1 week, 2 week, 1 month, 2 month) and final (T = 3 month) time point samples were analyzed immediately following sample collection. In addition, initial (T = 0) samples were frozen and stored at −70°C until thawed for secondary analyses at the end of the three month study. BHA10 IgG (8.7 mg/ml) was used as control and was similarly handled.
Protein aggregation or precipitation was monitored using SEC linked inline to a miniDawn light scattering detector and a rEX refractive index detector (SEC/LS, Wyatt Technology Corp., Santa Barbara, CA). Light scattering data were analyzed using the Astra V software provided by the manufacturer. Twenty-five microgram samples were injected onto a TSK-gel G3000SWXL analytical SEC column (Tosoh Biosciences, Montgomeryville, PA) at 5 µl/min using an Agilent 1100 HPLC system (Agilent Technologies, Santa Clara, CA) pre-equilibrated with 10 mM phosphate, 150 mM NaCl, 0.02% azide at pH 6.8.
Proteolysis and post-translational modifications were monitored using SDS-PAGE and liquid chromatography/intact mass spectral analyses (LC/MS, Agilent LC/MSD TOF coupled to an Agilent 1100 LC system via an electrospray interface). For SDS-PAGE analysis, 5 µg of protein was loaded per lane. For LC/MS analysis, T = 0, T = 1 month, and T = 3 month samples were analyzed in both the non-reduced and reduced form. Reduced samples were prepared by dilution into a 50 mM DTT/4 M guanidine hydrochloride buffer for 1 hour at 37°C. HPLC Buffer A and Buffer B were 0.03% TFA in H2O and 0.025% TFA in acetonitrile, respectively. The flow rate was 100 µl/min. 7.5 µg of each sample (reduced and non-reduced) was injected onto a 2.1 × 50 mm Vydac C4 column and analyzed by Agilent ESI-TOF. A bind-and-elute method was used for non-reduced samples while a gradient method was used for reduced samples. Spectra were obtained using the Analyst and deconvoluted using the MaxEnt1 software packages included with the instrumentation.
For freeze-thaw analyses one set of high and low concentration BsAb samples and the BHA10 IgG control were stored at 4°C for the duration of the study. The remaining samples were frozen at −70°C for a minimum of one hour. The samples were removed from the freezer and allowed to stand at room temperature until completely thawed. Samples were then mixed, one set of samples moved to 4°C storage for the remainder of the study, and the remaining samples refrozen. This freeze-thaw sequence was repeated two additional times. Percent monomers were analyzed by SEC/LS.
Lastly, BsAb solutions and the BHA10 IgG control were subjected to agitation stress on an orbital shaker at 650 rpm for 16 h at room temperature. Percent monomers were analyzed by SEC/LS.
Tumor cell proliferation assay.
The human tumor cell lines WiDr colon carcinoma (ATCC No. CCL-218), Me180 cervical carcinoma (ATCC No. HTB-33), and MDA-MB-231 breast carcinoma (generously provided by Dr. Dajun Yang, Ascenta Therapeutics, San Diego, CA) were cultured in MEM-Earles with 10% FCS, 2 mM L-Glutamine, 1X non-essential amino acids, 0.5 mM sodium pyruvate, and penicillin/streptomycin. Cell lines were rinsed once with PBS and cells released by digestion with trypsin. Cells were collected by centrifugation, resuspended in complete media, counted and seeded into 96-well tissue culture plates at 5,000 cells/well for WiDr and Me180; and 1,500 cell/well for MDA-MB-231. Human IFNγ (Biogen Idec, Corp.,) was added to the cell suspensions to result in a final cytokine concentration of 50–80 U/ml.Citation39 Fifty microliters of the cell + IFNγ suspension were mixed with 50 µl of 2X concentrated 3-fold serial dilutions of test antibodies prepared in complete media. The final concentrations of test antibodies typically ranged from 5,000 pM to 0.07 pM. Cells were grown for 4 d (WiDr and Me180) or 3 d (MDA-MB-231) at 37°C in a 5% CO2 humidified chamber and cell killing assessed by the addition of 20 µl/well Promega CellTiter 96 Aqueous One Solution Cell Proliferation Assay reagent (Promega Corporation, Madison, WI). Human umbilical vein endothelial cells (HUVEC) were used as control. Plates were read in a micotiter plate reader at 490 nm (Spectromax Plus, Molecular Devices, Sunnyvale CA). Data was graphed using Microsoft Excel (Microsoft Inc., Bellevue, WA).
In vivo PK analysis and tumor xenograft models.
Mice were maintained in accordance with the Biogen Idec Institutional Animal Care and Use Committee, and city, state and federal guidelines for the humane treatment and care of laboratory animals. For pharmacokinetic (PK) studies a single bolus injection of 10 mg/kg (1 mg/ml) BsAb diluted in PBS was administered intraperitoneally (ip) into male CB17-scid mice (Harlan Sprague-Dawley, Inc.). Mice were sacrificed at 0, 0.5, 2, 6, 24, 48, 72, 96, 168, 240 and 336 h post-injection using three mice per timepoint for each BsAb. Serum samples were prepared for analysis by ELISA assay to quantify levels of the BsAbs. For the standard Ig detection ELISA, plates were coated with goat-anti-human IgG, blocked with PBS/1% BSA, and dilutions of serum containing the BsAbs were serially diluted in PBS/1% BSA, added to the plates and incubated. Captured antibodies were detected with a goat-anti-human kappa chain-HRP-linked antibody. Pharmacokinetic properties were calculated using noncompartmental module of WinNonLin (Pharsight Corp., Mountain View, CA). For the antigen-specific ELISA, a sandwich ELISA was employed. 96-well microtiter Immulon II plates were coated with 2 µg/ml LTβR-Fc overnight at 4°C. Nonspecific sites were blocked with 0.5% Nonfat Dry Milk in PBS plus 0.01% Thimerosal for 1 hour at 37°C. Serial dilutions of serum samples were added to duplicate wells and incubated for 1 hour at 37°C. Plates were washed and 100 µl of 100 ng/ml TRAIL-R2/Fc/His6 (R&D Systems, Minneapolis, MN) was added to the wells and the plate was incubated for 1 h at 37°C. LTβR-Fc: BsAb: TRAIL-R2/Fc/His6 complexes were detected with Penta-His Horseradish Peroxidase Conjugate (QIAGEN, Cat. 34460) and developed with TMB Peroxidase Substrate (Kirdgaard and Perry Labs, Cat. 50-76-00). Plates were read on a SpectraMax platereader and data plotted with the SoftMax Pro software provided by the manufacturer (Molecular Devices Corp., Sunnyvale, CA).
For tumor xenograft studies, WiDr human colon carcinoma (2 × 106 cells per mouse) cells were implanted subcutaneously into the flanks of athymic nude mice. Mice bearing tumors of approximately 100 mg in size were randomly divided into six groups of ten mice. IP treatments were administered, beginning on day 13 post-implantation, as follows: (1) pyrogen-free PBS; (2) BHA10 IgG, 2 mg/kg, 2×/wk; (3) 14A2 IgG, 2 mg/kg, 2×/wk; (4) N-BsAb-SS/GS4, 2 mg/kg, 1×/wk; (5) C-BsAb-SS/GS4, 2 mg/kg, 1×/wk; (6) BHA10 IgG + 14A2 IgG, 1 mg/kg each, 2×/wk. Tumor sizes and body weights were recorded bi-weekly. Study was terminated when average tumor size of vehicle group reached approximately 2,000 mg. Tumor volume was calculated using the formula: (L × W2/2).
MDA-MB-231 human breast carcinoma cells (2 × 106 cells per mouse) were implanted subcutaneously into the flanks of athymic nude mice. The tumors were grown until day 13 at which point tumor-bearing mice with an average tumor size of approximately 168 mg were randomly assigned to treatment (N = 10) and vehicle control (N = 15) groups. Mice received antibodies and vehicle IP starting at day 13. Groups are shown as follows: (1) pyrogen-free PBS; 1×/wk; (2) BHA10 IgG, 2 mg/kg, 2×/wk; (3) 14A2 IgG, 2 mg/kg, 2×/wk; (4) C-BsAb-SS/GS4, 2 mg/kg, 1×/wk; (5) BHA10 IgG + 14A2 IgG, 1 mg/kg each, 2×/wk. Tumor sizes and body weights were recorded bi-weekly. Study was terminated when average tumor size of vehicle group reached approximately 2,800 mg.
Abbreviations
| TNF | = | tumor necrosis factor |
| TRAIL-R2 | = | TNF-related apoptosis inducing ligand receptor-2 |
| LTβR | = | lymphotoxin-β receptor |
| BsAb | = | bispecific antibody |
| Fc | = | fragment crystallizable |
| FcRn | = | neonatal Fc receptor |
| FAb | = | fragment antigen binding |
| scFv | = | single-chain fragment variable |
| VL | = | variable light |
| VH | = | variable heavy |
| CHO | = | chinese hamster ovary |
| ANS | = | 1-anilino-8-naphthaline sulfonate |
| DHFR | = | dihydrofolate reductase |
| mAbs | = | monoclonal antibodies |
| DSC | = | differential scanning calorimetry |
| SEC | = | size-exclusion chromatography |
| SPR | = | surface plasmon resonance |
Figures and Tables
Figure 1 Design and production of IgG-like BsAbs. (A and B), Schematic diagrams of N- and C-BsAbs designs and mammalian expression vectors used for producing IgG-like BsAbs. Detailed components of the expression vectors are shown at the bottom of (B). (C), Analytical size-exclusion chromatography profile of C-BsAb constructed with wild-type BHA10 scFv following expression in CHO cells and purification on Protein A.
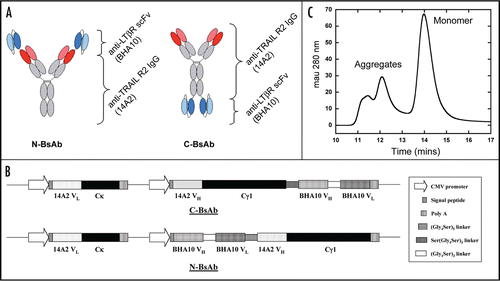
Figure 2 DSC analysis of BHA10 FAb and wild-type BHA10 scFv. Overlay of BHA10 FAb (dashed line) and wild-type scFv (solid line) thermograms. Unfolding transitions are measured at the endothermic peaks (transition midpoints) and reported as Tm values (°C).
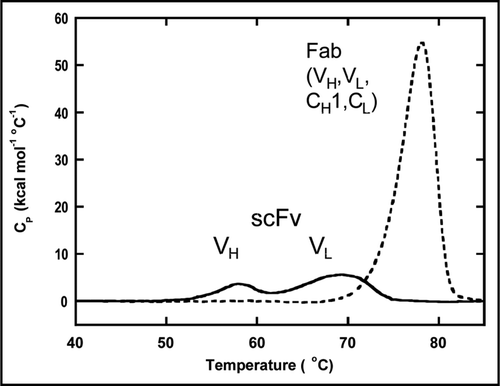
Figure 3 Molecular model and sequence of BHA10 scFv. (A) The scFv is in the VH□VL orientation. VH is shown in red, VL is shown in blue, and the Gly/Ser linker is shown in red. Paired cysteine substitutions are shown at VH and VL positions 44 and 100, and 105 and 43, respectively. The VH 105C is shown unpaired as it utilizes VH 44C. (B) Amino acid sequence of wild-type BHA10 scFv. The Gene III signal peptide is shown underlined, (Gly4Ser)3 linker is indicated in bold type, and the Enterokinase site, Myc and His tags are indicated in italics. Positions of cysteine substitutions are as follows—the VH substitution at position 44 is shown as a single underline and at position 105 as a double underline. The VL substitution at position 43 is shown as a single underline and at positions 100 and 105 as double underlines.
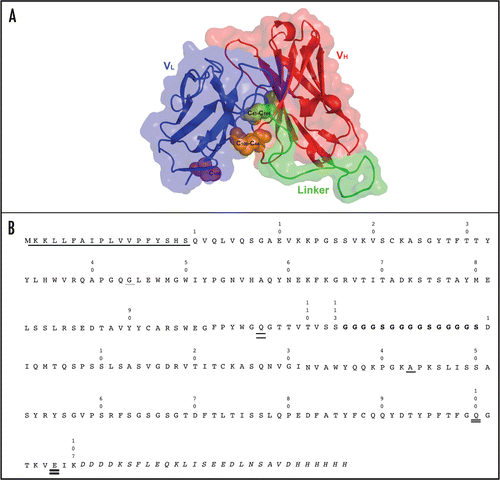
Figure 4 Characterization of stability-engineered BHA10 scFvs. (A) Binding activity of wild-type BHA10 scFv, stability-engineered BHA10 scFvs, and BHA10 FAb to LTβR was measured following thermal challenge. Binding profiles are normalized to 100% maximum binding. The temperature at which 50% binding activity is retained (T50, °C) is indicated. Samples were assayed in duplicate. (B) Temperature-dependent binding of the hydrophobic dye ANS to BHA10 scFvs. PBS, (○); wild-type BHA10 scFv (containing (Gly4Ser)3 linker), (●); BHA10-GS4 scFv (▴); and BHA10-SS/GS4 scFv (▪).
Figure 5 Design and expression of stability-engineered BsAbs. (A) Western blot analysis of BsAbs transiently expressed in CHO cells. Supernatant samples in the left panel are analyzed under non-reducing conditions and under reducing conditions in the right panel. Lane 1: MW marker, 2: C-BsAb, 3: C-BsAb-GS4, 4: C-BsAb-SS, 5: C-BsAb-SS/GS4, 6: N-BsAb, 7: N-BsAb-GS4, 8: N-BsAb-SS, 9: N-BsAb-SS/GS4. Arrow indicates presence of ∼55–60 kDa unidentified immunoreactive species. (B) Normalized SEC profiles of stability-engineered C-BsAbs following Protein A chromatography. Sharp peaks eluting between 13–15 min represent monomeric BsAb. Broad peaks eluting between ∼10.75–14 min represent aggregated protein. Blue dash-dot = C-BsAb, red dash = C-BsAb-SS, and green line = C-BsAb-SS/GS4.
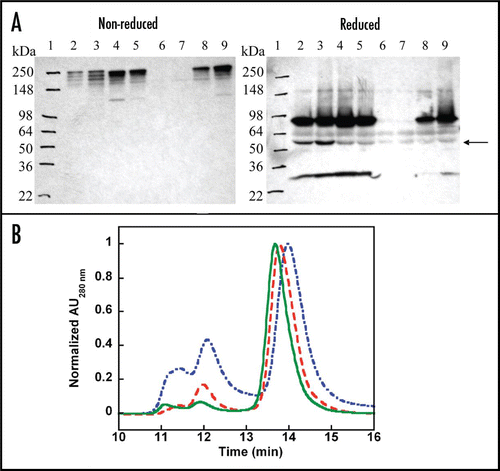
Figure 6 Intact mass analyses of N- and C-BsAb-SS/GS4. N- and C-BsAb-SS/GS4 stored for T = 0 and T = 3 months at 4°C were analyzed for proteolysis and post-translational modification by LC/MS. Upper, N-BsAb-SS/GS light and heavy chains; lower, C-BsAb-SS/GS light and heavy chains. Both light chain and heavy chain masses are consistent with theoretical calculations based upon protein primary sequences. No significant changes in mass as results of modifications were detected. Typical IgG biantennary glycans with variable numbers of terminal galactose were observed such as G0, G1 and G2.
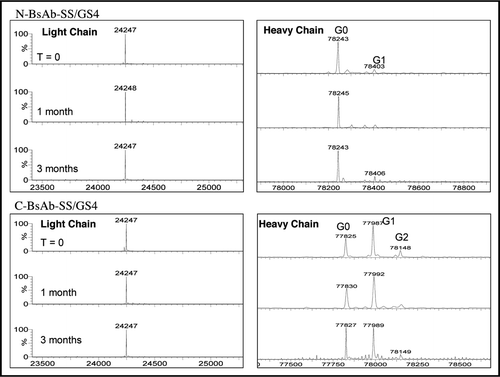
Figure 7 Dual-binding activity of stability-engineered BsAbs. Surface plasmon resonance analysis of stability-engineered BsAbs for dual-binding to TRAIL-R2 and LTβR. Biotinylated anti-His6 antibody was immobilized onto a streptavidin-labeled sensorchip followed by capture of TRAIL-R2-Fc-His6 (TRAIL-R2). (A) sensorgrams show subsequent additions of BsAbs and 0, 3, 10, 30 or 100 nM concentrations of sLTβR-Fc (LTβR). N-BsAb-SS/GS4 shown as dashed line and C-BsAb-SS/GS4 shown as solid line. (B) calculated kinetic rate constants.
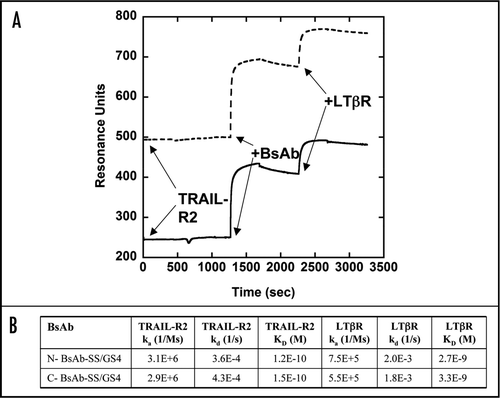
Figure 8 Inhibition of tumor cell line proliferation by stability-engineered BsAbs. Viability of human tumor cell lines and control HUVEC cells in response to treatment with BsAb and parent mAbs in a 96-well proliferation assay. (A) WiDr colon tumor cell line; (B) MDA-MB231 breast tumor cell line; (C) Me180 breast tumor cell line; and (D) HUVEC normal umbilical vein endothelial cells. Data is representative of triplicate experiments and shown as mean ± S.D.
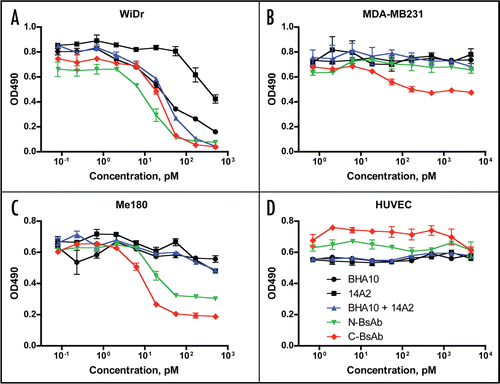
Figure 9 Binding of BsAbs to neonatal Fc receptor. Binding of various concentrations of biotinylated FcRn-Fc to fixed concentrations of immobilized N-BsAb-SS/GS4 BsAb (+), N-BSAb-SS/GS4 BsAb (□), hIgG1 (◆) and mIgG1 (▴) in an ELISA assay. Receptor complexes were washed with PBS, pH 6, detected by incubating with streptavidin-HRP conjugate and developed with peroxidase substrate. Plates were read at 450 nm. (A) mouse FcRn-Fc; and (B) human FcRn-Fc.
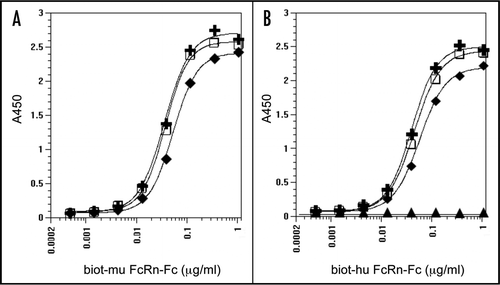
Figure 10 Inhibition of tumor growth by stability-engineered BsAbs. (A) WiDr and (B) MDA-MB231 tumor cells were implanted subcutaneously into immunocompromised mice. Treatment was initiated on day 13 when tumor size reached approximately 100 mm3. Vehicle control and individual or mixtures of mAbs BHA10 and 14A2 were administered by ip injection 2×/week throughout the study. BsAbs were administered by ip injection 1 ×/week throughout the study. Tumor growth curves are each derived from cohorts consisting of ten mice. Data is represented as mean tumor volume ± S.D.
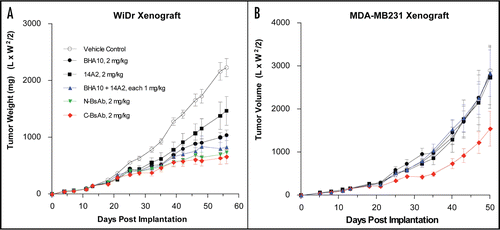
Table 1 Thermodynamic properties of BHA10 scFv and FAb proteins
Table 2 Recombinant IgG-like BsAbs
Table 3 Pharmacokinetic parameters
Acknowledgements
We would like to acknowledge the following people for providing humanized BHA10 and chimeric 14A2 plasmids: Kenneth Simon, Amie Carmillo, Paul Lyne and Jose Saldanha. We thank Dr. Mitchell Reff for critical review of the manuscript. We also thank members of the Cell Culture Development department and Mike Favis for scale-up cell culture, Ina Hughes for expertise in molecular biology, Gregg Hetu and Ellen Rohde for PK analysis, Rebecca Kelly and Kathleen Wortham for Oncopharmacology support, Irene Sizing for the 14A2 mAb, Jurg Tschopp and Pascal Schneider for recombinant TRAIL-R2 and Cheryl Wilson and Apinya Ngam for the early cytotoxicity assays.
Note
Supplementary materials can be found at: www.landesbioscience.com/supplement/MichaelsonMABS1-2-Sup.pdf
References
- Fischer N, Leger O. Bispecific antibodies: molecules that enable novel therapeutic strategies. Pathobiology 2007; 74:3 - 14
- Boerman OC, van Schaijk FG, Oyen WJ, Corstens FH. Pretargeted radioimmunotherapy of cancer: progress step by step. J Nucl Med 2003; 44:400 - 411
- Jain M, Kamal N, Batra SK. Engineering antibodies for clinical applications. Trends Biotechnol 2007; 25:307 - 316
- Kontermann RE. Recombinant bispecific antibodies for cancer therapy. Acta Pharmacol Sin 2005; 26:1 - 9
- Marvin JS, Zhu Z. Recombinant approaches to IgG-like bispecific antibodies. Acta Pharmacol Sin 2005; 26:649 - 658
- Worn A, Pluckthun A. Stability engineering of antibody single-chain Fv fragments. J Mol Biol 2001; 305:989 - 1010
- Ewert S, Huber T, Honegger A, Pluckthun A. Biophysical properties of human antibody variable domains. J Mol Biol 2003; 325:531 - 553
- Ewert S, Honegger A, Pluckthun A. Stability improvement of antibodies for extracellular and intracellular applications: CDR grafting to stable frameworks and structure-based framework engineering. Methods Enzymol 2004; 34:184 - 199
- Brockmann EC, Cooper M, Stromsten N, Vehniainen M, Saviranta P. Selecting for antibody scFv fragments with improved stability using phage display with denaturation under reducing conditions. J Immunol Methods 2005; 296:159 - 170
- Reiter Y, Brinkmann U, Lee B, Pastan I. Engineering antibody Fv fragments for cancer detection and therapy: disulfide-stabilized Fv fragments. Nat Biotechnol 1996; 14:1239 - 1245
- Buchsbaum DJ, Zhou T, Lobuglio AF. TRAIL receptor-targeted therapy. Future Oncol 2006; 2:493 - 508
- Lukashev M, LePage D, Wilson C, Bailly V, Garber E, Lukashin A, et al. Targeting the lymphotoxin-beta receptor with agonist antibodies as a potential cancer therapy. Cancer Res 2006; 66:9617 - 9624
- Rothlisberger D, Honegger A, Pluckthun A. Domain interactions in the Fab fragment: a comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. J Mol Biol 2005; 347:773 - 789
- Desplancq D, King DJ, Lawson AD, Mountain A. Multimerization behaviour of single chain Fv variants for the tumour-binding antibody B72.3. Protein Eng 1994; 7:1027 - 1033
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nature Rev Immunol 2007; 7:715 - 725
- Coloma MJ, Morrison SL. Design and production of novel tetravalent bispecific antibodies. Nat Biotechnol 1997; 15:159 - 163
- Lu D, Zhang H, Koo H, Tonra J, Balderes P, Prewett M, et al. A fully human recombinant IgG-like bispecific antibody to both the epidermal growth factor receptor and the insulin-like growth factor receptor for enhanced antitumor activity. J Biol Chem 2005; 280:19665 - 19672
- Merchant AM, Zhu Z, Yuan JQ, Goddard A, Adams CW, Presta LG, Carter P. An efficient route to human bispecific IgG. Nat Biotechnol 1998; 16:677 - 681
- Shen J, Vil MD, Jimenez X, Zhang H, Iacolina M, Mangalampalli V, et al. Single variable domain antibody as a versatile building block for the construction of IgG-like bispecific antibodies. J Immunol Methods 2007; 318:65 - 74
- Asano R, Watanabe Y, Kawaguchi H, Fukazawa H, Nakanishi T, Umetsu M, et al. Highly effective recombinant format of a humanized IgG-like bispecific antibody for cancer immunotherapy with retargeting of lymphocytes to tumor cells. J Biol Chem 2007; 282:27659 - 27665
- Wu C, Ying H, Grinnell C, Bryant S, Miller R, Clabbers A, Bose S, et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat Biotechnol 2007; 25:1290 - 1297
- Glockshuber R, Malia M, Pfitzinger I, Pluckthun A. A comparison of strategies to stabilize immunoglobulin Fv-fragments. Biochemistry 1990; 29:1362 - 1367
- Young NM, MacKenzie CR, Narang SA, Oomen RP, Baenziger JE. Thermal stabilization of a single-chain Fv antibody fragment by introduction of a disulphide bond. FEBS Lett 1995; 377:135 - 139
- Jung S, Honegger A, Plückthun A. Selection for improved protein stability by phage display. J Mol Biol 1999; 294:163 - 180
- Krauss J, Arndt MA, Martin AC, Liu H, Rybak SM. Specificity grafting of human antibody frameworks selected from a phage display library: generation of a highly stable humanized anti-CD22 single-chain Fv fragment. Protein Eng 2003; 16:753 - 759
- Holler N, Tardivel A, Kovacsovics-Bankowski M, Hertig S, Gaide O, et al. Two adjacent trimeric Fas ligands are required for Fas signaling and formation of a death-inducing signaling complex. Mol and Cell Biol 2003; 23:1428 - 1440
- Gajate C, Mollinedo F. Edelfosine and perifosine induce selective apoptosis in multiple myeloma by recruitment of death receptors and downstream signaling molecules into lipid rafts. Blood 2007; 109:711 - 719
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 1993; 234:779 - 815
- Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, Friesner RA. A hierarchical approach to all-atom protein loop prediction. Proteins 2004; 55:351 - 367
- Canutescu AA, Shelenkov AA, Dunbrack RL Jr. A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci 2003; 12:2001 - 2014
- Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy, minimization and dynamics calculations. J Comput Chem 1983; 4:187 - 217
- Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. Sequences of Proteins of Immunological Interest 1991; Washington DC, USA US Department of Health and Human Services
- Mage MG. Preparation of Fab fragments from IgGs of different animal species. Methods Enzymol 1980; 70:142 - 150
- Eldredge J, Berkowitz S, Corin AF, Day ES, Hayes D, Meier W, Strauch K, Zafari M, Tadi M, Farrington GK. Stoichiometry of LTbetaR binding to LIGHT. Biochemistry 2006; 45:10117 - 10128
- Yang J, Moyana T, MacKenzie S, Xia Q, Xiang J. One hundred seventy-fold increase in excretion of an FV fragment-tumor necrosis factor alpha fusion protein (sFV/TNFalpha) from Escherichia coli caused by the synergistic effects of glycine and triton X-100. Appl Enviro Microbiol 1998; 64:2869 - 2874
- Barnett RS, Limoli KL, Huynh TB, Ople EA, Reff ME. Antibody Expression and Engineering 1995; New York NY Oxford University Press
- Brezinsky SC, Chiang GG, Szilvasi A, Mohan S, Shapiro RI, MacLean A, et al. A simple method for enriching populations of transfected CHO cells for cells of higher specific productivity. J Immunol Methods 2003; 277:141 - 155
- Kim JK, Tsen MF, Ghetie V, Ward ES. Identifying amino acid residues that influence plasma clearance of murine IgG1 fragments by site-directed mutagenesis. Eur J Immunol 1994; 24:542 - 548
- Chicheportiche Y, Bourdon PR, Xu H, Hsu YM, Scott H, Hession C, et al. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J Biol Chem 1997; 272:32401 - 32410